

Abstract
MT1-MMP (membrane type 1 matrix metalloproteinase, or MMP-14) is a key enzyme in molecular carcinogenesis, tumour-cell growth, invasion and angiogenesis. Novel and potent MMP inhibitors with a mercaptosulphide zinc-binding functionality have been designed and synthesized, and tested against human MT1-MMP and other MMPs. Binding to the MT1-MMP active site was verified by the competitive-inhibition mechanism and stereochemical requirements. MT1-MMP preferred deep P1′ substituents, such as homophenylalanine instead of phenylalanine. Novel inhibitors with a non-prime phthalimido substituent had Ki values in the low-nanomolar range; the most potent of these inhibitors was tested and found to be stable against air-oxidation in calf serum for at least 2 days. To illustrate the molecular interactions of the inhibitor–enzyme complex, theoretical docking of the inhibitors into the active site of MT1-MMP and molecular minimization of the complex were performed. In addition to maintaining the substrate-specificity pocket (S1′ site) van der Waals interactions, the P1′ position side chain may be critical for the peptide-backbone hydrogen-bonding network. To test the inhibition of cell-mediated substrate cleavage, two human cancer-cell culture models were used. Two of the most potent inhibitors tested reached the target enzyme and effectively inhibited activation of proMMP-2 by endogenous MT1-MMP produced by HT1080 human fibrosarcoma cells, and blocked fibronectin degradation by prostate cancer LNCaP cells stably transfected with MT1-MMP. These results provide a model for mercaptosulphide inhibitor binding to MT1-MMP that may aid in the design of more potent and selective inhibitors for MT1-MMP.
Keywords: cell-mediated substrate cleavage, enzyme inhibition kinetics, inhibitor–enzyme active-site interaction, matrix metalloproteinase (MMP), molecular modelling, novel synthetic inhibitor
Abbreviations: cdMT1-MMP, catalytic domain of membrane type 1 matrix metalloproteinase; ConA, concanavalin A; Dpa, N-3-(2,4-dinitrophenyl)-L-2,3-diaminopropionyl; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); ECM, extracellular matrix; FN, fibronectin; GFP, green fluorescent protein; GPI, glycosylphosphatidylinositol; Mca, (7-methoxycoumarin-4-yl)acetyl-; MMP, matrix metalloproteinase; MMPI, MMP inhibitor; MT1-MMP, membrane type 1 MMP; MT1–GFP, MT1-MMP complexed to GFP; proMMP-2, progelatinase A; TCEP, tris-(2-carboxyethyl)phosphine; TIMP-2, tissue inhibitor of metalloproteinases-2
INTRODUCTION
MMPs (matrix metalloproteinases) are a family of zinc endo-peptidases that play prominent roles during normal and pathological ECM (extracellular matrix) remodelling events, including cancer progression [1]. MT-MMPs (membrane type-MMPs) are a unique subset tethered to the cell membrane by a transmembrane domain or GPI (glycosylphosphatidylinositol) anchor [2]. The first identified MT-MMP, MT1-MMP, has been shown to play a central role in tumour-cell invasion and migration [3–6]. It hydrolyses constituents of the ECM directly [7–9] and activates proMMP-2 (progelatinase A) efficiently; activated MMP-2 cleaves the basement membrane type IV collagen [10–13]. Because MT1-MMP is highly localized at the leading edge of invading cancer cells and is a powerful proteolytic enzyme, it may be a good target for inhibition [14].
Synthetic MMPIs (MMP inhibitors) have been in development for more than a decade. Potent MMPIs have three general requirements: a functional group capable of co-ordinating to the enzyme active site Zn(II), at least one functional group participating in hydrogen bonding with the enzyme, and one or more side chains participating in favourable van der Waals interactions, particularly at the substrate specificity pocket (S1′ site) [15]. The most potent inhibitors reported have used a hydroxamic acid zinc-chelating group. Although this functionality affords good MMPI potency, many reports suggest the need for other functional groups owing to problems with oral bioavailability and toxicity [16–18]. Other MMPI functional groups include thiol, carboxylate, phosphinate and sulphodi-imine. Mercaptosulphide inhibitors that have been developed and characterized for several MMPs exhibit IC50 and Ki values comparable with those of well-known hydroxamic acids [19–23]. Mercaptosulphide peptidomimetic inhibitors are distinct from the hydroxamate peptidomimetics; they have different zinc-co-ordinating functionality, and the favoured stereochemistry at the P1′ site is an unnatural D-amino-acid. In the present study, the inhibition characteristics of MT1-MMP with novel mercaptosulphide inhibitors were determined, and a model of the mercaptosulphide inhibitor complexed to MT1-MMP is proposed to provide a rational approach towards inhibitor design. Furthermore, the efficacy of potent new MMPIs was tested in live cells, using two different models of substrate cleavage mediated by human cancer cells expressing endogenous and transfected functional MT1-MMP respectively.
EXPERIMENTAL
Materials
The fluorescence-quenched peptide substrate, Mca [(7-methoxycoumarin-4-yl)acetyl]-Pro-Leu-Gly-Leu-Dpa [N-3-(2,4-dinitrophenyl)-L-2,3-diaminopropionyl]-Ala-Arg-NH2, was purchased from Bachem. The mercaptosulphide inhibitors were synthesized and characterized as described previously [19,21,23,24]. The recombinant cdMT1-MMP (catalytic domain of MT1-MMP) was provided by Professor Harald Tschesche (Bielefeld University, Eastern Westphalia, Germany) [22,25]. MMP-1, -2, -3, -7 and -9 were described previously [21]. FITC was purchased from Pierce Biotechnology, and human FN (fibronectin) was from Invitrogen. All standard chemicals were purchased from Fisher Scientific with the exception of Ellman's reagent/DTNB [5,5′-dithiobis-(2-nitrobenzoic acid)] and ConA (concanavalin A), which were from Sigma, and TCEP [tris-(2-carboxyethyl)-phosphine], which was from Calbiochem. Human fibrosarcoma cell line HT1080 and human prostate cancer cell line LNCaP were purchased from ATCC (Manassas, VA, U.S.A.) and were maintained in Dulbecco's modified Eagle's medium (Invitrogen).
Inhibitor-concentration determination
The reduced mercaptosulphide inhibitor concentration was determined with DTNB [20,26]. Absorbance was measured with a Shimadzu UV-260 UV–visible spectrophotometer with a 2 nm slit-width. Alternatively, for inhibitors with a phthalimido substituent, the total concentration of inhibitor was determined by absorbance at 219 nm. The molar absorption coefficient, ϵ219, was determined for a standard, N-phthaloyl-L-phenylalanine, by measuring absorbance for concentrations up to 40 μM of four separate stock solutions. The ϵ219 was determined to be (3.30±0.08)×104 M−1·cm−1.
Enzyme kinetics
Kinetic assays were performed with the peptide substrate Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (1 μM) as described previously [27,28]. The assays were performed in 50 mM Hepes buffer, pH 7.5, with 10 mM CaCl2, 0.2 M NaCl and 0.05% (w/v) Brij-35 (polyoxyethylene dodecyl ether) at 25 °C. Active cdMT1-MMP concentrations were determined by titration with standardized preparations of recombinant TIMP-2 (tissue inhibitor of metalloproteinases-2) [22]. For the inhibition assays, the inhibitors were incubated with enzyme for 15–30 min before adding substrate, to ensure equilibrium conditions. For those inhibitors with a phthalimido substituent, 1 mM TCEP (a reducing agent) was included to ensure complete reduction of the inhibitor to the free thiol. No effect on enzyme activity was noted for concentrations of TCEP up to 1 mM. The inhibitor dissociation constant, Ki, was determined as described previously [29] using the Morrison equation [30]:
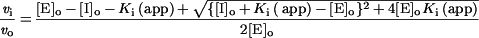 |
(1) |
where vi and vo are the initial rates with and without inhibitor respectively, [E]o and [I]o are the initial enzyme and inhibitor concentrations respectively, and Ki (app) is the apparent Ki, which is equal to the true Ki under the conditions of substrate concentration ([S])<<Km (the Michaelis constant) for competitive inhibition according to eqn (2):
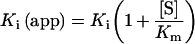 |
(2) |
Inhibitor stability
The stability of the thiol was determined using the inhibition assay to quantify the concentration of active inhibitor. Selected inhibitors were incubated in calf serum at 37 °C. Aliquots were removed periodically, diluted with the assay buffer (described above) to 25% serum, and the inhibition was tested against cdMT1-MMP. Control assays containing the same amounts of serum with no inhibitor were performed to obtain relative rates.
Modelling studies
The co-ordinates from the X-ray crystal structure of the cdMT1-MMP–TIMP-2 complex were downloaded from the Brookhaven Protein Data Bank (accession number 1BUV) [31]. MacroModel version 7.0 was used to remove the TIMP-2 molecule, and the inhibitors were theoretically docked into the active site of cdMT1-MMP by the following procedure: the MMP-1–hydroxamic acid inhibitor complex (PDB accession number 1HFC) [32] was modified at the inhibitor zinc-co-ordinating group to a mercaptosulphide functionality; the modified structure was energy-minimized according to procedures described below; the imidazolyl nitrogen atoms co-ordinated to zinc of the minimized structure were superimposed with those of cdMT1-MMP; and MMP-1 was deleted to leave the inhibitor bound in the cdMT1-MMP active site. Both sulphurs of the mercaptosulphide functionality were co-ordinated to zinc with zero-order bonds. The rationale for this zinc chelation was based primarily on earlier studies of the inhibition of MMP-1, -2, -3, -7, -8 and -9 with a mercaptosulphide inhibitor modified with an oxygen in place of the sulphide, which significantly reduced inhibitor potency. The mercaptosulphide inhibitor had IC50 values of 52, 1.4, 250, 33, 3.6 and 1.2 nM respectively, and the analogous inhibitor with oxygen had IC50 values of 1700, 330, 29000, 4800, 910 and 260 nM respectively (M. A. Schwartz, Y. Jin, D. R. Hurst and Q.-X. Sang, unpublished work).
Energy minimizations of the inhibitor and all enzyme residues within 7 Å (1 Å=0.1 nm) were performed with MacroModel version 7.0. Briefly, the AMBER force field [33,34], modified to include parameters for Zn and Ca ([35–37]; M. A. Schwartz, Y. Jin, D. R. Hurst and Q.-X. Sang, unpublished work), was used with the PRCG (Polak–Ribiere conjugate gradient) method with a 0.05 gradient threshold in a GB/SA (generalized Born solvent/accessible surface area) continuum water solvent. A global minimization of the cdMT1-MMP–inhibitor complex was carried out using the MCMM (Monte Carlo multiple minimum) method (1000 steps), in which the inhibitor torsion angles at all relevant bonds were randomly varied and then minimized.
Gelatin substrate zymography and densitometry analyses of the bands
The procedures were reported in our previous papers [38,39]. In brief, HT1080 cells were pre-treated with different doses of the novel MMPIs in serum-free medium at 37 °C for 1 h, followed by stimulation with ConA (20 μg/ml) at 37 °C for 18 h. The spent conditioned media were collected and examined by gelatin zymography. The effect of the novel MMPI on proMMP-2 activation by MT1-MMP was documented by densitometry analysis for 62 kDa fully active MMP-2 using AlphaImager™ 2200 with Alpha-EaseFC software (AlphaInnotech).
FITC–FN degradation assay
To observe cell-mediated protein-substrate degradation and cell migration, FN was conjugated to FITC in borate-buffered saline (pH 10) for 30 min at room temperature (approx. 23 °C). After extensive dialysis against PBS, FITC-labelled FN was coated on to glass coverslips by incubation with glutaraldehyde-cross-linked gelatin for 1 h at room temperature [39]. Human prostate cancer cell line LNCaP cells stably transfected with MT1–GFP (MT1-MMP–green fluorescent protein) cDNA construct were generated as described previously [39]. LNCaP cells expressing the MT1–GFP chimaera were plated onto FITC–FN coverslips and incubated with vehicle control (DMSO), TIMP-2, YHJ-132 or YHJ-294-2 at different concentrations in serum-free medium at 37 °C for 18 h. The cells were then fixed with 4% (w/v) para-formaldehyde in PBS and examined under a fluorescent microscope (Olympus, IX-70). The substrate degradation was demonstrated by loss of fluorescence of FITC-labelled substrate and cell migration was determined by observing the tracks of digested substrate on the same slide. MT1–GFP-expressing cells digested FN and migrated over digested substrate. The dark lines on the green FITC–FN background indicate substrate degradation and cell migration. The cells are also green because of the GFP expression together with MT1-MMP.
RESULTS
Mechanism of interaction
The inhibitors (Table 1) were designed with a zinc-co-ordinating functionality and would be predicted to bind in the active site groove of MMPs based on structures of similar hydroxamate inhibitors complexed with MMP-1. However, detailed inhibition mechanisms have not been determined experimentally, and alternative modes of inhibition must be considered. To verify a competitive mechanism of inhibition, a prototypical inhibitor, MAG-182, was assayed with cdMT1-MMP at several substrate concentrations. A linear form of the Morrison equation derived by Henderson [40] (eqn 3) was applied:
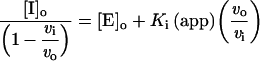 |
(3) |
where [I]o and [E]o are the total inhibitor and enzyme concentrations, and vi and vo are the rates with and without inhibitor respectively. The Morrison plot and Henderson plot are shown in Figure 1. The linear increase in Ki(app) with increasing substrate concentration is indicative of competitive inhibition, as shown by eqn (2). Additionally, a well-known ACE (angiotensin- converting enzyme) inhibitor, captopril {N-[(S)-3-mercapto-2-methylpropionyl]-L-proline}, which is also a thiol compound, was used as a negative control and found to have a Ki value of 4 mM. Therefore the mercaptosulphide inhibitors are interacting exclusively at the enzyme active site.
Table 1. Inhibition constants for inhibition of MT1-MMP and other MMPs by mercaptosulphide inhibitors.
Stereochemistry (*) for the Cα to SH is always listed first. Data were reported in a [22] and b [21]. iBu, isobutyl; PhMe, phenylmethyl; PhEt, phenethyl; PhtN, phthalimido; PhtNMe, phthalimidomethyl; PhtNBu, phthalimidobutyl; PhtNPr, phthalimidopropyl; PhtNPe, phthalimidopentyl; PhtNEt, phthalimidoethyl; PhtNEtNH, phthalimidoethylimino; PMP, paramethoxyphenyl.
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki (nM) | ||||||||||||
| Compound | Identity | * | R | R1 | R2 | R3 | MT1-MMP | MMP-1 | MMP-2 | MMP-3 | MMP-7 | MMP-9 |
| 1a | MAG-42 | R | H | iBu | 27 | 52 | 0.28 | 250 | 56 | 0.43 | ||
| 1b | MAG-58 | S | H | iBu | 160 | 400 | 34 | 3600 | 50 | 17 | ||
| 1c | MAG-148 | R | H | PhMe | 10000 | 12000 | 1400 | 83000 | 7300 | 1500 | ||
| 1d | MAG-133 | S,R | Me | iBu | 18a | 13 | 0.35 | 46 | 15 | 0.21 | ||
| 1e | MAG-128 | R,R | Me | iBu | 70a | 200 | 110 | 400 | 45 | 18 | ||
| 1f | MAG-292 | S,R | PhtNEt | iBu | 4.5 | 0.95 | 0.77 | 22 | 15 | 0.09 | ||
| 1g | MAG-254 | S,R | PhtNBu | iBu | 5.5 | 20 | 0.2 | 10 | 13 | 0.11 | ||
| 2a | MAG-182 | S,R | iBu | PhMe | Me | 24a,b | 49b | 1.1b | 470b | 40b | 0.57b | |
| 2b | MAG-181 | R,S | iBu | PhMe | Me | 260a,b | 680b | 85b | 2500b | 710b | 44b | |
| 2c | YHJ-73 | S,R | PhEt | iBu | PMP | 16b | >12000b | 20b | 100b | 1000b | 8.6b | |
| 2d | YHJ-72 | R,S | PhEt | iBu | PMP | 380b | >12000b | 930b | 150b | 5500b | 180b | |
| 3a | YHJ-294-2 | S,R | NH2 | iBu | PhMe | Me | 13a,b | 100b | 6.1b | 360b | 26b | 1.2b |
| 3b | YHJ-294-1 | R,S | NH2 | iBu | PhMe | Me | 3000a,b | 5200b | 430b | >40000b | 3500b | 550b |
| 3c | YHJ-265 | 1:1 | Me | iBu | PhMe | Me | 6.2 | 99 | 14 | 990 | 91 | 5.7 |
| 3d | YHJ-96 | 1:1 | PhtNMe | iBu | PhMe | Me | 70 | 110 | 17 | 300 | 50 | 4.9 |
| 3e | YHJ-97 | 1:1 | PhtNPr | iBu | PhMe | Me | 6 | 75 | 8.5 | 31 | 12 | 3.9 |
| 3f | YHJ-44 | 1:1 | PhtNPe | iBu | PhMe | Me | 7 | 52 | 1.7 | 1.9 | 11 | 0.98 |
| 3g | YHJ-223 | 1:1 | PhtNEtNH | PhEt | PhMe | Me | 3.7 | 190 | 1.8 | 13 | 250 | 0.35 |
| 3h | YHJ-224 | 1:1 | PhtNEtNH | PhEt | iBu | PMP | 3.1 | >3000 | 31 | 1.5 | 76 | 4.6 |
| 3i | YHJ-132 | S,R | PhtNEtNH | iBu | PhMe | Me | 1.2 | 8.8 | 0.7 | 6 | 6.5 | 1.1 |
| 3j | YHJ-133 | R,S | PhtNEtNH | iBu | PhMe | Me | 430 | 1900 | 90 | 1100 | 670 | 122 |
Figure 1. Competitive inhibition mechanism.

(A) A typical inhibition curve fitted with the Morrison equation (eqn 1) is shown for MAG-182. (B) The inhibition assay was also performed with substrate concentrations of 1, 10 and 24 μM and plotted according to the Henderson equation (eqn 3), described in the text. (C) A linear increase in the slope, Ki (app), is indicative of competitive inhibition, according to eqn (2).
The competitive mechanism of inhibition does not distinguish between primed and unprimed binding. Members of the MMP family are known to have either a deep or a shallow S1′ pocket that is easily probed by modifying the P1′ substituent. Shallow-pocket MMPs, MMP-1 and -7, have Ki values that are an order of magnitude higher compared with deep-pocket MMPs, such as MT1-MMP, for inhibitors with an extended-chain P1′ substituent. The inhibition by MAG-182 and YHJ-73 is compared with MMP-1 and -7 and MT1-MMP in Table 1. Respective Ki values for MMP-1 are 49 nM and 12 μM; for MMP-7, 40 nM and 1 μM; and for MT1-MMP, 24 nM and 16 nM [21]. The significant decrease in inhibition potency with a homophenylalanine side chain, compared with leucine, against MMP-1 and -7 demonstrates that the P1′ substituent is interacting at the S1′ pocket.
To ensure that the binding interactions are specific, the stereochemical dependence of inhibition was probed. Variation in the stereochemistry of the inhibitor near the zinc-co-ordinating functionality and at the P1′ side chain led to variation in inhibition. The Cα to SH favours the absolute S configuration (compare MAG-133 with MAG-128), and the derivatives containing a cyclopentyl or pyrrolidinyl ring significantly favour the S,R configuration (Cα, Cβ to SH respectively; compare MAG-182 with MAG-181, YHJ-73 with YHJ-72, and YHJ-294-2 with YHJ-294-1). The stereochemical requirements at the P1′ site favour the absolute R configuration corresponding to the unnatural D-amino-acid derivative (compare MAG-42 with MAG-58). These results confirm that stereochemistry is important for inhibitor potency and provide additional support for the inhibitor interacting specifically and exclusively at the active site of the enzyme.
Probing the non-prime site
From the crystal structure of cdMT1-MMP, the non-prime site does not have well-defined pockets comparable with S1′. However, it was found that a phthalimido substituent (Figure 2) added to the Cα to SH of MAG-42 (MAG-292) increased the potency by a factor of five (Table 1). Compared with a methyl group (MAG-133), the potency is increased by a factor of four. To probe the possibility of a specific interaction with cdMT1-MMP, the tether length attaching the phthalimido substituent was increased by two carbons (MAG-254). No change was noted in the Ki value, suggesting that the increase in potency is because of non-specific hydrophobic interactions.
Figure 2. Novel mercaptosulphide inhibitors containing a phthalimido substituent.

YHJ-130 is the 1:1 mixture of YHJ-132 and YHJ-133, as shown in Table 1.
A new series of mercaptosulphide inhibitors that incorporate a pyrrolidine ring at the zinc-co-ordinating functionality was designed to increase water-solubility and possibly provide structural features that would allow for greater selectivity of inhibition (Table 1, compound 3) [21,23]. These derivatives displayed identical stereochemical requirements for potency with the previously designed mercaptosulphides (compounds 1 and 2). A phthalimido substituent was also added, with varying tether lengths, to the N-acetyl of these compounds, with no change in potency (YHJ-265, -97 and -44). However, a 10-fold increase in Ki was noted for the N-phthalimidomethylacetyl (YHJ-96). This suggests that YHJ-96 is sterically hindered by the short attachment of the phthalimido substituent, or possibly that π–π interactions between the phthalimido and an aromatic residue of the enzyme affect inhibitor binding. Incorporating a urea group (YHJ-132, -133, -223 and -224) did not affect the Ki, although the water-solubility would be significantly increased.
The mercaptosulphide inhibitors may be readily air-oxidized in buffer and typically lose their effectiveness as MMPIs over time. Stability experiments with the non-cyclic mercaptosulphide, MAG-42, in buffer showed complete loss of inhibitor activity within 45 min at 25 °C. Incorporation of the cyclopentyl ring at the mercaptosulphide functionality, MAG-182, increased inhibitor stability by approx. 6 h. Pyrrolidinyl derivatives of these inhibitors have improved water-solubility and may be candidates for in vitro cell and in vivo animal studies (M. A. Schwartz, Y. Jin, D. R. Hurst and Q.-X. Sang, unpublished work). Moreover, the phthalimidoethylurea derivative, YHJ-132, which is the most potent of these inhibitors towards cdMT1-MMP, was stable for at least 2 days in calf serum at 37 °C (M. A. Schwartz, Y. Jin, D. R. Hurst and Q.-X. Sang, unpublished work). The simple urea derivative, YHJ-294-2, was stable for approx. 4–6 h. MAG-42 and MAG-182 were tested in calf serum and were found to have stability similar to that found in the buffer experiments.
Proposed model
These mercaptosulphide inhibitors appear to be structurally similar to some well-characterized hydroxamate peptidomimetic MMPIs and might be predicted to interact correspondingly; however, the mercaptosulphides are distinct from the hydroxamate peptidomimetics with regard to the zinc-co-ordinating functionality and the favoured unnatural D-amino-acid stereochemistry at the P1′ site. Molecular modelling was employed for structural analysis of the binding of the mercaptosulphide inhibitors into the cdMT1-MMP active site. The enzyme–inhibitor complexes were globally minimized so that interactions important for inhibitor potency could be identified. Rational explanations for the disparity between Ki values of structurally similar inhibitors were sought and identified.
The three general requirements for MMPI potency are achieved with mercaptosulphide inhibitors: a zinc-binding functionality, multiple hydrogen bonds and favourable van der Waals interactions with the S1′ pocket. The cdMT1-MMP complexed with the inhibitor MAG-182 is illustrated in Figure 3. The inhibitor interacts with the primed side of the enzyme active site, with the P1′ leucine side chain oriented with one terminal methyl in line with the S1′ pocket. Four hydrogen bonds between the peptide backbone of the mercaptosulphide inhibitors and the enzyme active site were identified. The P1′ carbonyl hydrogen bonds with the NH of Leu199, the P2′ NH with the carbonyl of Pro259, the P2′ carbonyl with the NH of Tyr261 and the terminal NH with the carbonyl of Gly197. Importantly, the Phe198 between the hydrogen-bonding residues Gly197 and Leu199 is co-ordinated to a structurally important calcium ion [31]. This severely limits the flexibility of these residues.
Figure 3. A model for inhibitor binding.

MAG-182 is shown with key enzyme residues that are within hydrogen-bonding distance. The side chains of the inhibitor maintain important van der Waals interactions with the S1′ and S2′ pockets.
Substitution at the P1′ site demonstrated the largest difference in Ki values. Even though a phenylalanine side chain would be predicted to show potency similar to that of a leucine side chain based on previous substrate studies [41], comparison of the inhibition by two such inhibitors (MAG-42 and -148) actually showed more than a 350-fold difference in Ki. These two inhibitors complexed with cdMT1-MMP were superimposed (Figure 4A). The inhibitors overlay quite well, with the exception of the P1′ carbonyl, which is a key hydrogen-bonding group with the NH of Leu199. The N to O distance is 3.2 and 3.6 Å for MAG-42 and -148 respectively. The disruption of this hydrogen bond is owing to the restricted orientation of the phenylalanine side chain by the well-defined S1′ pocket, along with the restricted flexibility of Leu199 due to co-ordination of the adjacent residue, Phe198, to a structural calcium ion. The respective inhibitor with a homophenylalanine side chain superimposed with MAG-42 maintains all of the necessary hydrogen-bonding interactions (Figure 4B). The C1 phenyl ring atom is located in the same position as the terminal methyl carbon that is pointing in the direction of the S1′ pocket. Therefore there is no distortion of the inhibitor backbone, and all necessary interactions are maintained. The distortion of the backbone in MAG-148 is a result of the steric restriction of the phenyl ring in the S1′ pocket. Disruption of one hydrogen bond, with all other interactions being the same, could account for a free energy change of the order of 8–21 kJ/mol. The free energy is related to Ki by eqn (4):
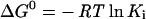 |
(4) |
and a 10-fold difference in Ki corresponds to a free energy change of 5.69 kJ/mol, 100-fold corresponds to 11.38 kJ/mol, and 1000-fold corresponds to 17.07 kJ/mol. A reasonable explanation for the 350-fold difference in Ki between MAG-42 and -148 is disruption of a single hydrogen bond. This is the first report to provide a rationale for the large disparity between these side chains in terms of either substrate specificity or inhibition studies.
Figure 4. Modification of the P1′ substituent.

The superimposed complexes of cdMT1-MMP with inhibitors modified at the P1′ site reveal reasons for the significant Ki differences. The inhibitors are shown as black or grey sticks, the enzyme residues as wire, and the catalytic zinc is black CPK (Corey–Pauling–Koltun). (A) MAG-42 (black) and -148 (grey) are superimposed. A key hydrogen bond with Leu199 and MAG-42 has an N to O distance of 3.2 Å. This hydrogen bond is not achieved by MAG-148, with an N to O distance of 3.6 Å. (B) MAG-42 (black) and an inhibitor modified with a phenethyl P1′ substituent (grey) are superimposed. With this P1′ substituent, all interactions important for inhibitor potency are conserved.
The 10-fold difference in Ki for YHJ-96 and -97 was also very intriguing. These inhibitors differ only in the tether length for the attachment of the phthalimido substituent at the non-prime site. Modelling of YHJ-97 showed an edge-to-face aromatic–aromatic interaction between the phthalimido ring and Phe260 that was not identified for YHJ-96 (Figure 5). The YHJ-97 phthalimido ring is oriented towards the centre of the phenyl ring of Phe260, with a distance of 5.5 Å between the two phenyl ring centroids. The preferential distance between phenyl ring centroids is 4.5–7 Å for this type of interaction and may contribute up to 8.4 kJ/mol of free energy [42], which correlates well with the 10-fold difference in Ki between these inhibitors.
Figure 5. Edge-to-face aromatic–aromatic interaction.

(A) The model of inhibitor YHJ-97 (purple stick) is shown complexed with cdMT1-MMP. The phenyl ring centroid-to-centroid distance between the phthalimido and Phe260 is 5.5 Å. Zinc is shown as magenta CPK (Corey–Pauling–Koltun) with the three co-ordinating histidines as blue wire. (B) The model of inhibitor YHJ-96 (beige stick) is shown complexed with cdMT1-MMP. The phthalimido substituent does not interact with any residues of cdMT1-MMP.
Inhibition by MMPIs of MT1-MMP-mediated substrate cleavage in cells
Two potent broad-spectrum and relatively stable novel MMPIs, YHJ-132 and YHJ-294-2, have been tested in two different human cancer-cell culture systems to examine their inhibition efficacy in live cells. The first model is the human fibrosarcoma HT1080 cell culture system. HT1080 cells produce endogenous active MT1-MMP upon treatment with 20 μg/ml ConA. The endogenous MT1-MMP can cleave and activate endogenous proMMP-2 to form the 64 kDa intermediate. This activation was blocked by these two inhibitors in a dose-dependent manner, as shown by the accumulation of proMMP-2 zymogen (Figure 6). The dynamic concentration changes of the 64 kDa intermediate were contributed by both MT1-MMP, which converted 72 kDa zymogen into the 64 kDa intermediate, and self-catalysed intermolecular cleavage of the 64 kDa enzyme to the 62 kDa active MMP-2. At an inhibitor concentration of 200 nM, the generation of the 64 kDa intermediate was partially blocked, and that of the 62 kDa active MMP-2 was almost completely blocked by these two inhibitors. Normally, the 64 kDa intermediate is quickly converted into the 62 kDa active form; at 100 nM of each of these two inhibitors, this conversion was effectively blocked, but the inhibition of the conversion of the 72 kDa zymogen into the 64 kDa species was incomplete (Figure 6). From these results, it appears that the 64 kDa form is ‘stabilized’ by these inhibitors, indicating that these two inhibitors are less effective on MT1-MMP than on MMP-2, which is consistent with the enzyme-inhibition parameters, demonstrating that these two inhibitors are slightly less potent against MT1-MMP than MMP-2 (Table 1). The Ki values for YHJ-132, YHJ-294-2 and some other mercaptosulphide inhibitors against MMP-1, 2, -3, -7, -9 and MT1-MMP are shown in Table 1.
Figure 6. Dose-dependent inhibition of proMMP-2 activation by two novel MMPIs in a cell culture system as shown by a representative gelatin zymogram and densitometry analyses using AlphaImager™ 2200.

The conditioned media of HT1080 cells treated with or without different doses of the novel MMPIs plus 20 μg/ml ConA were examined by gelatin zymography. The molecular masses of latent, intermediate and fully activated MMP-2 were demonstrated as 72 kDa pro-MMP-2, 64 kDa int-MMP-2, and 62 kDa act-MMP-2 respectively. Lane 1, HT1080 control; lane 2, HT1080 with ConA; lanes 3–5, HT1080 with ConA and 10, 100 or 200 nM YHJ-132 respectively; lanes 6–8, HT1080 with ConA and 10, 100 or 200 nM YHJ-294-2 respectively.
The second model utilizes the human prostate cancer cell line LNCaP cells expressing stably transfected MT1–GFP chimaera. These cells were plated on to FITC–FN glass coverslips. YHJ-132 and YHJ-294-2 partially or completely inhibited MT1-MMP- and perhaps other MMP-induced substrate degradation at 5 μM and 10 μM, but were much less effective in blocking cell migration (Figure 7). TIMP-2, at 1 μM, completely inhibited cell-mediated substrate cleavage and partially impeded cell migration. These results are consistent with our previous report that MMPIs did not effectively stop cell migration, owing to MMP-independent migration mechanisms such as phagocytosis-mediated cell movement and migration [39]. These data demonstrate that these novel synthetic inhibitors are able to reach MT1-MMP and other MMPs produced by live cells and block their substrate digestion.
Figure 7. Inhibition of cell-mediated substrate degradation by two novel MMPIs.

LNCaP cells stably transfected with the MT1–GFP chimaera were plated on to FITC–FN coverslips and incubated with vehicle control (DMSO), TIMP-2, YHJ-132 or YHJ-294-2 at different concentrations in serum-free medium at 37 °C for 18 h. The cells were then fixed and examined under fluorescence microscopy. The substrate degradation was demonstrated by loss of fluorescence of FITC-labelled substrate, and cell migration was determined by observing the tracks of digested substrate on the same slide. MT1–GFP-expressing cells digested FN and migrated over digested substrate. The dark lines on the green FITC–FN background indicate substrate degradation and cell migration. The cells are also green because of the GFP expression together with MT1-MMP. The positive control, 1 μM TIMP-2, completely blocked the FN degradation and partially inhibited cell migration. New MMPIs, YHJ-132 and YHJ-294-2, partially or completely inhibited MT1-MMP-induced substrate degradation at 5 μM and 10 μM, but were much less effective in stopping cell migration.
DISCUSSION
In the present study, the inhibition profile, mechanisms and requirements for potency of mercaptosulphide inhibitors against the target enzyme, cdMT1-MMP, have been investigated. Synthetic MMPIs have been developed and characterized extensively with collagenase 1 and 2 (MMP-1 and -8), gelatinase A and B (MMP-2 and -9), stromelysin 1 (MMP-3), and matrilysin (MMP-7), but few inhibition studies have included MT1-MMP. The most potent inhibitors developed by other researchers contain a hydroxamic acid zinc-chelating group, and these compounds have been the lead agents for clinical trials. The results of some clinical trials have been disappointing for many reasons; for example, the trial design may not have been optimal and the hydroxamate inhibitors may not be specific [43–47]. There is an urgent need for developing MMPIs with novel zinc-binding groups [48–50]. Mercaptosulphide inhibitors have been developed and characterized for many MMPs and exhibit IC50 and Ki values approaching those of well-characterized hydroxamates [19–23]. The detailed mechanisms of interaction of these inhibitors with a MMP have not been previously elucidated.
The mercaptosulphide inhibitors were demonstrated to bind reversibly and competitively to MMPs. In agreement with other MMP family members, there is strong stereoselectivity at the P1′ and zinc-binding groups. This observation correlates with structural studies that indicate that MMPs have a highly conserved active site [51]. Stereochemistry at the P1′ site favours the absolute R configuration corresponding to the unnatural D-amino-acid derivative. Around the mercaptosulphide zinc-co-ordinating functionality, the S,R stereochemistry is favoured (Cα, Cβ to thiol). The non-prime site was probed with a phthalimido substituent. The inhibition potency of inhibitors containing this substituent was comparable with, and in some cases improved upon, that of similar inhibitors without the non-prime phthalimido. An exception was noted with YHJ-96, which was 10-fold less potent than YHJ-97. These inhibitors differ only in the tether length for the attachment of the phthalimido (methyl and propyl respectively). The S1′ pocket is the most well-defined pocket and is deep in MT1-MMP compared with other MMPs. This pocket may simply accommodate long hydrophobic groups at the inhibitor P1′ site and therefore a phenylalanine side-chain inhibitor should be approximately equal in potency when compared with leucine. However, MAG-148, which contains a P1′ phenylalanine side chain, was found to be less potent (more than 350-fold) than the corresponding inhibitor MAG-42 with a leucine side chain. This result could not be predicted from the descriptions posted in the literature regarding inhibitor binding. It was therefore necessary to evaluate the experimental results with molecular modelling to propose rational explanations for the requirements of inhibitor potency.
The overall inhibition by the mercaptosulphide compounds of cdMT1-MMP follows the general requirements of other MMP-family members, with the exception of YHJ-96. Although the non-prime site is not of particular importance for selectivity, the 10-fold difference in Ki between YHJ-96 and -97 is interesting. An aromatic–aromatic interaction was identified between YHJ-97 and Phe260 that was not seen with YHJ-96. This would explain the 10-fold difference in potency against cdMT1-MMP. MMP-3 was the only other MMP that showed this behaviour. It is not clear whether MMP-3 interacts with these inhibitors in a similar fashion; however, Cohen and colleagues recently reported π–π stacking interactions at the non-prime site for the inhibitor futoenone complexed with MMP-3 [52]. This inhibitor is selective for MMP-3 over other MMPs owing to a unique tyrosine residue. The cdMT1-MMP Phe260 appears to be located at the S2′ site. Therefore the phthalimido substituent in YHJ-97 may be interacting at the S2′ site and not in the non-prime site as would have been predicted. Although YHJ-97 is not selective for cdMT1-MMP compared with other MMPs, the 10-fold preference of cdMT1-MMP for YHJ-97 over YHJ-96 could be explained rationally with modelling. Extension of the tether length to five carbons, as in YHJ-44, did not affect inhibitor potency compared with YHJ-97.
The S1′ pocket has been the most-characterized site in MMPs, especially with regards to inhibition studies. Although MT1-MMP is known to have a deep S1′ pocket, the phenylalanine side-chain inhibitor, MAG-148, was more than 350-fold less potent compared with the leucine side-chain inhibitor, MAG-42. Four hydrogen bonds between the peptide backbone of mercaptosulphide inhibitors and the enzyme active site appear to be necessary for inhibition potency. Disruption of one of these bonds, with all other interactions being the same as predicated, as in the case of MAG-148, could account for a free energy change of the order of 8–21 kJ/mol. The cdMT1-MMP active site is rigid at the Leu199 hydrogen-bonding site owing to the structural calcium metal that is co-ordinated to the adjacent residue, Phe198. This calcium-binding site is highly conserved among the MMPs and may be the reason why MAG-148 is not favoured in any of the deep-pocket MMPs tested. The present paper is the first report to provide a rationale for the large disparity between these side chains in terms of either substrate specificity or inhibition potency.
A new series of inhibitors incorporating a pyrrolidinyl ring at the zinc-binding functionality (Table 1, compound 3) has been developed recently [21,23]. These compounds have increased water-solubility, while maintaining inhibitor potency. The stereochemical requirements of these inhibitors are the same as for the previously designed inhibitors (Table 1, compounds 1 and 2) and demonstrate binding in the same orientation. The increased water-solubility may correspond to an improved oral bioavailability that must be considered for future drug design. Therefore these compounds may be ideal candidates for future in vivo cancer-cell invasion and metastasis studies.
The prominent roles of MMPs in diseases have led to aggressive targeting of these enzymes by pharmaceutical companies. Despite all the advancements in MMPI research, and particularly with hydroxamic acids, clinical trials have been disappointing. Recent papers suggest the need for specific targeting of MT1-MMP in cancer invasion [5,6]. To increase our understanding of MMPIs, and particularly mercaptosulphide inhibitors, we have characterized them with a potential target, MT1-MMP, in enzyme-inhibition kinetic and mechanistic studies. Moreover, we have demonstrated that these inhibitors have reached their target enzyme on the cell surface and blocked the substrate cleavage by MT1-MMP in two different human cancer-cell culture systems, with functional endogenous and exogenous MT1-MMP expression respectively. Owing to the exceptionally significant roles that MT1-MMP plays, not only in cancer-cell invasion and growth [1–8], but also in conferring tumorigenicity on non-malignant epithelial cells [53,54], it is essential to make efforts to develop novel potent and selective inhibitors to target MT1-MMP. Our studies might identify enzyme–inhibitor interactions that assist the rational design of new generations of compounds with increased potency, selectivity, stability and water-solubility.
Acknowledgments
This work was supported in part by a predoctoral fellowship award from D. O. D./U.S. Army Breast Cancer Research Program DAMD17-00-1-0243 (to D. R. H.); grants from D. O. D./U.S. Army Prostate Cancer Research Program DAMD17-02-1-238, National Institutes of Health (NIH) CA78646, and Florida State University Research Foundation, and a Florida State University Developing Scholar Award (to Q.-X. A. S.); grants from D. O. D. Prostate Cancer Program DAMD17-02-1-0109 and the Carol M. Baldwin Breast Care Foundation (to J. C.); and grants from the Molecular Design and Synthesis Research Foundation (to M. A. S.). We thank Professor Harald Tschesche of the University of Bielefeld, Dr Henning Birkedal-Hansen of the NIH, and Dr Harold van Wart of Roche BioSciences, for providing us with invaluable reagents. We also appreciate the valuable discussions and critical reading of the manuscript by Dr Hyun Park, Robert Newcomer and Shelbourn Kent.
References
- 1.McCawley L. J., Matrisian L. M. Matrix metalloproteinases: they're not just for matrix anymore! Curr. Opin. Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez-Barrantes S., Bernardo M., Toth M., Fridman R. Regulation of membrane type-matrix metalloproteinases. Semin. Cancer Biol. 2002;12:131–138. doi: 10.1006/scbi.2001.0421. [DOI] [PubMed] [Google Scholar]
- 3.Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature (London) 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 4.Hotary K., Allen E., Punturieri A., Yana I., Weiss S. J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 2000;149:1309–1323. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotary K. B., Allen E. D., Brooks P. C., Datta N. S., Long M. W., Weiss S. J. Membrane type I matrix metalloproteinase usurps tumour growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 6.Ueda J., Kajita M., Suenaga N., Fujii K., Seiki M. Sequence-specific silencing of MT1-MMP expression suppresses tumour cell migration and invasion: importance of MT1-MMP as a therapeutic target for invasive tumours. Oncogene. 2003;22:8716–8722. doi: 10.1038/sj.onc.1206962. [DOI] [PubMed] [Google Scholar]
- 7.Ohuchi E., Imai K., Fujii Y., Sato H., Seiki M., Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997;272:2446–2451. doi: 10.1074/jbc.272.4.2446. [DOI] [PubMed] [Google Scholar]
- 8.Pei D., Weiss S. J. Transmembrane-deletion mutants of the membrane-type matrix metalloproteinase-1 process progelatinase A and express intrinsic matrix-degrading activity. J. Biol. Chem. 1996;271:9135–9140. doi: 10.1074/jbc.271.15.9135. [DOI] [PubMed] [Google Scholar]
- 9.d'Ortho M. P., Will H., Atkinson S., Butler G., Messent A., Gavrilovic J., Smith B., Timpl R., Zardi L., Murphy G. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur. J. Biochem. 1997;250:751–757. doi: 10.1111/j.1432-1033.1997.00751.x. [DOI] [PubMed] [Google Scholar]
- 10.Strongin A. Y., Collier I., Bannikov G., Marmer B. L., Grant G. A., Goldberg G. I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 11.Butler G. S., Butler M. J., Atkinson S. J., Will H., Tamura T., van Westrum S. S., Crabbe T., Clements J., d'Ortho M.-P., Murphy G. The TIMP2 membrane type 1 metalloproteinase “receptor” regulates the concentration and efficient activation of progelatinase A: a kinetic study. J. Biol. Chem. 1998;273:871–880. doi: 10.1074/jbc.273.2.871. [DOI] [PubMed] [Google Scholar]
- 12.Kinoshita T., Sato H., Okada A., Ohuchi E., Imai K., Okada Y., Seiki M. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J. Biol. Chem. 1998;273:16098–16103. doi: 10.1074/jbc.273.26.16098. [DOI] [PubMed] [Google Scholar]
- 13.Zucker S., Drews M., Conner C., Foda H. D., DeClerck Y. A., Langley K. E., Bahou W. F., Docherty A. J. P., Cao J. Tissue inhibitor of metalloproteinase-2 (TIMP-2) binds to the catalytic domain of the cell surface receptor, membrane type 1-matrix metalloproteinase 1 (MT1-MMP) J. Biol. Chem. 1998;273:1216–1222. doi: 10.1074/jbc.273.2.1216. [DOI] [PubMed] [Google Scholar]
- 14.Nakahara H., Howard L., Thompson E. W., Sato H., Seiki M., Yeh Y., Chen W.-T. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7959–7964. doi: 10.1073/pnas.94.15.7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitaker M., Floyd C. D., Brown P., Gearing J. H. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 16.Skotnicki J. S., Zask A., Nelson F. C., Albright J. D., Levin J. I. Design and synthetic considerations of matrix metalloproteinase inhibitors. Ann. N.Y. Acad. Sci. 1999;878:61–72. doi: 10.1111/j.1749-6632.1999.tb07674.x. [DOI] [PubMed] [Google Scholar]
- 17.Lauer-Fields J. L., Fields G. B. Matrix metalloproteinase inhibitors and cancer. Exp. Opin. Ther. Patents. 2000;10:1873–1884. [Google Scholar]
- 18.Hodgson J. Remodeling MMPIs. BioTechnology. 1995;13:554–557. doi: 10.1038/nbt0695-554. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz M. A., van Wart H. E. Mercaptosulfide metalloproteinase inhibitors. U.S. Pat. 5455262. 1995 [Google Scholar]
- 20.Sang Q.-X., Jia M.-C., Schwartz M. A., Jaye M. C., Kleinman H. K., Ghaffari M. A., Luo Y.-L. New thiol and sulfodiimine metalloproteinase inhibitors and their effect on human microvascular endothelial cell growth. Biochem. Biophys. Res. Commun. 2000;274:780–786. doi: 10.1006/bbrc.2000.3212. [DOI] [PubMed] [Google Scholar]
- 21.Park H. I., Jin Y., Hurst D. R., Monroe C. A., Lee S., Schwartz M. A., Sang Q.-X. The intermediate S1′ pocket of the endometase/matrilysin-2 active site revealed by enzyme inhibition kinetic studies, protein sequence analyses, and homology modeling. J. Biol. Chem. 2003;278:51646–51653. doi: 10.1074/jbc.M310109200. [DOI] [PubMed] [Google Scholar]
- 22.Hurst D. R., Schwartz M. A., Ghaffari M. A., Jin Y., Tschesche H., Fields G. B., Sang Q.-X. Catalytic- and ecto-domains of membrane type 1-matrix metalloproteinase have similar inhibition profiles but distinct endopeptidase activities. Biochem. J. 2004;377:775–779. doi: 10.1042/BJ20031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz M. A., Jin Y., Hurst D. R., Sang Q.-X. Substituted heterocyclic mercaptosulfide inhibitors. U.S. Pat. pending PCT 2004/031847. 2004 [Google Scholar]
- 24.Jin Y., Ghaffari M. A., Schwartz M. A. A practical synthesis of differentially-protected cis-1,2-cyclopentanedithiols and cis-3,4-pyrrolidinedithiols. Tetrahedron Lett. 2002;43:7319–7321. [Google Scholar]
- 25.Lichte A., Kolkenbrock H., Tschesche H. The recombinant catalytic domain of membrane-type matrix metalloproteinase-1 (MT1-MMP) induces activation of progelatinase A and progelatinase A complexed with TIMP-2. FEBS Lett. 1996;397:277–282. doi: 10.1016/s0014-5793(96)01206-9. [DOI] [PubMed] [Google Scholar]
- 26.Riddles P. W., Blakeley R. L., Zerner B. Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid) – a re-examination. Anal. Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 27.Knight C. G., Willenbrock F., Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 28.Park H. I., Ni J., Gerkema F. E., Liu D., Belozerov V. E., Sang Q.-X. Identification and characterization of human endometase (matrix metalloproteinase-26) from endometrial tumour. J. Biol. Chem. 2000;275:20540–20544. doi: 10.1074/jbc.M002349200. [DOI] [PubMed] [Google Scholar]
- 29.Park H. I., Turk B. E., Gerkema F. E., Cantley L. C., Sang Q.-X. Peptide substrate specificities and protein cleavage sites of human endometase/matrilysin-2/matrix metalloproteinase-26. J. Biol. Chem. 2002;277:35168–35175. doi: 10.1074/jbc.M205071200. [DOI] [PubMed] [Google Scholar]
- 30.Morrison J. F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Catalan C., Bode W., Huber R., Turk D., Calvete J. J., Lichte A., Tschesche H., Maskos K. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 1998;17:5238–5248. doi: 10.1093/emboj/17.17.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spurlino J. C., Smallwood A. M., Carlton D. D., Banks T. M., Vavra K. J., Johnson J. S., Cook E. R., Falvo J., Wahl R. C., Pulvino T. A., et al. 1.56 A structure of mature truncated human fibroblast collagenase. Proteins. 1994;19:98–109. doi: 10.1002/prot.340190203. [DOI] [PubMed] [Google Scholar]
- 33.Weiner S. J., Kollman P. A., Case D. A., Singh U. C., Ghio C., Alagona G., Profeta S., Weiner P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984;106:765–784. [Google Scholar]
- 34.Weiner S. J., Kollman P. A., Nguyen D. T., Case D. A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 35.Toba S., Damodaran K. V., Merz K. M. Binding preferences of hydroxamate inhibitors of the matrix metalloproteinase human fibroblast collagenase. J. Med. Chem. 1999;42:1225–1234. doi: 10.1021/jm980577f. [DOI] [PubMed] [Google Scholar]
- 36.Guida W. C., Bohacek R. S., Erion M. D. Probing the conformational space available to inhibitors in the thermolysin active site using Monte Carlo/energy minimization techniques. J. Comput. Chem. 1992;13:214–228. [Google Scholar]
- 37.Hoops S. C., Anderson K. W., Merz K. M. Force field design for metalloproteins. J. Am. Chem. Soc. 1991;113:8262–8270. [Google Scholar]
- 38.Li H., Bauzon D. E., Xu X., Tschesche H., Cao J., Sang Q. A. Immunological characterization of cell-surface and soluble forms of membrane type 1 matrix metalloproteinase in human breast cancer cells and in fibroblasts. Mol. Carcinog. 1998;22:84–94. [PubMed] [Google Scholar]
- 39.Cao J., Kozarekar P., Pavlaki M., Chiarelli C., Bahou W. F., Zucker S. Distinct roles for the catalytic and hemopexin domains of membrane type 1-matrix metalloproteinase in substrate degradation and cell migration. J. Biol. Chem. 2004;279:14129–14139. doi: 10.1074/jbc.M312120200. [DOI] [PubMed] [Google Scholar]
- 40.Henderson P. J. A linear equation that describes the steady-state kinetics of enzymes and subcellular particles interacting with tightly bound inhibitors. Biochem. J. 1972;127:321–333. doi: 10.1042/bj1270321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mucha A., Cuniasse P., Kannan R., Beau F., Yiotakis A., Basset P., Dive V. Membrane type-1 matrix metalloprotease and stromelysin-3 cleave more efficiently synthetic substrates containing unusual amino acids in their P1′ positions. J. Biol. Chem. 1998;273:2763–2768. doi: 10.1074/jbc.273.5.2763. [DOI] [PubMed] [Google Scholar]
- 42.Burley S. K., Petsko G. A. Aromatic–aromatic interaction: a mechanism of protein structure stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 43.Zucker S., Cao J., Chen W.-T. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 44.Brown P. D. Ongoing trials with matrix metalloproteinase inhibitors. Exp. Opin. Invest. Drugs. 2000;9:2167–2177. doi: 10.1517/13543784.9.9.2167. [DOI] [PubMed] [Google Scholar]
- 45.Hoekstra R., Eskens F. A. L. M., Verweij J. Matrix metalloproteinase inhibitors: current developments and future perspectives. Oncologist. 2001;6:415–427. doi: 10.1634/theoncologist.6-5-415. [DOI] [PubMed] [Google Scholar]
- 46.Coussens L. M., Fingleton B., Matrisian L. M. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 47.Overall C. M., López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 48.Brown S., Bernardo M. M., Li Z.-H., Kotra L. P., Tanaka Y., Fridman R., Mobashery S. Potent and selective mechanism-based inhibition of gelatinases. J. Am. Chem. Soc. 2000;122:6799–6800. [Google Scholar]
- 49.Schroder J., Henke A., Wenzel H., Brandstetter H., Stammler H. G., Stammler A., Pfeiffer W. D., Tschesche H. Structure-based design and synthesis of potent matrix metalloproteinase inhibitors derived from a 6H-1,3,4-thiadiazine scaffold. J. Med. Chem. 2001;44:3231–3243. doi: 10.1021/jm010887p. [DOI] [PubMed] [Google Scholar]
- 50.Puerta D. T., Cohen S. M. Examination of novel zinc-binding groups for use in matrix metalloproteinase inhibitors. Inorg. Chem. 2003;42:3423–3430. doi: 10.1021/ic026029g. [DOI] [PubMed] [Google Scholar]
- 51.Bode W., Fernandez-Catalan C., Tschesche H., Grams F., Nagase H., Maskos K. Structural properties of matrix metalloproteinases. Cell. Mol. Life Sci. 1999;55:639–652. doi: 10.1007/s000180050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puerta D. T., Schames J. R., Henchman R. H., McCammon J. A., Cohen S. M. From model complexes to metalloprotein inhibition: a synergistic approach to structure-based drug discovery. Angew. Chem. Int. Ed. 2003;42:3772–3774. doi: 10.1002/anie.200351433. [DOI] [PubMed] [Google Scholar]
- 53.Soulié P., Carrozzino F., Pepper M. S., Strongin A. Y., Poupon M. F., Montesano R. Membrane-type-1 matrix metalloproteinase confers tumorigenicity on nonmalignant epithelial cells. Oncogene. 2005;24:1689–1697. doi: 10.1038/sj.onc.1208360. [DOI] [PubMed] [Google Scholar]
- 54.Golubkov V. S., Boyd S., Savinov A. Y., Chekanov A. V., Osterman A. L., Remacle A., Rozanov D. V., Doxsey S. J., Strongin A. Y. Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J. Biol. Chem. 2005;280:25079–25086. doi: 10.1074/jbc.M502779200. [DOI] [PubMed] [Google Scholar]