

Abstract
Current studies provide evidence that proteins are initial targets of ROS (reactive oxygen species) in biological systems and that the damaged proteins can in turn damage other cell constituents. This study was designed to test the possibility that protein radicals generated by ROS can oxidize GSH and assess the probability of this reaction in vivo by measurement of the rate constant of this reaction. Lysozyme radicals were generated by hydroxyl and azide radicals in steady-state gamma ray radiolysis. In the absence of dioxygen, a range of protein carbon-centred amino acid radicals were produced by the hydroxyl radicals, and defined tryptophan radicals by the azide radicals. In the presence of dioxygen, each carbon-centred radical was converted to a protein peroxyl radical. Each of the peroxyl radicals was able to oxidize a molecule of GSH, regardless of its location in the protein. The peroxyl radicals were 10 and 20 times more effective GSH oxidants than the carbon-centred radicals produced randomly in the lysozyme, or the defined tryptophan lysozyme radicals respectively. We obtained for the first time the rate constant of reaction between a protein free-radical and GSH. Lysozyme tryptophan carbon radicals generated by nanosecond pulse radiolysis and flash photolysis oxidized GSH with a rate constant of (1.05±0.05)×105 M−1·s−1. Overall, the results are consistent with the hypothesis that protein radicals may be important intermediates in the pathway linking oxidative stress and damage in living organisms and emphasize the strongly enhancing role of dioxygen in this process.
Keywords: flash photolysis, glutathione, peroxyl radicals, protein radicals, pulse radiolysis, rate constant
Abbreviations: LZTrp•, lysozyme tryptophan radicals; PROS, partly reduced oxygen species; SOD, superoxide dismutase
INTRODUCTION
Living organisms are constantly exposed to ROS (reactive oxygen species) more correctly designated here as PROS (partly reduced oxygen species) formed as by-products of normal respiration [1], metabolism and autoxidation of xenobiotics [2], or resulting from stresses such as excessive exercise, trauma, ischaemia/reperfusion, or infection. In addition, environmental factors such as heat, freezing, radiation, toxins and ultrasound generate PROS [3]. Within limits, the formation of PROS is essential to maintain cell homoeostasis [4], achieved by a system of antioxidant defences maintaining a delicate balance between too little and too much PROS. Excessive production of PROS results in oxidative stress, leading to activation of specific signalling pathways and general damage, which can result in disease or death [5–7].
Attempts to modify the effects of PROS in vivo require knowledge of the chain of reactions which they initiate in cells. The biologically important PROS have been identified and the chemistry of their reactions with many potential target molecules is also well understood [8]. However, the pathway connecting the formation of PROS with endpoint biological damage is far from clear, largely because the identity of the first critical molecular targets of the PROS is not established. These targets must react efficiently with the PROS and must themselves be crucial for the survival of the cell, or have the ability to transmit the initial molecular damage to other cell components. There is general agreement that the main candidate for initial target molecules are lipids, DNA and proteins [8]. For the first of these, there is now strong evidence that lipid oxidation can often be dissociated from critical biological damage, as for example in stressed neocortical synapses [9], myocardial dysfunction [10], injured hepatocytes [11], acute pancreatitis [12] and in unchanged radiation sensitivity of cells with enhanced unsaturated fatty acid content [13]. In the case of DNA, although it is clear that it is a vital molecule whose integrity must be maintained, it is also believed to be a secondary rather than a primary target of PROS. Thus DNA was not the initial site of damage in hepatocytes [14], in mouse myeloma cells [15] and in T7 phage [16], whereas studies have demonstrated strong protection of chromatin and operon DNA by the associated proteins [17,18].
By contrast, there is evidence that proteins are significant targets for PROS in vivo, and are likely to be the first major class of molecules attacked. Increased levels of protein carbonyls, a common measure of protein oxidation, were found in early stages of exposure of different biological systems to PROS, with the antioxidants present unable to prevent this process [14,19,20]. Importantly, elevated levels of protein oxidation products were measured in tissues of aged humans and animals, as well as in those suffering from Alzheimer's and Parkinson's diseases, amyotrophic lateral sclerosis, advanced atherosclerotic plaques and diabetes, suggesting that changes in brain function may be related to progressive oxidation of critical proteins [21–26]. Although these results provide strong evidence of damage to proteins, for many years proteins were not considered to be significant targets of PROS, because there was no evidence that they could pass the damage on to other molecules. This was provided in the last dozen years by the discovery of reactive hydroperoxides and hydroxylated phenylalanines in proteins attacked by many of the biologically significant PROS. The altered proteins could damage DNA, enzymes, antioxidants and lipids, generate a range of further reactive radicals and modulate apoptosis, so that it is now clear that oxidized proteins containing reactive groups constitute a new form of PROS [27–33]. Evidence for the formation of protein peroxides in biological systems exposed to PROS was obtained in cultured cells, in advanced atherosclerotic plaques and in LDL [15,34–36].
In most cases the protein peroxides were generated in reactions between protein radicals and oxygen [28]:
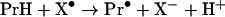 |
(1) |
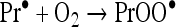 |
(2) |
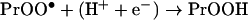 |
(3) |
where X• is a one-electron oxidant such as a hydroxyl (HO•), peroxyl (XOO•) or a thiyl (XS•) radical [31,37] and Pr• and PrOO• are the protein carbon and oxygen-centred radicals. It is plausible to suggest that the protein radicals responsible for the formation of the hydroperoxides may themselves constitute a significant biological hazard. Such radicals would be more powerful oxidizing agents than the relatively stable protein peroxides and their reactions should be significantly faster. However, although there have been many reports of the formation of protein radicals by radiolysis or photolysis and the end-products have been well characterized (reviewed in [38]), the potential for protein radicals to cause biological damage has not been evaluated. Estimates of this potential require identification of the protein radicals generated by physiologically relevant PROS and measurements of the kinetics of their reactions with significant biological target molecules. In this study, we have measured the oxidation of the important antioxidant and regulator of cell function, GSH, by carbon- and oxygen-centred lysozyme free radicals. We also measured the rate constant of the reaction of GSH and LZTrp• (lysozyme tryptophan radicals) by fast-kinetic methodology. The results show that protein radicals can damage a molecule important for the preservation of cell homoeostasis and that this process is significantly enhanced by oxygen.
EXPERIMENTAL
Materials
Crystalline preparations of lysozyme were supplied by Sigma (St. Louis, MO, U.S.A.) or Fluka (Buchs, Switzerland) and were used as received. Bovine erythrocyte SOD (superoxide dismutase), 7500 units/mg of protein) was from Sigma. Acetyl tyrosine and tryptophan amides from Bachem (Bubendorf, Switzerland) were of at least 99% purity. All other chemicals from Sigma or Fluka were of at least analytical reagent quality. Water was purified in a Millipore Q system and solutions were freed of air and saturated with N2O or argon by at least 3 cycles of evacuation by a membrane pump, followed by shaking under the desired gas at atmospheric pressure in a 1 cm×1 cm quartz cell equipped with a gas-tight valve. Glassware used in radiolysis was cleaned in warm concentrated nitric acid and rinsed in pure water.
Flash photolysis and pulse radiolysis
Laser flash photolysis experiments employed an Applied Photophysics (Leatherhead, U.K.) laser kinetic spectrophotometer (LKS 50) equipped with a Quantel Brilliant B Nd:YAG laser tuned to deliver 5 ns pulses at 266 nm. The signals were recorded and analysed by fast-kinetic spectrophotometry. Electron pulses of 2 MeV energy and 20–50 ns duration were generated by a Febetron 705 accelerator (Titan, San Leandro, CA, U.S.A.). A 300 μl capacity quartz cell was used and a flow system ensured that each sample was exposed only to a single electron pulse [39,40].
Gamma ray radiolysis
The Macquarie University 60Co source, delivering an energy dose of 44 Gy/min (Gy=1 J/kg), was used. Where required, the solutions were freed of dioxygen and presaturated with N2O in round-bottom flasks by shaking or gentle bubbling under a blanket of N2O for >1 h. The solutions were buffered with 10 mM phosphate buffer pH 7.4, and equilibrated with washed Chelex 100 resin. Oxidation of GSH by the radiation-generated radicals was measured by the 5,5′-dithiobis-(2-nitrobenzoic)acid (Ellman's reagent) at 412 nm, with the pH raised to 8.0 by the addition of 20 mM phosphate. The molar absorption coefficient of solutions containing high concentrations of lysozyme was 1.47×104 M−1·cm−1 rather than the lower value reported previously [41]. The coefficient was not affected by the presence of 0.1 M azide and tests with catalase showed that none of the measurements were altered by the radiation-generated H2O2.
RESULTS
Pulse radiolysis
Irradiation of dilute solutions with nanosecond pulses of high energy electrons, coupled to fast analysis of the transient spectra generated, has been widely used to study the formation and reactions initiated by radicals. Since the energy of the electrons is partitioned according to the relative masses of components of the irradiated system, in dilute aqueous solutions the energy is effectively deposited in the aqueous component, producing several decomposition products:
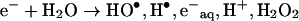 |
(4) |
The amounts of these products generated by a given amount of energy absorbed are accurately known [8]. However, formation of a range of species able to initiate a variety of processes represents a complication in the derivation of the mechanism of reactions taking place, particularly for complex macromolecules such as proteins [38]. This problem can be overcome by selective removal or conversion of the reactive species generated, thus reducing the number of possible reaction pathways and products. In the presence of N2O the hydrated electrons are converted to hydroxyl radicals and the low H• yield has no effect on the amounts of products [8]:
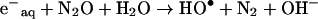 |
(5) |
Because of their high reactivity, the HO• radicals have been widely used to generate other radicals, whose reactions can be studied in turn. In the case of proteins, the effects of direct exposure to HO• have been extensively studied and include chain breakage, crosslinking and the formation of carbonyl groups, peroxides and hydroxylated amino acids (reviewed in [38]). However, direct generation of protein radicals (Pr•, PrOO•) by HO• was not used in this study for measurements of reaction kinetics. The high reactivity and low discrimination of the HO•, result in formation of a range of radical-centres on the protein with different yields, lifetimes and accessibility to reactants. Thus although in a preliminary study we measured the rate constants of reactions of 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulphonic)acid (ABTS) with radicals generated by HO• on small proteins, reactions of proteins of 20 kDa or more could not be analysed by simple kinetic models (J. L. Gebicki and J. M. Gebicki, unpublished work). We have therefore made use of the selective radical probe approach, where the HO• is converted to the azide radical (N3•) [42]. This selectively and rapidly oxidizes tryptophan residues in many proteins [42–45], including lysozyme (LZTrpH), where additional reaction with LZTyrOH makes an approx. 10% contribution to the disappearance of N3• [43,45]:
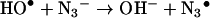 |
(6) |
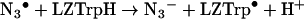 |
(7) |
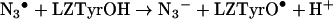 |
(8) |
The rate constants at near neutral pH are k6=1.2×1010, k7 ≈ 2×109 and k8=1.0×108 (all in units of M−1·s−1) [42,43,46]. The absorbance of solutions saturated with N2O, containing 2 mg/ml (140 μM) lysozyme, 10 mM phosphate buffer (pH 7.4) and 0.1 M sodium azide is shown in Figure 1 at different times following the radiation pulse. The changes of particular interest were near 510 and 400 nm, which are characteristic of the tryptophan and tyrosyl radicals respectively [43,44]. The spectra show formation of LZTrp• at 510 nm in the first 20 μs after pulse, followed by decay. In contrast, the LZTyrO• radical (≈400 nm) did not form immediately, but only in the period corresponding to the decay of the LZTrp•. This is consistent with studies demonstrating intramolecular first-order radical transfer between these residues in lysozyme and other proteins [43–45]. In the presence of 2.5 mM GSH, when more than 92% of the azide radicals were scavenged by the lysozyme, neither amino acid radical was detecTable 2 ms after the pulse (Figure 1).
Figure 1. Time-resolved absorbance spectrum of lysozyme solutions irradiated with 50 ns pulses of 2 MeV electons.

Radiation doses were 35 Gy/pulse. The solutions were saturated with N2O and contained 0.14 mM lysozyme, 10 mM phosphate buffer pH 7.4 and 0.1 M NaN3. Each spectrum had 512 data points which are presented as averages of groups of 9 for clarity.
Earlier studies of the rates of intramolecular Trp•→TyrO• transfer in lysozyme showed a marked increase as the pH was lowered from 7.4 to 5.2, attributed to protonation of Glu35 and a conformational change [45]. Results shown in Figure 2 were recorded at 510 nm, the peak absorbance of the LZTrp•. They confirm the increased rate of radical decay as the pH was lowered from 7.4 to 5.2. The accelerating effect of 2.5 mM GSH on the initial decay at either pH confirmed that the GSH repaired the protein radical [29,47]:
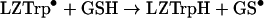 |
(9) |
Previous studies have shown that reaction (9) probably involved LZTrp62 as it is the most solvent-accessible Trp residue in lysozyme, with smaller contributions from Trp123 and Trp63 [45]. However, the rate constant of the repair reaction (k9) could not be calculated from pulse radiolysis results, because the high GSH/lysozyme ratios required would divert some of the HO• and N3• from the protein to GSH, affecting the measurements.
Figure 2. The effect of GSH and pH on the kinetics of growth and decay of Trp•.

The solutions and pulse conditions were as for those in Figure 1 with absorbance changes followed at 510 nm.
Flash photolysis
The problem of competing reactions was overcome by the application of flash photolysis. This technique offers the advantage of selective photo-ionization of aromatic amino acids in proteins to radicals and the possibility of measurement of their reactions with solutes, providing the solute has no significant absorbance at the wavelength of the exciting laser. Transient absorbance changes in solutions containing 30 μM lysozyme and up to 0.45 M tert-butanol buffered with 10 mM phosphate near neutral pH were recorded following the laser flash. The alcohol was included in some experiments to scavenge any HO• generated, but had no effect on the reaction kinetics. The results of a typical set of experiments are shown in Figure 3. Unlike the electron pulse, the flash resulted in immediate formation of absorbance near 390 nm, caused at least in part by LZTyrO• (λmax=405 nm) as well as of LZTrp• (λmax=510 nm). While the 390 nm absorbance was stable over 2 ms, the LZTrp• decayed within that time. This showed that the rate of any spontaneous decay of the LZTyrO• was either slow, or almost balanced by the transfer of the unpaired electrons from LZTrp• to LZtyrOH. It is possible to make an estimate of the approximate numbers of LZTrp• and LZTyrO• radicals generated by the flash. Using molar absorption coefficients of 2600 and 1800 M−1·cm−1 for the respective species [45], the flash led to the formation of one LZTrp• in the 11 lysozyme molecules present and one LZTyrO• in 13. It is not possible to determine which of the 6 tryptophans and 3 tyrosines in hen-egg lysozyme were photoionized by the flash, because all should be accessible to the 266 nm photons. Photolysis of lysozyme solutions in the presence of GSH did not interfere with the excitation of the protein residues, because at the concentrations used GSH is transparent at 266 nm.
Figure 3. Time-resolved absorbance spectrum of lysozyme solutions following a light flash.

The 6 ns 266 nm flash was produced by a laser delivering 9±1 mJ of energy. The solutions contained 30 μM lysozyme, 450 mM tert-butanol and 2 mM phosphate buffer pH 7.0.
The accelerating effect of different concentrations of GSH on the rates of decay of the LZTrp• (Figure 4) was used to calculate the rate constant of reaction (9) (Table 1). The result is in excellent agreement with the value measured for reaction of GSH with the model for LZTrp•, the N-Ac-Trp•-NH2 radical, generated similarly by flash photolysis in the presence of 2 mM GSH (Figure 4 and Table 1).
Figure 4. The relationship between the pseudo first order rate constant (kobs) and concentration of GSH in reaction with Trp•.

The radical was generated by flash photolysis of solutions of 28 μM lysozyme, 100 mM tert-butanol, GSH and 10 mM phosphate, pH 7.4. Radical decay was followed at 510 nm. ○ Shows the rate constant for reaction of GSH with N-Ac-Trp-NH2.
Table 1. Rate constants for reactions of protein and amino acid radicals with GSH.
Rate constants units are M−1·s−1 for the bimolecular and s−1 for unimolecular reactions.
| Rate constant | ||
|---|---|---|
| Reaction | pH 5.2 | pH 7.0–7.4 |
| GSH+LZTrp• | (1.2±0.2)×105 | (1.05±0.05)×105 |
| GSH+N-Ac-Trp••-NH2 | (7.8±0.4)×104 | (1.1±0.3)×105 |
| GSH+N-Ac-TyrO•-NH2 | (3±2)×103 | (2.4±1.6)×103 |
| LZTrp•→LZTyrO• | (4.4±0.7)×103 | (5.3±1.3)×102 |
| LZTrp•→LZTyrO• | 8.3×103* | 5.0125×102* |
*Rate constants quoted are from reference [40].
Attempts to measure the rate constant of the reaction
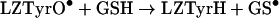 |
(10) |
by following the decay of 390 nm absorbance maximum in the presence of GSH were not successful. This peak (Figure 3) was consistently shifted from the 405 nm maximum reported for TyrO•, generated by pulse radiolysis either free or in a protein [42,44,48], indicating the presence of other unidentified transients. In addition, analysis of the kinetics of the decay of the presumed LZTyrO• peak at 390 nm in the presence of GSH gave a much higher value for the bimolecular rate constant than that of the free N-Ac-TyrO•-NH2 (Table 1), which is unlikely to be correct.
Gamma irradiation
As outlined above, measurements of the rate constant of reaction (9) imposed constraints on the design of the experiments, which might influence any conclusions on the biological relevance of the value of k9. Thus the use of N3•, rather than the PROS generally held responsible for oxidative stress, could give a false impression of the mode of more common interactions between protein radicals and GSH in vivo. This problem was addressed by comparing the amounts of GSH oxidized by lysozyme radicals generated by N3• or HO• under N2O or air. The N3• were generated by steady-state radiolysis in a gamma source of 0.1 M azide solutions saturated with N2O, as in the fast-kinetic experiments. The HO• were produced similarly, but without the azide. The solutions were buffered with 10 mM phosphate pH 7.4 and contained 1.0 mM lysozyme and 50 μM GSH. The results under air were obtained from 6 experiments (Table 2). The 18 irradiations under N2O initially gave variable results because traces of dioxygen in the protein-rich samples led to increased losses of GSH. The results listed are averaged from the 3 experiments which produced the lowest and constant amounts of GSH oxidized.
Table 2. Oxidation of GSH by radiation-generated radicals.
In all cases the initial oxidizing radical was the hydroxyl group. Where indicated, this was converted to the azide radical. G-values give the number of GSH molecules oxidized per 100 eV of energy absorbed by the solution.
| Atmosphere | Azide | Primary radical | Principal protein radical(s) | Protein GSH radicals oxidized (nM/Gy) | GSH oxidized (nM/Gy) | GSH oxidized per LZ | Dioxygen enhancement factor | G-value |
|---|---|---|---|---|---|---|---|---|
| Air | − | HO• | LZOO• | 279 | 325±2 | 1.16 | 11 | 3.1 |
| Air | + | N3• | LZTrpOO• | 279 | 332±6 | 1.19 | 22 | 3.2 |
| N2O | − | HO• | LZ•/LZTrp• | 570 | 63±5 | 0.11 | − | 0.6 |
| N2O | + | N3• | LZTrp• | 570 | 31±3 | 0.05 | − | 0.3 |
In the absence of azide and dioxygen, the radiation-generated HO• attacked the lysozyme to produce carbon-centred protein radicals, LZ•. Since the G-value for the HO• is 2.7 radicals/100 eV [8], the 44 Gy radiation doses generated 12.3 μM HO•. Each of these was scavenged by the lysozyme, generating one protein radical per 81 molecules, which then oxidized the GSH:
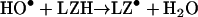 |
(11) |
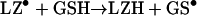 |
(12) |
Using azide, all HO• were scavenged to produce 12.3 μM N3• in reaction (6), followed by formation of LZTrp• in reaction (7), with some LZTyrO• generated in reaction (8) and in long-range electron transfer from LZTrp• [43]. Oxidation of GSH or other substrates can then be potentially achieved in reactions similar to (9) and (10).
Dioxygen is expected to react rapidly with carbon-centred species such as LZ• [8], although the relevant rate constants have not been measured for protein radicals. The resultant peroxyl radicals are likely to have different reactivities towards GSH than LZ• and LZTrp•. This was tested by comparing the amounts of GSH oxidized by lysozyme radicals generated by radiolysis of solutions saturated with N2O or air (Table 2). Under N2O and in the absence of azide the only radicals generated were HO•, so that the protein radicals consisted of undefined carbon-centred LZ• (reaction 11) which oxidized GSH in reaction (12). With azide, the HO• were converted to N3• and the GSH was oxidized only by LZTrp•. No LZTyrO• was formed by long-range electron transfer from LZTrp• in the presence of GSH, because under the conditions of these experiments reaction (9) was faster than the transfer (Table 1). Under air saturation, equal amounts of HO• and superoxide (O2•−) radicals were generated by the radiation [8]. The former reacted with the lysozyme to produce LZ• and LZTrp• radicals, whose most likely fate was conversion to the corresponding peroxyl radicals (reaction 13). The GSH was then oxidized in reaction (14) when azide was absent and in reaction (15) in its presence:
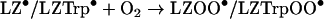 |
(13) |
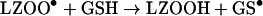 |
(14) |
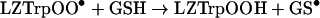 |
(15) |
The possibility that the O2•− could affect the overall course of these reactions was tested by irradiating solutions of 1 mM lysozyme and 50 μM GSH in 10 mM phosphate (pH 7.4) in the absence or presence of 370 units of SOD per ml. Separate tests showed that the activity of SOD was fully protected by the lysozyme. The radiation led to oxidation of 11.56±0.14 μM GSH in the absence of SOD and 11.61±0.20 μM in its presence, thus showing no reaction with O2•−. Since under these conditions the amount of HO• formed was 11.34 μM evidently each HO• produced a protein radical (reactions 11 and 13), each of which in turn oxidized one GSH molecule. The results (Table 2) show that the presence of dioxygen strongly enhanced the amounts of GSH oxidized by the protein radicals, with factors of about 10 and 20 per protein radical generated by HO• or N3•, respectively. Table 2 also identifies the protein radicals principally responsible for the oxidation of GSH under the different experimental conditions.
DISCUSSION
General
Several considerations underpinned the choice of lysozyme in this study. The protein is readily available in pure crystalline form, its primary and tertiary structures are known in detail [49] and the formation and intramolecular reactions of lysozyme radicals generated by radiolysis or photolysis have been the subject of detailed studies. In a number of these studies, formation of lysozyme radicals by pulse radiolysis was achieved under the conditions followed in this study, with selective oxidation of LZTrpH by the azide radicals [43–45]. Comparison of the results shown in Figure 1 with published spectra allowed the identification of the lysozyme radicals generated by N3• and confirmed the occurrence of long-range electron transfer from LZTrp• to LZTyrOH [43–45]. The effect of GSH demonstrated its ability to repair the protein radicals (Figure 1), with further measurements (Table 1) suggesting that the reduction was in reaction with LZTrp• (9) rather than LZTyrO• (10), because the rate of intramolecular electron transfer from LZTrp• is much slower than its reaction with GSH. Studies by the Bobrowski group [45] have shown that of the six tryptophan residues in lysozyme, Trp62, Trp63 and Trp123 are the main targets of N3• and are the main contributors to the radical transfer to 2 of the 3 TyrOH in the protein [45]. Our results also show that radical transfer is responsible for only a part of the LZTrp• decay; although the molar absorption coefficients of the Trp• and TyrO• are similar [50], clearly more of the former was lost in a given time than the latter generated (Figure 1).
Investigation of the fate of LZTrp• and LZTyrO• in the absence of GSH was beyond the scope of this study and was unlikely to be successful because most of the stable end products would remain as part of the damaged lysozyme and could not survive the necessary protein hydrolysis [38]. In the present study, lysozyme was oxidized only by either HO• or N3• radicals. Of the other potential reactants (reaction 4), under N2O the electrons were converted to HO• in reaction (5) and under air to O2•−, which does not produce protein radicals in the absence of transition metals [31]. The highest radiation dose was 88 Gy, generating 24.6 μM HO• or N3•, so that in 1 mM lysozyme solutions only 1 in 40 protein molecules was attacked by a single radical, with practically no chance of a multiple attack. The detection and characterization of 1 altered amino acid in the 5000 released from hydrolysed lysozyme presents a difficult challenge, in previous studies measurement of amino acid losses was confined to individual amino acids in radical-oxidized proteins exposed to much higher radiation doses (reviewed in [38]).
Although the finding of protein hydroperoxides in cultured cells exposed to PROS shows that protein radicals must have been generated in reactions (1)–(3) [15,36], there are no techniques capable of directly measuring the rate constants of their reactions owing to the many potential targets present in such complex systems. However, quantitative studies suggest that results obtained under biochemically defined conditions can be used to distinguish which reactions are likely to occur in biological systems and which are not. Thus results of studies on the formation of free- and peptide-bound Trp• and TyrO• and on the mechanism and products of their decay have proved to be directly relevant to proteins, although present techniques do not allow measurements at the higher level of complexity in cells.
The reaction of tryptophan with HO• leads to addition of the radical to the indole ring [51] followed partly by dehydration and peroxidation and partly by reaction with dioxygen to give indoyl and peroxyl radicals [52]. Stable products detected in several studies consisted mainly of N-formylkyurenine, kyurenine, 5-hydroxytryptophan, 3-indoleacetic acid, 3-indole propionic acid and organic peroxides [52–54]. Importantly, similar results were reported for peptides containing tryptophan, suggesting that the decay of the tryptophan radicals in proteins would follow similar routes [54–56]. In tyrosine, the principal reaction product with HO• was 3,4-dihydroxyphenylalanine (dopa), regardless of the presence of dioxygen [57,58]. In the presence of dioxygen, the yields of dopa were equal to the loss of the amino acid and to the amount of HO• generated, demonstrating that dopa was virtually the sole stable product [59]. Analogous results were obtained with peptides of glycine and tryptophan, again showing that tryptophan was the main residue attacked. The reaction of N3• with glycyltyrosine in the absence of dioxygen produced 2,2′-diphenol in reactions inhibited by cysteine or dioxygen [60]. As in the case of Trp radicals, these results suggest that similar reactions could occur in proteins and there are many reports linking the presence of di-tyrosine with protein oxidation in vitro and in vivo (summarized in [38]).
In the presence of GSH at millimolar concentrations, LZTrp• and LZTyrO• would be repaired in reactions (9) and (10), as shown for a number of other reducing agents [61], so that the normal products of the amino acid radiolysis would not form. The fate of the GSH thiyl radicals formed in these reactions depends on the presence of dioxygen [62]. In its absence, the main product will be GSSG, although recent results show that abstraction of hydrogen atoms from the lysozyme, which would regenerate the GSH, may be possible [63].
Kinetics
The use of flash photolysis potentially allowed measurements of the rate constants of reactions between the lysozyme tryptophan and tyrosine radicals with GSH by following their decay at the characteristic absorbance peaks of 510 and 405 nm [43,44]. Calculation of the rate constant of reaction (9) gave values of (1.2±0.2)×105 M−1·s−1 and (1.05±0.05)×105 M−1·s−1 at pH 5.2 and 7.0 respectively (Table 1). Although these are not high for radical reactions, in view of the commonly high concentrations of GSH in living organisms, reaction with protein radicals could result in oxidation of significant amounts of GSH under conditions of severe oxidative stress. The strictly bimolecular mode of the LZTrp•–GSH reaction shows that either several LZTrp• were generated, reacting with GSH with the same rate constants, or that the oxidation of GSH was dominated by one radical in the protein. The latter seems most likely, because the reaction requires GSH access to the radical. The close agreement between the rate constants for the oxidation of GSH by free or lysozyme tryptophan radicals (Figure 4, Table 1) demonstrates that any steric constraints imposed by the location of the reactive tryptophan in the protein had no effect on the reaction velocity. Extension of this argument to LZTyO• suggests that it too should react with GSH with a rate constant similar to the k=2.4×103 M−1·s−1 measured for N-Ac-TyrO•-NH2 (Table 1). However, the rate of decay of the absorbance near 400 nm in the presence of GSH, which should reflect the presence of tyrosyl radical, gave an apparent rate constant for reaction (10) of 2.9×105 M−1·s−1 (results not shown). This is unlikely to be correct for the LZTyrO• reaction with GSH alone, in view of the much lower value for N-Ac-Tyr•-NH2 (Table 1). Even if the rates of reaction for protein-bound and free tyrosine radicals with GSH were very different, the former (reaction 10) is likely to be slower, not faster, than that of the free amino acid radical because of steric inhibition imposed by the protein environment. This and the occurrence of the maximum absorbance at 390 nm, rather than at the 405 nm characteristic of the LZTyrO• suggests that other transient species besides this radical were generated by the flash.
Comparison of the initial rates of loss of LZTyr• generated by pulse radiolysis (Figure 2) and by flash photolysis (Figure 4) in the absence of GSH shows significant differences in the decay mechanisms, with the decay at pH 7.4 slow, not first order and strongly dependent on the radiation dose. It appears that the LZTyr• disappeared not only by electron transfer from LZTrp•→LZTyrO•, but also in reaction with other reactive species generated on the lysozyme, the concentrations of which would be dose-dependent and the identity of which is not known. Such species can be generated by 0.5% of the HO• which escaped scavenging by the azide. In flash photolysis, the loss of LZTyr• was biphasic with a high initial rate followed by a slower decay. The fast component was bimolecular with a rate constant of 3.5×106 M−1·s−1. This was not affected by GSH and not observed in measurements of the decay of N-Ac-Trp•-NH2 radicals. Although we cannot assign a mechanism to this process, it is likely to involve intramolecular recombination of LZTrp• and LZTyrO• generated simultaneously by photoionization. In the presence of GSH, the slower part of the decay had second order kinetics, allowing the measurement of the rate constant of reaction (9) (Table 1).
Gamma irradiation
In steady-state radiolysis, the lysozyme was oxidized either by the HO• or by N3• radicals. Of the other potential reactants (reaction 4), the electrons were converted to HO• under N2O in reaction (5) and to O2•− under air, which does not produce protein radicals.
In the absence of O2
In steady-state irradiation experiments, the relative concentrations of the solutes ensured selective formation of primary and secondary radicals. In the absence of azide, over 98% of the hydroxyl radicals (reaction 4) reacted with the 1 mM lysozyme (reaction 11, k11=3.1×1010 M−1·s−1) with insignificant direct reaction with the 50 μM GSH (k=1.4×1010 M−1·s−1) [46]. Reactions of HO• with proteins have been extensively studied over several decades (reviewed in [38]). The randomly-generated HO• initially oxidizes the amino acids located at the protein surface to produce carbon-centred protein radicals with additional damage to α-carbon sites on the backbone. In the absence of reactive solutes, these radicals undergo a variety of intramolecular reactions, depending on their nature and location, including long-range electron transfer to new sites, so that reactions of protein radicals with solutes may not involve the amino acids initially attacked by HO• [43–45]. For this reason the identities of the LZ• radicals generated by HO• and reacting with GSH could not be determined (Table 2), but their variety may not be extensive, especially with only 1 in 40 protein molecules affected. Since under N2O all the HO• produced by the 88 Gy radiation dose generated 50.2 μM LZ• radicals, the 5.5 μM GSH oxidized (Table 2) show that only about 11% of the protein radicals took part in oxidation. The rest would be expected to decay by chain processes leading to less reactive radicals or non-radical products.
More precise protein radical identification was made when N3• was the oxidant. The 0.1 M azide scavenged HO• (reaction 6, k6=1.2×1010 M−1·s−1), eliminating direct reaction with lysozyme. All the N3• then oxidized the protein (reaction 7, k7=9.6×108 M−1·s−1) because of the low concentration of GSH and its relatively slow reaction with N3• (k=9.5×106 M−1·s−1) [64]. GSH was oxidized by the selectively produced LZTrp• (reaction 7), with an approx. 10% contribution from LZTyrO• [45]. Current information does not allow estimation of the relative contributions of these radicals to GSH oxidation in steady-state radiolysis but the results (Table 2) show that 49.2 μM LZTrp•/LZTyrO• radicals generated, oxidized only 2.7 μM GSH, an efficiency of 5%. The LZTrp• radicals reacted simultaneously with GSH, transferred electrons to LZTyrOH and also clearly decayed in other reactions, since they contributed only approx. 50% to the formation of LZTyrO• [45]. Evidently a large proportion of the Trp• and TyrO• and other carbon-centred radicals located on the lysozyme decayed rapidly in the condensed amino acid environment within the protein molecule before reacting with the GSH. The nature of the end products of this decay cannot be determined in practice because of the low numbers of protein radicals generated, the probable range of products and the fact that they are unlikely to survive the harsh conditions of protein hydrolysis.
In the presence of O2
Comparison of the amounts of GSH oxidized by the protein radicals in the presence and absence of O2 demonstrates the crucial enhancement role of this element conferred by its ability to trap and stabilize radical centres (Table 2). In air-saturated solutions, all of the 24.6 μM lysozyme radicals generated by HO• or N3• reacted with GSH, giving enhancement factors per lysozyme radical of about 10 and 20 respectively for oxygenated versus dioxygen-free systems (Table 2). In the case of protein oxidation by the HO•, the peroxyl radicals are precursors of protein hydroperoxides, known to form in lysozyme and other proteins (reactions 1–3, where X•=HO•) [27,28]. However, the amounts of GSH oxidized by the protein radicals are surprisingly high and show that not only the peroxyl radicals involved in hydroperoxide formation were effective. The G-values for hydroperoxide groups in proteins are typically about 1-OOH per 100 eV of absorbed energy, whereas those of GSH oxidized are close to 3 (Table 2). This is similar to the G-value of 2.7 for the lysozyme radicals initially generated, demonstrating virtually 100% efficiency of the reaction chain HO•/N3•→LZ•→LZOO•→(−GSH). In the presence of dioxygen, the possibility of additional contribution to GSH oxidation, by superoxide radicals formed in fast reaction between the hydrated electrons (reaction 4) and O2, has to be considered. In these experiments, 24.6 μM O2•− was generated by the radiation, which is disproportionate to O2 and H2O2 at pH 7 with k=2×105 M−1·s−1, giving a rate of O2•− disappearance of 1.3×10−4 M·s−1 [65]. Since the rate constant of reaction of O2•− with GSH is about 200 M−1·s−1 [66], the rate of loss of 50 μM GSH in reaction with O2•− was 2.5×10−7 M·s−1, approx. 500 times slower than disproportionation, so that O2•− should not contribute to the loss of GSH. This was confirmed in experiments comparing the amounts of GSH oxidized by 1 min irradiations in the absence and presence of SOD, which as shown in the Results section were identical.
The effectiveness of protein peroxyl radicals in oxidizing GSH shows that the reaction of dioxygen with the lysozyme carbon-centred radicals formed initially outcompeted alternative decay pathways. This suggests that the protein radical/dioxygen reactions are fast and/or that the peroxyl radicals formed are relatively stable. To our knowledge this important information is not available. A limited number of measurements, carried out with free aliphatic amino acid radicals, gave rate constants for reaction with dioxygen of ≈1×109 M−1·s−1, typical of other carbon-centred radicals, with smaller values of about 5×106 M−1·s−1 for the Trp• and 2×105 M−1·s−1 for TyrO• [67,68]. All of the common amino acid radicals except Cys, Met, Ser and Thr react with dioxygen, since they form stable hydroperoxides [28]. The rate constants of these reactions are unknown and they are likely to be modified for amino acid radicals located in proteins. We plan to carry out these measurements in the next phase of this study.
The close correspondence between the amounts of GSH oxidized by LZOO•, where the exact identity and location of the peroxyl radical are unknown, and the defined LZTrpOO• was not expected (Table 2). Although HO• has the ability to generate the tryptophan radical, its high reactivity and low discrimination is expected to result in formation of a wide range of initial carbon-centred radicals by abstraction of H atoms from the protein backbone and amino acid side chains and by addition to the aromatic residues [38]. It appears from the results in Table 2 that each HO• generated a protein peroxyl radical, each in turn oxidizing one GSH molecule, or that the HO• initiated radical chains, resulting in formation of several radicals in each protein molecule [38]. In the case of N3• no such radical chains have been reported and apparently every LZTrp• formed initially, reacted to form a LZTrpOO• which oxidized the GSH stoichiometrically. It seems likely that either a combination of these factors resulted in similar rates of oxidation of the GSH, or that the tryptophan residues acted as major sink for the amino acid radicals initially produced by the HO• at the protein surface.
Although our study involved chemically simple systems, the results provide mechanistic support for the damage-enhancing dioxygen effect well known in radiation biology [8]. Here both the physiologically possible extremes, from complete anoxia to saturation with air, were investigated. The amounts of GSH oxidized by lysozyme radicals in the presence of 10–50 μM dioxygen, typical of living tissues, were not measured. However, when the last trace of O2 was not removed from the solutions, GSH oxidation was enhanced and close to the extent typical of the air-saturated solutions, suggesting that stabilization of the carbon-centred protein radicals generated in reaction (13) by dioxygen can out-compete alternative decay pathways.
Biological relevance
This study was designed as part of a broader series of tests of the hypothesis that protein radicals, formed in reactions with biologically significant partly reduced oxygen species, can in turn inactivate important cell constituents. A crucial aspect of this test is the estimate of the probability of the occurrence of such reactions in vivo. As a typical soluble, compact and well characterized protein, lysozyme was a suitable model molecule for a large number of other cell proteins. Importantly, there is much detailed information from earlier studies on the interaction between lysozyme and a range of radicals, producing defined lysozyme radicals [42–44]. However, the biological relevance of the previous studies is limited, because formation of the defined protein radicals required the use of agents not generated in vivo (azide or bromine) and the absence of dioxygen. We are aware of only one report of the rate constant of reaction between a protein radical and a cell molecule, in which Santus et al. [69] calculated a value of 8×107 M−1·s−1 for oxidation of ascorbate by lysozyme Trp• at pH 6.2 [69]. The measurements were carried out in the absence of O2, which is likely to affect the kinetics of protein radical reactions and yields of products, as shown for GSH in Table 2.
Given cellular concentrations of ∼30 μM dioxygen and 5–10 mM GSH, the pseudo-first order rate constant of LZTrpOO• peroxyl formation, k14′, is 6×104 s−1, while that of repair of tryptophan by GSH, k9′, is 1×103 s−1. These numbers are to be compared with the intramolecular rate of electron transfer from tyrosine to the tryptophan radical of 5×103·s−1. Thus because the primary damage is not repaired in time by GSH in the presence of O2, peroxyl radicals are the major initial reaction products. These are rapidly reduced to hydroperoxides by GSH, or they may oxidize a nearby amino acid, which reacts with oxygen etc. Thus GSH does stop the peroxidation chain, but from our results one does not know how many cycles have been completed. Protein hydroperoxides decompose in the presence of transition metals to give new radicals, including oxidizing alkoxyl radicals, which are even more reactive than the peroxyl [30,70]. Furthermore, GSH radicals themselves are reactive [37].
In general terms, this study supports the hypothesis that proteins are likely to be important intermediates in the transmission of the damaging potential of PROS to biologically significant molecules. In living organisms, molecules such as DNA, GSH, ascorbate, NADH, lipids and enzymes are protected from direct PROS attack by the wide variety and quantity of alternative targets. It appears that the most abundant of these targets, the proteins, can act as agents of damage, rather than as protective molecules neutralizing the effects of PROS in vivo. Discovery of compounds able to inhibit reactions of protein radicals in living organisms may therefore lead to the alleviation of many of the well-documented biological consequences of oxidative stress.
Acknowledgments
This study was supported by research grants from Macquarie University (Sydney, Australia) and from the Eidgenossische Technische Hochschule (Zurich, Switzerland).
References
- 1.Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B., Gutteridge J. M. C. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B. The biological significance of oxygen-derived species. In: Valentine J. S., Foote C. S., Greenberg A., Liebman J. L., editors. Active Oxygens in Biochemistry. London: Blackie Academic & Professional; 1995. pp. 313–335. [Google Scholar]
- 4.Finkel T., Holbrook N. J. Oxidants, oxidative stress and the biology of aging. Nature (London) 2000;408:239–246. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 5.Sies H., editor. London: Academic Press; 1991. Oxidative Stress. [Google Scholar]
- 6.Floyd R. A. Role of oxygen free radicals in carcinogenesis. FASEB J. 1990;4:2587–2597. [PubMed] [Google Scholar]
- 7.Diplock A. T., Charleux J.-L., Crozier-Willi G., Kok F. J., Rice-Evans C., Roberfroid M., Stahl W., Vina-Ribes J. Functional food science and defence against reactive oxygen species. British J. Nutr. 1998;80(Suppl. 1):S77–S112. doi: 10.1079/bjn19980106. [DOI] [PubMed] [Google Scholar]
- 8.Von Sonntag C. London: Taylor & Francis; 1987. The Chemical Basis of Radiation Biology. [Google Scholar]
- 9.Hensley K., Carney J. M., Mattson M. P., Aksenova M., Harris M., Wu J. F., Floyd R. A., Butterfield D. A. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durot I., Maupoil V., Pousard B., Cordelet C., Vergely-Vandriesse C., Rochette L., Athias P. Oxidative injury of isolated cardiomyocytes: dependence of free radical species. Free Radical Biol. Med. 2000;29:846–857. doi: 10.1016/s0891-5849(00)00382-8. [DOI] [PubMed] [Google Scholar]
- 11.Caraceni P., Yao T., Esposti S. D, Gasbarrini A., Bowie B. T., Zern M., Borle A. B., Van Thiel D. H. Effect of vitamin E on reoxygenation injury experienced by isolated hepatocytes. Life Sci. 1994;55:1427–1432. doi: 10.1016/0024-3205(94)00757-8. [DOI] [PubMed] [Google Scholar]
- 12.Reinheckel T., Nedelev B., Prause B., Augustin W., Schulz H.-U., Lippert H., Halangk W. Occurrence of oxidatively modified proteins: an early event in acute pancreatitis. Free Radical Biol. Med. 1998;24:393–400. doi: 10.1016/s0891-5849(97)00271-2. [DOI] [PubMed] [Google Scholar]
- 13.Wolters H., Konings A. W. T. Radiation effects on membranes. III. The effect of X irradiation on survival of mammalian cells substituted by polyunsaturated fatty acids. Radiat. Res. 1982;92:474–482. [PubMed] [Google Scholar]
- 14.Caraceni P., De Maria N., Ryu H. S., Colantoni A., Roberts L., Maidt M. L., Pye Q., Bernardi M., Van Thiel D. H., Floyd R. A. Proteins but not nucleic acids are molecular targets for the free radical attack during reoxygenation of rat hepatocytes. Free Radical Biol. Med. 1997;23:339–344. doi: 10.1016/s0891-5849(96)00571-0. [DOI] [PubMed] [Google Scholar]
- 15.Du J., Gebicki J. M. Proteins are major initial cell targets of hydroxyl free radicals. Int. J. Biochem. Cell Biol. 2004;36:2334–2343. doi: 10.1016/j.biocel.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 16.Samuni A., Chevion M., Czapski G. Role of copper and the O2−• in the radiation-induced inactivation of T7 bacteriophage. Radiat. Res. 1984;99:562–572. [PubMed] [Google Scholar]
- 17.Ljungman M., Hanawalt P. C. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinogenesis. 1992;5:264–269. doi: 10.1002/mc.2940050406. [DOI] [PubMed] [Google Scholar]
- 18.Begusova M., Sy D., Charlier M, Spotheim-Maurizot M. Radiolysis of nucleosome core DNA: a modelling approach. Int. J. Radiat. Biol. 2000;76:1063–1073. doi: 10.1080/09553000050111532. [DOI] [PubMed] [Google Scholar]
- 19.Ciolino H. P., Levine R. L. Modification of proteins in endothelial cells during oxidative stress. Free Radical Biol. Med. 1997;22:1277–1282. doi: 10.1016/s0891-5849(96)00495-9. [DOI] [PubMed] [Google Scholar]
- 20.Reinheckel T., Noack H., Lorenz S., Wiswedel I., Augustin W. Comparison of protein oxidation and aldehyde formation during oxidative stress in isolated mitochondria. Free Radical Res. 1998;29:297–305. doi: 10.1080/10715769800300331. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield D. A., Subramaniam R., Cole P., Yatin S., Hensley K., Hall N. C., Carney J. Electron paramagnetic resonance studies of amyloid β-peptide and ischemia-reperfusion associated oxygen free radicals and the membrane damage they cause: relevance to Alzheimer's disease and stroke. In: Thomas C. E., Kalyanaraman B., editors. Oxygen Radicals and the Disease Process. Amsterdam: Harwood Academic Publishers; 1997. pp. 41–64. [Google Scholar]
- 22.Beal M. F. Oxidatively modified proteins in aging and disease. Free Radical Biol. Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 23.Merker K., Sitte N., Grune T. Hydrogen peroxide-mediated protein oxidation in young and old human MRC-5 fibroblasts. Arch. Biochem. Biophys. 2000;375:50–54. doi: 10.1006/abbi.1999.1657. [DOI] [PubMed] [Google Scholar]
- 24.Bruce D., Fu S., Armstrong S., Dean R. T. Human apo-lipoprotein B from normal plasma contains oxidized peptides. Int. J. Biochem. Cell Biol. 1999;31:1409–1420. doi: 10.1016/s1357-2725(99)00107-7. [DOI] [PubMed] [Google Scholar]
- 25.Smith C. D., Carney J. M., Starke-Reed P. E., Oliver C. N., Stadtman E. R., Floyd R. A., Markesbery W. R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stadtman E. R. Metal ion-catalysed oxidation of proteins: Biochemical mechanism and biological consequences. Free Radical Biol. Med. 1990;9:315–325. doi: 10.1016/0891-5849(90)90006-5. [DOI] [PubMed] [Google Scholar]
- 27.Simpson J. A., Narita S., Gieseg S., Gebicki S., Gebicki J. M. Long-lived reactive species on free-radical-damaged proteins. Biochem. J. 1992;282:621–624. doi: 10.1042/bj2820621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gebicki S., Gebicki J. M. Formation of peroxides in amino acids and proteins exposed to oxygen free radicals. Biochem. J. 1993;289:743–749. doi: 10.1042/bj2890743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irvin J. A., Ostdal H., Davies M. J. Myoglobin-induced oxidative damage: evidence for radical transfer from oxidized myoglobin to other proteins and antioxidants. Arch. Biochem. Biophys. 1999;362:94–104. doi: 10.1006/abbi.1998.0987. [DOI] [PubMed] [Google Scholar]
- 30.Davies M. J., Fu S., Dean R. T. Protein hydroperoxides can give rise to reactive free radicals. Biochem. J. 1995;305:643–649. doi: 10.1042/bj3050643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebicki J. M. Protein hydroperoxides as new reactive oxygen species. Redox Rep. 1997;3:99–110. doi: 10.1080/13510002.1997.11747096. [DOI] [PubMed] [Google Scholar]
- 32.Gebicki S., Gebicki J. M. Crosslinking of DNA and proteins induced by protein hydroperoxides. Biochem. J. 1999;338:629–636. [PMC free article] [PubMed] [Google Scholar]
- 33.Dean R. T., Fu S., Stocker R., Davies M. J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu S., Davies M. J., Stocker R., Dean R. T. Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaques. Biochem. J. 1998;333:519–525. doi: 10.1042/bj3330519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruce D., Fu S., Armstrong S., Dean R. T. Human apo-lipoprotein B from normal plasma contains oxidized peptides. Int. J. Biochem. Cell Biol. 1999;31:1409–1420. doi: 10.1016/s1357-2725(99)00107-7. [DOI] [PubMed] [Google Scholar]
- 36.Gieseg S., Duggan S., Gebicki J. M. Peroxidation of proteins before lipids in U937 cells exposed to peroxyl radicals. Biochem. J. 2000;350:215–218. [PMC free article] [PubMed] [Google Scholar]
- 37.Nauser T., Pelling J., Schöneich C. Thiyl radical reaction with amino acid side chains: Rate constants for hydrogen transfer and relevance for posttranslational protein modification. Chem. Res. Toxicol. 2004;17:1323–1328. doi: 10.1021/tx049856y. [DOI] [PubMed] [Google Scholar]
- 38.Davies M. J., Dean R. T. Oxford: Oxford University Press; 1997. Radical-Mediated Protein Oxidation. [Google Scholar]
- 39.Nauser T., Koppenol W. H. The rate constant of the reaction of superoxide with nitrogen monoxide: Approaching the diffusion limit. J. Phys. Chem. A. 2002;106:4084–4086. [Google Scholar]
- 40.Nauser T., Buhler R. E. Pivalic acid as combined buffer and scavenger for studies of cloud water chemistry with pulse radiolysis. J. Chem. Soc. Faraday Trans. 1994;90:3651–3656. [Google Scholar]
- 41.Ellman G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 42.Land E. J., Prutz W. A. Reaction of azide radicals with amino acids and proteins. Int. J. Radiat. Biol. 1979;36:75–83. doi: 10.1080/09553007914550831. [DOI] [PubMed] [Google Scholar]
- 43.Butler J., Land E. J., Prutz W. A., Swallow A. J. Charge transfer between tryptophan and tyrosine in proteins. Biochim. Biophys. Acta. 1982;705:150–162. [Google Scholar]
- 44.Weinstein M., Alfassi Z. B., DeFilippis M. R., Klapper M. H., Faraggi M. Long range electron transfer between tyrosine and tryptophan in hen egg-white lysozyme. Biochim. Biophys. Acta. 1991;1076:173–178. doi: 10.1016/0167-4838(91)90262-x. [DOI] [PubMed] [Google Scholar]
- 45.Bobrowski K., Holcman J., Poznanski J., Wierzchowski K. L. Pulse radiolysis studies of intramolecular electron transfer in model peptides and proteins. Biophys. Chem. 1997;63:153–166. doi: 10.1016/s0301-4622(96)02226-0. [DOI] [PubMed] [Google Scholar]
- 46.Buxton G. V., Greenstock C. L., Helman W. P., Ross A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O-) in aqueous solution. J. Phys. Chem. Ref. Data. 1988;17:513–886. [Google Scholar]
- 47.Sturgeon B. E., Sipe H. J., Barr D. P., Corbett J. T., Martinez J. G., Mason R. P. The fate of the oxidizing tyrosyl radical in the presence of glutathione and ascorbate. J. Biol. Chem. 1998;273:30116–30121. doi: 10.1074/jbc.273.46.30116. [DOI] [PubMed] [Google Scholar]
- 48.Grossweiner L. I., Kaluskar A. G., Baugher J. F. Flash photolysis of enzymes. Int. J. Radiat. Biol. 1976;29:1–16. doi: 10.1080/09553007614551511. [DOI] [PubMed] [Google Scholar]
- 49.Imoto T., Johnson L. N., North A. C. T., Phillips D. C., Rupley J. A. Vertebrate lysozymes. In: Boyer P. D., editor. The Enzymes. New York: Academic Press; 1972. pp. 665–868. [Google Scholar]
- 50.Bensasson R. V., Land E. J., Truscott T. G. Oxford: Oxford University Press; 1993. Excited States and Free Radicals in Biology and Medicine. [Google Scholar]
- 51.Armstrong R. C., Swallow A. J. Pulse- and gamma-radiolysis of aqueous solutions of tryptophan. Radiat. Res. 1969;40:563–579. [PubMed] [Google Scholar]
- 52.Josimovic Lj., Jankovic I., Jovanovic S. V. Radiation induced decomposition of tryptophan in the presence of oxygen. Radiat. Phys. Chem. 1993;41:835–841. [Google Scholar]
- 53.Stulik K., Pacakova V., Weingart M., Vlasakova V. High-performance liquid chromatography of tryptophan and its radiation products using UV photometric and voltammetric detection. J. Chromatogr. 1986;354:449–453. [Google Scholar]
- 54.Winchester R. V., Lynn K. R. X- and γ-radiolysis of some tryptophan dipeptides. Int. J. Radiat. Biol. 1970;17:541–548. doi: 10.1080/09553007014550681. [DOI] [PubMed] [Google Scholar]
- 55.Steinhart H. Stability of tryptophan in peptides against oxidation and irradiation. Adv. Exp. Med. Biol. 1991;241:29–40. doi: 10.1007/978-1-4684-5952-4_3. [DOI] [PubMed] [Google Scholar]
- 56.Pruetz W. A., Land E. J. Charge transfer in peptides. Pulse radiolysis investigation of one-electron reactions in dipeptides of tryptophan and tyrosine. Int. J. Radiat. Biol. 1979;36:513–520. doi: 10.1080/09553007914551301. [DOI] [PubMed] [Google Scholar]
- 57.Lynn K. R., Purdie J. W. Some pulse and gamma radiolysis studies of tyrosine and its glycyl peptides. Int. J. Radiat. Biol. 1976;8:685–689. [Google Scholar]
- 58.Maskos Z., Rush J. D., Koppenol W. H. The hydroxylation of phenylalanine and tyrosine. A comparison with salicylate and tryptophan. Arch. Biochem. Biophys. 1992;296:521–529. doi: 10.1016/0003-9861(92)90606-w. [DOI] [PubMed] [Google Scholar]
- 59.Cudina I., Josimovic L. The effect of oxygen on the radiolysis of tyrosine in aqueous solutions. Radiat. Res. 1987;109:206–215. [PubMed] [Google Scholar]
- 60.Pruetz W. A., Butler J., Land E. J. Phenol coupling initiated by one-electron oxidation of tyrosine units in peptides and histone. Int. J. Radiat. Biol. 1983;44:183–196. doi: 10.1080/09553008314550981. [DOI] [PubMed] [Google Scholar]
- 61.Hoey B. M., Butler J. The repair of oxidized amino acids by antioxidants. Biochim. Biophys. Acta. 1984;791:212–218. doi: 10.1016/0167-4838(84)90011-6. [DOI] [PubMed] [Google Scholar]
- 62.Wardman P., von Sonntag C. Kinetic factors that control the fate of thiyl radicals in cells. Methods Enzymol. 1995;251:31–45. doi: 10.1016/0076-6879(95)51108-3. [DOI] [PubMed] [Google Scholar]
- 63.Nauser T., Schoneich C. Thiyl radicals abstract hydrogen atoms from the αC-H bonds in model peptides: absolute rate constants and effect of amino acid structure. J. Am. Chem. Soc. 2003;125:2042–2043. doi: 10.1021/ja0293599. [DOI] [PubMed] [Google Scholar]
- 64.Abedinzadeh Z., Gardès-Albert M., Ferradini C. One-electron oxidation of glutathione by azide radicals in neutral medium: A gamma and pulse radiolysis study. Radiat. Phys. Chem. 1991;38:1–5. [Google Scholar]
- 65.Bielski B. H. J., Allen A. O. Mechanism of disproportionation of superoxide radicals. J. Phys.Chem. 1977;81:1048–1050. [Google Scholar]
- 66.Jones C. M., Lawrence A., Wardman P., Burkitt M. J. Kinetics of superoxide scavenging by glutathione: an evaluation of its role in the removal of mitochondrial superoxide. Biochem. Soc. Trans. 2003;37:1337–1339. doi: 10.1042/bst0311337. [DOI] [PubMed] [Google Scholar]
- 67.Jovanovic S. V., Simic M.-G. Repair of tryptophan radicals by antioxidants. J. Free Radical Biol. Med. 1985;1:125–129. doi: 10.1016/0748-5514(85)90016-9. [DOI] [PubMed] [Google Scholar]
- 68.Candeias L. P, Wardman P., Mason R. P. The reaction of oxygen with radicals from oxidation of tryptophan and indole-3-acetic acid. Biophys. Chem. 1997;67:229–237. doi: 10.1016/s0301-4622(97)00052-5. [DOI] [PubMed] [Google Scholar]
- 69.Santus R., Patterson L. K., Hug G. L., Bazin M., Maziere J.-C., Morliere P. Interactions of superoxide anion with enzyme radicals: kinetics of reaction with lysozyme tryptophan radicals and corresponding effects on tyrosine electron transfer. Free Radical Res. 2000;33:383–391. doi: 10.1080/10715760000300921. [DOI] [PubMed] [Google Scholar]
- 70.Koppenol W. H. Oxyradical reactions: From bond-dissociation energies to reduction potentials. FEBS Lett. 1990;264:165–167. doi: 10.1016/0014-5793(90)80239-f. [DOI] [PubMed] [Google Scholar]