

Abstract
There is accumulating evidence that many ribosomal proteins are involved in shaping rRNA into their functionally correct conformations through RNA-protein interactions. Moreover, although rRNA seems to play the central role in all aspects of ribosome function, ribosomal proteins may be involved in facilitating communication between different functional regions in ribosome, as well as between the ribosome and cellular factors. In an effort to more fully understand how ribosomal proteins may influence ribosome function, we undertook large-scale mutational analysis of ribosomal protein L3, a core protein of the large subunit that has been implicated in numerous ribosome-associated functions in the past. A total of 98 different rpl3 alleles were genetically characterized with regard to their effects on killer virus maintenance, programmed −1 ribosomal frameshifting, resistance/hypersensitivity to the translational inhibitor anisomycin and, in specific cases, the ability to enhance translation of a reporter mRNA lacking the 5′ 7mGppp cap structure and 3′ poly(A) tail. Biochemical studies reveal a correlation between an increased affinity for aminoacyl-tRNA and the extent of anisomycin resistance and a decreased peptidyltransferase activity and increased frameshifting efficiency. Immunoblot analyses reveal that the superkiller phenotype is not due to a defect in the ability of ribosomes to recruit the Ski-complex, suggesting that the defect lies in a reduced ability of mutant ribosomes to distinguish between cap+/poly(A)+ and cap−/poly(A)− mRNAs. The results of these analyses are discussed with regard to how protein-rRNA interactions may affect ribosome function.
Early in the history of modern yeast genetics, two mutants were independently identified based on separate phenotypes: resistance to the peptidyltransferase inhibitors trichodermin and anisomycin (tcm1-1) (25, 47) and inability to maintain the M1 killer virus (mak8-1 [maintenance of killer]) (63). Subsequently, they were shown to be allelic to RPL3, the gene that encodes ribosomal protein L3, a 44-kDa essential ribosomal protein in the large subunit of the ribosome (16, 64). Independently, ribosome reconstitution experiments demonstrated that L3 is one of a few proteins essential for peptidyltransferase center (PTC) activity (49), and L3 and L24 are the only proteins capable of initiating in vitro assembly of the Escherichia coli 50S ribosomal subunit (35). These early findings implicated L3 as a central component of peptidyltransfer activity and ribosome assembly and as an important factor in the interaction between host cells and viruses.
Previous studies demonstrating that peptidyltransferase inhibitors had antiviral activities by promoting altered efficiencies of programmed −1 ribosomal frameshifting (−1 PRF) (11) provoked more in-depth characterization of the mak8-1 mutants. An initial study (i) demonstrated that increased −1 PRF efficiency constituted the basis for the loss of killer phenotype in mak8-1 cells, (ii) showed these cells were immune to further changes in −1 PRF by anisomycin or sparsomycin, two drugs that were previously shown to alter −1 PRF efficiencies, and (iii) identified that the original mak8-1 allele consisted of the double rpl3 mutation W255C P257T (39). Notably, mutations in the same vicinity of the L3 proteins of E. coli and Brachyspira spp. also promoted resistance to tiamulin, a pleuromutilin antibiotic inhibitor of peptidyltransfer (4, 44). In a subsequent study, three additional single mutations were identified that were unable to maintain the killer virus (W255C, P257T, and I282T) (33). The inability of the yeast L3 mutants to propagate the killer virus was due to increased efficiencies in−1 PRF, and this correlated with decreased peptidyltransferase activities in ribosomes isolated from mak8-1 cells (33). Importantly, the correlation between peptidyltransferase activity and −1 PRF efficiencies is not specific to mak8-1: these two properties also correlated in mutants of RPL41 and RPD3 (31, 33). A subsequent publication employed pharmacogenetic, biochemical, and molecular methods to follow up on these mutants and demonstrated that small changes in the primary sequence of L3 resulted in significant changes in the structure of the large subunit rRNA, which in turn promoted increased affinities for both aminoacyl (aa)- and peptidyl-tRNAs (41).
In the course of these studies, it became apparent that screening for loss of the killer virus was in some way serving to limit the initial pool of mutants that could be used for more detailed structure/function analysis of L3. To address this, a library of randomly generated rpl3 alleles was screened first for resistance to anisomycin, resulting in the identification of 54 candidates representing 31 new unique alleles, and ensuing site-directed mutagenesis studies resulted in the production and characterization of an additional 43 new rpl3 alleles. In addition to the previously described coupled killer virus maintenance (Mak−) and anisomycin-resistant (Anr) phenotypes of L3 mutants, we have identified three new phenotypic expressions. These are (i) the uncoupling of the virus maintenance and drug resistance defects, (ii) the observation that the phenotypes promoted by single amino acid substitutions can differ from those promoted by combinations thereof, and (iii) the emergence of a class of phenotypes new to rpl3 alleles, the “superkillers” (Ski−). Further investigations into this class of mutants revealed that they most resembled the ski2/ski3/ski6/ski7 class of mutants: M1 double-stranded RNA (dsRNA) viral copy numbers were elevated and translation of cap−/poly(A)− mRNAs was derepressed. This is the first time that a ribosomal protein has been implicated in this type of translational discrimination. Structural mapping of mutated yeast L3 amino acids onto the Haloarcula marismortui large-subunit X-ray crystal structure revealed that most of the mutants correspond to positions of L3 that have been proposed to interact with bases in 23S rRNA (29). A detailed structure/function analysis is proposed as the basis for follow-up biochemical and molecular studies.
MATERIALS AND METHODS
Strains, plasmids, genetic manipulation, and media.
E. coli DH5α was used to amplify plasmid DNA. Transformation of yeast and E. coli and the YPAD, synthetic drop out medium (H-), and 4.7 MB plates for testing the killer phenotype were as previously reported (13). Restriction enzymes were obtained from Promega (Madison, WI), MBI Fermentas (Vilnius, Lithuania), BRL-Life Technologies (Gaithersburg, MD), and Roche Applied Science (Indianapolis, IN). The QuikChange XL II site-directed specific mutagenesis kit and GeneMorph II random mutagenesis kit were obtained from Stratagene (La Jolla, CA). The FluoReporter lacZ/galactosidase quantitation kit was obtained from Molecular Probes (Eugene, OR). Macrogen Inc. (Seoul, South Korea) performed DNA sequence analysis. Oligonucleotide primers were purchased from IDT (Coralville, IA).
The yeast strains used in this study are listed in Table 1. Strain RW1906 and the RNA polymerase I (Pol I)-driven lacZ reporter plasmid p599 (RDN1-lacZ) (65) were generous gifts from Reed Wickner. pTI25 (PGK1-lacZ) was previously described (12). The rpl3-gene disruption (rpl3Δ) strain JD1090 was constructed as described elsewhere (33). Plasmids for expression of dual luciferase frameshift reporters were as previously described (22).
TABLE 1.
Yeast strains used in this study
| Strain | Characteristics |
|---|---|
| RW1906 | MATaleu2 mak8-1 |
| 5X47 | MATa/MATα his1/+ trp1/+ ura3/+ K− |
| JD111 | MATα ura3-52 lys2-801 trp1Δ leu2 his3 [L-AHNB M1] |
| JD1090 | MATα ura3-52 lys2-801 trp1Δ leu2 his3 RPL3::HIS3 pJD166.ura [L-AHNB M1] |
| JD1123 | MATα lys2 ura3 ho::LYS2 leu2::hisG his4b xrn1Δ::Tn10luk [L-AHN M1] |
| JD2 | MATα ura3 trp1 ade8 ski2-2 [L-AHN M1] |
| JD19 | MATα ura3 leu2 ade2 PEP4::HIS3 NUC1::LEU2 ski3 [L-AHN M1] |
Preparation and screening of a library of rpl3 mutants.
A library of plasmid-borne rpl3 mutants was constructed using the error-prone PCR and gap repair method (34). Mutagenizing primers (70 nucleotides) for PCR were designed to be complementary to 5′ and 3′ untranslated regions of RPL3 and included translational start and stop codons (forward, 5′-TCTTTACTCATTATTTCATTTCGGTTTTGTCATCTCTAGAACAACACAGTTACTACAGAATTCAATCATG-3′; reverse, 5′-ATGCTCAATTAAAAAATGATTTATTTTCTAACAAAACTTCGCGGCCGCTCTAGAACTAGTGGATCCCTTA-3′). Random mutagenesis was performed with the GeneMorph II random mutagenesis kit with template concentrations optimized to generate one to four mutations per RPL3 coding sequence. Plasmid pJD225 (33) was digested with EcoRI and BamHI. The linearized plasmid lacking the RPL3 coding sequence was purified by Tris-acetate-EDTA-agarose gel electrophoresis and cotransformed with the random PCR-mutagenized RPL3 coding sequences into JD1090 cells. This approach was optimized to yield approximately 800 colonies per plate. Controls using either gapped vector or PCR products alone yielded 20 and 0 colonies, respectively. After 3 days growth on selective medium (H-trp), cells that had lost the wild-type RPL3-containing plasmids were selected by replica plating onto 5-fluoroorotic acid (5-FOA)-containing medium (45). Colonies were subsequently replica plated onto H-trp plates containing anisomycin (50 μg/ml) and grown for 3 days. Approximately 2 × 104 colonies were screened. Plasmids were rescued from anisomycin-resistant cells into E. coli and reintroduced into JD1090 cells. Colonies were replica plated onto 5-FOA followed by anisomycin plates, and only those that were confirmed as anisomycin resistant were selected as new rpl3 alleles.
Assays for the killer phenotypes, measurement of frameshifting, and viral dsRNA analyses.
The killer virus assay was carried out as previously described (12). Briefly, yeast colonies were replica plated to 4.7MB plates newly seeded at an optical density at 595 nm (OD595) of 0.5 of the 5 × 47 killer indicator strain per plate. After 2 to 3 days at 20°C, killer activity was scored as a zone of growth inhibition around the Killer+ colonies. To monitor programmed −1 frameshifting using the dual luciferase reporter plasmids, glass beads were used to prepare lysates from cells expressing the 0-frame, or −1 (L-A-derived) dual luciferase plasmids (22). After clarification of the lysates by centrifugation, typically 5 μl was used in a total volume of 100 μl of dual luciferase assay reagents (Promega, Madison WI), and Renilla and firefly luciferase activities were quantitated using a TD20/20 luminometer (Turner Designs, Sunnyvale, CA). Frameshifting efficiencies were calculated by dividing the firefly/Renilla luminescence ratios from lysates of cells expressing the −1 PRF test reporters by the same ratio obtained from lysates of cells expressing the zero-frame control reporter. All assays were replicated enough times to achieve >95% confidence levels, and statistical analyses were performed as previously described (24). Extractions of total nucleic acids to detect the presence of L-A and M1 dsRNAs were performed as previously described (13).
β-Galactosidase assays.
Yeast cells containing p599, in which the lacZ gene is transcribed from the yeast RNA Pol I promoter (65), were grown overnight in 2 ml of H-ura medium and collected by centrifugation. The OD595 readings were taken, and protein extraction and quantitation were performed as described previously (22). Equal volumes (10 μl) of protein extracts were used for β-galactosidase activity determinations using the FluoReporter lacZ/galactosidase kit as recommended by the manufacturer. Control experiments were done with cells expressing lacZ under control of an RNA Pol II promoter in pTI25. β-Galactosidase activities were normalized to total protein amounts. The ratios of β-galactosidase activities generated from p599 divided by those from pTI25 were used to monitor the abilities of mutant ribosomes to translate mRNAs lacking 5′ 7mGppp caps and poly(A) tails.
Isolation of ribosomes.
Yeast ribosomes were isolated using a modification of a previously published protocol (58). Briefly, cells were grown in YPAD medium to an OD595 of 0.8, collected by centrifugation, and washed twice with cold 0.9% KCl. Cells were suspended to concentrations of 1 g/ml in buffer A [20 mM Tris-HCl, pH 7.5 at 4°C, 5 mM Mg(CH3COO)2, 50 mM KCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM 1,4-dithioerythritol (DTE)], and one-half volume of glass beads (0.5 mm) were added. Cells were disrupted at 4°C with a Mini BeadBeater for 2 min set at maximum speed. Extracts were transferred to 4-ml centrifuge tubes, and glass beads were washed twice with 1 ml of buffer A. Washes were combined with extracts and spun in an MSL-50 rotor (Beckman) for 25 min at 20,000 rpm. Supernates were transferred to 4-ml polycarbonate tubes containing 1 ml of a cushion of buffer B [20 mM Tris-HCl, pH 7.5 at 4°C, 5 mM Mg(CH3COO)2, 0.5 M KCl, 25% glycerol, 1 mM PMSF, 1 mM DTE], and ribosomes were sedimented by centrifugation for 2 h at 50,000 rpm using the MSL-50 rotor. Fines from ribosome pellets were gently washed off with buffer C [50 mM Tris-HCl, pH 7.5 at 4°C, 5 mM Mg(CH3COO)2, 50 mM NH4Cl, 0.1 mM PMSF, 0.1 mM DTE, 25% glycerol]. Ribosomes were suspended in buffer C at concentrations of 2 to 10 pmol/μl (1 OD260 = 20 pmol) and stored frozen at −80°C.
Purification of aminoacyl-tRNA synthetases.
Aminoacyl-tRNA synthetases were purified as previously described with minor modifications (61). Two pounds of frozen cake yeast (Geroge R. Ruhl & Son, Inc., Hanover, MD) were placed into 500 ml of buffer A (0.2 M Tris-base, 0.3 M NH4Cl, 20 mM MgSO4, 1 mM EDTA, 0.15 M dextrose) and allowed to ferment overnight. Cells were disrupted by three passages through an ice-cooled Microfluidaser at ∼18,000 lb/in2, cell debris was removed by centrifugation at 4°C in a Beckman JLA rotor at 10,000 rpm for 30 min, and 800 ml of supernatant was obtained. Fines and nucleic acids were precipitated by addition of polyethylenimine (1.73 g/lb. = 4.32 g/liter of lysate) over a period of 5 min with slow stirring, and precipitates were removed by centrifugation 4°C using a GSA rotor at 9,000 rpm for 40 min. Proteins in supernatants were precipitated by addition of 472 g of ammonium sulfate per liter of extract (70% saturation), and precipitates were collected by centrifugation in a GSA rotor at 12,000 rpm for 45 min at room temperature. The pellet from this step was suspended in 43.75 ml of buffer C (30 mM potassium phosphate, pH 7.2, 1 mM EDTA, 1 mM DTE, 0.01 mM PMSF) per 100 g of pellet and subsequently dialyzed in 2 liters of buffer C overnight with two changes of buffer, after which the extract was clarified by centrifugation in a GSA rotor at 12,000 rpm for 45 min at 4°C. Supernatant was diluted 2.5 times with buffer C and fractionated through a Sephadex CM50 column equilibrated with buffer C. The column was washed with buffer D (30 mM potassium phosphate, pH 7.2, 1 mM EDTA, 0.01 mM PMSF, 10% glycerol) with 50 mM KCl. The proteins were eluted from the column using a series of step gradients composed of buffer D containing 150 mM, 300 mM, and 500 mM KCl. The material eluted by buffer D with 150 mM KCl contains phenylalanyl-tRNA synthetase activity. Proteins were precipitated by addition of 472 g/liter of ammonium sulfate, and pellets were suspended in buffer D containing 50 mM KCl. Extracts were dialyzed against 1 liter with two changes of buffer D50 for 10 h, after which they were clarified by centrifugation in a GSA rotor at 12,000 rpm for 45 min at 4°C. The obtained preparation of aa-tRNA synthases was aliquoted and flash frozen in liquid nitrogen.
Synthesis of aminoacyl-tRNA and acetylated aminoacyl-tRNA.
Yeast phenylalanyl-tRNAs were aminoacylated by scaling up a previously described method (61). The reaction mix (5 ml) contained 300 mM Tris-HCl, pH 7.6, 100 mM KCl, 20 mM MgCl2, 0.4 mM ATP, 40 μM [14C]Phe [496 mCi/mM], plus 5 mg of tRNA-Phe and 475 μl of aminoacyl-tRNA synthases. Reaction mixtures were incubated for 30 min at 30°C, and proteins were removed by extraction with acid-phenol-chloroform. [14C]Phe-tRNA was separated from uncharged tRNA and free [14C]Phe by high-performance liquid chromatography (HPLC) as previously described (59) with the following modifications. Samples were loaded onto a 4.6- by 250-mm JT Baker wide-pore butyl column equilibrated with buffer A (20 mM NH4Cl, 10 mM MgCl2, 400 mM NaCl; pH 5.0) at 1 ml/min. The column was washed with 10 ml of buffer A, conditions under which free phenylalanine and aminoacyladenylate are eluted from the column. Uncharged tRNAs were eluted by isocratic elution of 19 ml at 15% of buffer B (20 mM NH4Cl, 10 mM MgCl2, 400 mM NaCl, 60% methanol; pH 5.0). [14C]Phe-tRNA was eluted using a step gradient to 100% of buffer B. Elution of aminoacyl-tRNA was monitored by OD260/280 readings, and [14C]Phe-tRNA concentrations and specific activities were determined. The presence of aminoacyl-tRNA in the eluted material was confirmed by gel filtration through G-25 spin columns and by nonenzymatic hydrolysis of diester bonds at basic pH (27). Ac-[14C]tRNA was obtained in a similar manner. Yeast phenylalanyl tRNA was charged with [14C]Phe as above and extracted with phenol. The [14C]Phe-tRNA was acetylated by addition of 64 μl of anhydride at 15-min intervals for 1 h on ice (59). The reaction mix was clarified by centrifugation at 15,000 rpm for 3 min, and Ac-[14C]Phe-tRNA was purified by HPLC as described above.
Characterization of peptidyltransferase activity.
Peptidyltransfer assays performed essentially as previously described (15). Complex C was formed in 200 μl of binding buffer (80 mM Tris-HCl, pH 7.4, 160 mM ammonium chloride, 11 mM magnesium acetate, 2 mM spermidine, and 6 mM β-mercaptoethanol), containing 0.4 mM GTP, 120 pmol ribosomes, 0.4 mg/ml poly(U), and 100 pmol Ac-[14C]Phe-tRNA. These mixtures were incubated for 16 min at 30°C and then placed on ice. The complex was mixed with 2 ml of binding buffer, filtered through a Millipore HA filter, and washed three times with 4 ml of binding buffer. Complex C was extracted off the filter disk as described previously (55). Briefly, the filter disks containing the adsorbed complex C were shaken gently for 30 min at 5°C in binding buffer containing 0.05% of Zwittergent 3-12 (1.8 ml per disk). Extracted complex C was kept on ice prior to use in puromycin reactions. For each reaction mixture, 0.9 ml of complex C extract was preincubated at 30°C for 5 min, and reactions were initiated by adding puromycin in 100 μl of binding buffer to final concentrations of 2 mM. Aliquots of 100 μl were removed, and reactions were terminated at the indicated time intervals by addition of 100 μl of 1.0 N NaOH. Reaction products were extracted with 0.5 ml of ethyl acetate, and radioactivity was determined by scintillation counting. For each sample, a 90-μl aliquot of initial reaction mixture was also transferred to scintillation vials, and total radioactivity (No) was determined. Controls without puromycin were included in each experiment, and the values obtained were subtracted. The percent of the bound Ac-[14C]Phe-tRNA that was converted to Ac-[14C]Phe-puromycin (x) was corrected with the complex C stability factor A (A = C/Co, where C and Co are the amounts of surviving complex C in binding buffer at the end of each incubation period and at zero time) and extent factor α (determined if complex C were allowed to react completely; Co = αNo), as described previously (15, 54). The value of x′ (x′ = x/A2) was obtained for various time intervals and fitted into the integrated form of a first-order reaction, such as Kobs · t = ln[100/(100 − x′)], which represents a straight line. The slope of this line gives the value of Kobs, the apparent rate constant of entire course of reaction at a given concentration of puromycin.
Aminoacyl-tRNA binding studies.
Aminoacyl-tRNA binding to the A-site of the ribosome was carried out as previously described (14) in reaction mixtures (50 μl) containing 80 mM Tris-HCl, pH 7.4, 160 mM NH4Cl, 11 mM Mg(CH3COO)2, 6 mM β-mercaptoethanol, 0.4 mM GTP, and 2 mM spermidine, 0.4 μg/ml of poly(U), and 12 to 25 pmol of ribosomes. Reaction mixtures were preincubated with uncharged tRNA (4:1 tRNA/ribosomes) at 30°C for 15 min to ensure full occupation of P- and E-sites by uncharged tRNA, after which various amounts of (4 to 264 pmol) of [14C]Phe-tRNA were added. Reaction mixtures were incubated at 30°C for an additional 15 min to allow formation of [14C]Phe-tRNA-80S-poly(U) complexes. Aliquots were then applied onto nitrocellulose membranes, filters were washed with 2 ml of binding buffer, and radioactivity was measured by scintillation counting. Background levels of radioactivity were determined using a blank sample and subtracted from test samples. The data were plotted onto a double reciprocal Scatchard plot according to the equation v/[tRNA] = nKa − vKa, where v is the number of bound tRNA molecules per ribosome, [tRNA] is the concentration of unbound tRNA, n is the number of binding sites, and Ka is the apparent association constant. Plots for different mutants were normalized for the percentage of active ribosomes in each preparation, and Ka values were determined as the slopes of linear regression trend lines (46).
Immunoblot analyses.
Cells harboring wild-type or selected Ski- rpl3 alleles were transformed with either a myc-tagged SKI2 clone expressed from pAJ160 (5) or a FLAG-tagged SKI7 clone expressed from pAJ1203 (A. Wang and A. Johnson, unpublished data), and ribosomes were isolated as described above in the presence of cycloheximide (100 μg/ml). Ribosomal proteins were fractionated through 10% polyacrylamide gel electrophoresis (PAGE) gels and electrotransferred to Sequi-Blot polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Nonspecific binding sites were blocked in using a 1% solution (wt/vol) of nonfat dry milk in phosphate-buffered saline (PBS)-0.1% Tween 20. Membranes were incubated at 4°C overnight with anti-FLAG M2 or anti-myc antibodies (1:1,000; Stratagene) to detect the FLAG-tagged Ski7p or myc-tagged Ski2p. Parallel membranes were probed with an anti-L3 monoclonal antibody (1:2,000; provided by Jonathan Warner) to detect L3 as a loading control. Membranes were washed extensively with PBS-Tween 20 and then incubated with a goat anti-mouse immunoglobulin G (IgG; H+L)-horseradish peroxidase (HRP) conjugate (1:5,000; Bio-Rad) for 3 h at 25°C. After extensive washing, membranes were treated with the Western Lightning chemiluminescence reagent (Perkin-Elmer) and exposed to Biomax ML film (Kodak). Ribosomes, isolated from JD1090 strain expressing vectors alone, were used to identify nonspecific anti-FLAG and anti-myc reactive species.
Computational analysis of ribosome structure.
The cryo-electron microscopy (cryo-EM) reconstruction of Saccharomyces cerevisiae ribosomal proteins threaded onto the X-ray crystal structure of the H. marismortui 50S ribosomal subunit (29) was visualized using the Swiss PDB viewer (http://www.rcsb.org/pdb/).
RESULTS
Identification of new mak8 alleles by random mutagenesis.
The original mak8-1 allele contained two mutations, W255C and P257T, and cells expressing this form of the L3 protein had increased −1 PRF efficiencies, were not able to maintain the killer virus, and were resistant to anisomycin (39). Subsequent studies identified one additional mak8 allele, I282T, and characterized the separate W255C and P257T alleles with regard to−1 PRF and drug resistance phenotypes, as well as biochemical analyses of their peptidyltransferase activities (33, 41). These studies were particularly informative, because they established linkages between genetic phenotypes and biochemical parameters. However, the limited number of mutant alleles precluded a detailed analysis of L3 structure/function. To address this deficiency, a large-scale mutagenesis project involving both random and site-specific PCR mutagenesis was devised. Mutants were primarily screened for resistance to anisomycin based on the rationale that this approach would yield more alleles, including mutations at multiple positions within the protein, at least some of which would be accompanied by translational defects.
To this end, the coding region of RPL3 was subjected to random PCR mutagenesis (average, 1 to 4 mutations/PCR product) and screened for resistance to anisomycin as described in Materials and Methods. In a primary screen of >20,000 colonies, 54 anisomycin-resistant mutants were identified (A1 to A54) (Table 2). Approximately 35% of the mutants were lethal, as determined by their inability to grow in the presence of 5-FOA (data not shown). The RPL3-TRP1 plasmids were rescued from the 54 anisomycin-resistant colonies, passaged through E. coli, reintroduced into rpl3Δ cells, and rescored for their abilities to promote anisomycin resistance. The mutations responsible for conferring this phenotype were identified by DNA sequence analysis of the clones, resulting in the identification of 31 new rpl3 alleles. In addition, the previously identified W255C and I282T alleles were also independently isolated in this screen. Spot assays of 10-fold dilutions were used to determine relative degrees of anisomycin resistance, an example of which is shown in Fig. 1A. Wild-type cells were given the score of 1+, and the most resistant alleles (both of which contained the W255C mutation) had scores of 4+. The mutants were also scored with respect to their killer virus maintenance phenotypes. Nine of the alleles promoted rapid loss of killer (K−), 9 promoted slow loss of the phenotype (Ksl), and Killer maintenance was unaffected by 14 alleles. Interestingly, two of the alleles conferred the Ski− phenotype (K++), a novel observation for a ribosomal protein. Programmed −1 ribosomal frameshifting efficiencies were also determined for the K− and Ksl alleles. Frameshifting efficiencies were significantly elevated by 10 of these mutants, decreased by 1, and unaffected by the remaining 5. These data are summarized in Table 2.
TABLE 2.
rpl3 alleles obtained from the primary screen
| Primary mutant | Mutation | Anisomycin resistancea | Killerb | % −1 PRFc |
|---|---|---|---|---|
| WT | 1+ | K+ | 8.8 ± 0.6 | |
| mak8-1 | W255C, P257T | 4+ | K− | 14.5 ± 0.4 |
| W255C, A3 | W255C | 4+ | K− | 14.2 ± 0.3 |
| P257T | P257T | 3+ | K− | 13.9 ± 0.4 |
| I282T, A9, A19, A30, A41, A43, A49, A52 | I282T | 2+ | K− | 13.2 ± 0.3 |
| A1 | L387F | 2+ | K− | 11.3 ± 0.8 |
| A2 | Y49F, A107T | 3+ | K+ | ND |
| A4 | G225S, Y283C, T344S | 3+ | K++ | ND |
| A5 | S347F, K357M, K384T | 3+ | Ksl | 9.0 ± 0.7 |
| A6 | H256Q, I282T, L387F | 2+ | Ksl | 8.9 ± 0.6 |
| A7 | S2T, F16I, I278T, I282T, F365Y, K385T | 2+ | K− | 8.1 ± 0.8 |
| A8 | V312I, K357E, I282T | 2+ | K+ | ND |
| A10 | I282T, M261I | 3+ | K+ | ND |
| A11 | H198T, I282T | 2+ | K+ | ND |
| A12 | K66E, I282T, R348G, R369S | 2+ | K+ | ND |
| A13 | I282T, Y343N | 2+ | K+ | ND |
| A14 | T81A, I282T, A285V | 2+ | K+ | ND |
| A15 | I282T, E316V | 3+ | K+ | ND |
| A16, A23 | T54A, L99F, I282T | 3+ | K+ | ND |
| A18 | V76I, L171W, I282T | 2+ | K+ | ND |
| A20 | P18S, G141R, I282T, L387F | 3+ | K+ | ND |
| A22 | F67L, I282T | 2+ | K+ | ND |
| A27 | K201E, N212S, A267S, I282T, L387F | 2+ | Ksl | 11.3 ± 0.8 |
| A29 | L17S, H259L, I282T | 2+ | K− | 12.1 ± 0.8 |
| A31 | L171W, I282T | 2+ | K+ | ND |
| A36 | V90A, W122G, I282T, L338S | 2+ | K+ | ND |
| A37 | I282T, K367M | 2+ | K++ | ND |
| A38 | I282T, T382S | 2+ | Ksl | 17 ± 0.6 |
| A42 | E227V, I282T, T382S | 3+ | Ksl | 16.4 ± 0.2 |
| A44 | G15C, I282T | 2+ | K− | 11.6 ± 0.8 |
| A45 | I282T, L387F | 3+ | Ksl | 7.3 ± 0.2 |
| A48 | K120N, I282T | 3+ | Ksl | 7.9 ± 0.4 |
| A50 | R196L, I282T, F298I | 3+ | Ksl | 14.6 ± 0.8 |
| A53 | H273Q, I282T, K333E | 3+ | Ksl | ND |
| A54 | I282T, D299G | 3+ | K− | ND |
Nomenclature used to describe relative resistance to anisomycin refers to Fig. 1, where 1+ is wild type and additional pluses indicate the relative ability of 10-fold dilutions of cells to grow.
Killer phenotype of the mutants. K+ is wild type, K++ represents the superkiller (Ski−) phenotype, Ksl indicates slow but eventual killer loss, and K− means complete inability to maintain the phenotype.
FIG. 1.

Anisomycin resistance phenotypes of sample rpl3 alleles. Ten-fold dilutions of cells harboring the indicated rpl3 alleles were spotted onto complete synthetic medium lacking tryptophan alone, or the same medium containing anisomycin (50 μg/ml), and were incubated at 30°C for 3 days. A. Scoring of representative primary mutants. Strains and anisomycin resistance scores are indicated. B. Anisomycin enhances growth of the T344S allele.
T282I reversion alleles.
The striking feature of new mutants was that 49 of 54 contained the previously identified I282T mutation (26 different alleles) (Table 2). Oligonucleotide site-directed mutagenesis was used to revert this residue back to Ile in 23 of the alleles to evaluate the contribution of the accompanying mutations to the observed phenotypes (unpublished data). One of these mutants was lethal (V90A+W122G+L338S), and all but one of the remaining revertants (K367M) lost their resistance to anisomycin (shown in Table 3, below), an observation that suggests that I282T was a critical contributor to anisomycin resistance (though increased levels of anisomycin resistance were observed for this mutation in combination with changes at other positions) (Table 2). However, 12 of reversion mutants lost the ability to maintain killer virus (Table 3), even though the original combinations of these mutations with I282T (which by itself is K−), were K+. The efficiencies of −1 PRF were all significantly elevated in the K− T282I reversion alleles (Table 3), suggesting that the I282T mutation may moderate the severity of mutations at other positions. We hypothesize that in screening for anisomycin resistance, we collected the set of mutants that are responsible for drug resistance while simultaneously selecting for the healthiest mutants, which were in turn Killer+. The decoupling of killer maintenance defects from anisomycin resistance makes this class of mutants of particular interest for further analysis.
TABLE 3.
Single mutantsa
| Allele | Original mutant | Anisomycin | Killer | % −1 PRF |
|---|---|---|---|---|
| WT | 1+ | K+ | 8.8 ± 0.6 | |
| S2T | A7 | 3+ | K+ | ND |
| G15C | A44 | 1+ | K− | 14.4 ± 0.7 |
| F16I | A7 | 2+ | K+ | ND |
| L17S | A29 | 2+ | K++ | ND |
| P18S | A20 | 3+ | K++ | ND |
| Y49F | A2 | Hypersensitive | K++ | ND |
| T54A | A16,A23 | 2+ | K++ | ND |
| L99F | A16,A23 | 1+ | K++ | ND |
| A107T | A2 | 2+ | K+ | ND |
| W122G | A36 | 1+ | K++ | ND |
| G141R (Lethal) | A20 | NA | NA | NA |
| H198T | A11 | 1+ | K− | 15.5 ± 0.4 |
| G225S | A4 | 2+ | K++ | ND |
| W255C | A3, prev. study | 4+ | K− | 14.2 ± 0.3 |
| H256Q | A6 | 2+ | K+ | ND |
| P257T | Previous study | 3+ | K− | 13.9 ± 0.4 |
| M261I | A10 | 2+ | K− | 11.3 ± 0.7 |
| I282T | Many alleles | 2+ | K− | 13.2 ± 0.3 |
| Y283C | A4 | 1+ | K++ | ND |
| V312I | A8 | 2+ | K+ | ND |
| E316V | A15 | 1+ | K− | 15.2 ± 0.8 |
| L338S | A36 | 1+ | K++ | ND |
| Y343N | A13 | 1+ | K− | 11.9 ± 0.8 |
| T344S | A4 | Growth enhanced | K++ | ND |
| S347F | A5 | 2+ | K+ | ND |
| K357E | A8 | 2+ | K+ | ND |
| F365Y | A7 | 1+ | K++ | ND |
| K367M | A37 | 2+ | K+ | ND |
| R369S | A12 | 2+ | K++ | ND |
| K384T | A5 | 2+ | K++ | ND |
| K385T | A7 | 3+ | K+ | ND |
| L387F | Many alleles | 2+ | K− | 11.3 ± 0.8 |
Tabulation of the anisomycin resistance or sensitivity, killer maintenance, and programmed −1 ribosomal frameshifting profiles of all rpl3 alleles harboring single amino acid changes. Phenotypic scoring and nomenclature are as described for Table 1. NA, not applicable.
Characterization of single mutations.
Reversion of I282T back to wild type was also helpful insofar as it aided in weeding out changes that did not promote mutant phenotypes. The remaining multiple mutants that retained phenotypes after this reversion analysis were selected to test the effects of specific, single mutations at the primary amino acid level. The properties of 32 single mutants are summarized in Table 3. One (G141R, derived from mutant A20) was inviable, and single amino acid substitution mutants not affecting anisomycin resistance and killer virus maintenance defects are not shown. Interestingly, examples of mutants having opposing anisomycin phenotypes were obtained. For example, Y49F was anisomycin hypersensitive (data not shown), while high concentrations of anisomycin actually enhanced growth of the T344S allele (Fig. 1B). Analysis of the data in Table 3 also reveals that responses to anisomycin are separable from frameshifting and killer virus maintenance phenotypes, suggesting that these two phenotypes can be used as tools to dissect different functional aspects of the ribosome. Notably, different single mutations conferred phenotypes opposite those observed when they occurred in combination with other mutations. For instance, although I282T by itself conferred both the Anr and K− phenotypes, combining this with mutations at other positions, e.g., in A10 (M261I and I282T), A11 (H198T and I282T), A13 (Y343N and I282T), and A15 (E316V and I282T), resulted in cells that were able to maintain the killer phenotype (K+) despite remaining Anr. Furthermore, the single mutants based on these double mutants, i.e., M261I, H198T, Y343N, and E316V, were all K− Ans. Similarly, mutations of single amino acids alone that remained K+ promoted K− phenotypes when in combination with other changes (e.g., V76I plus L171W, T81A plus A295V, T54A plus L99F, and V312I plus K357E).
As noted above, different rpl3 alleles promoted either killer loss (Mak−) or superkiller (Ski−) phenotypes, examples of which are shown in Fig. 2A. This is the first observation that mutations of a ribosomal protein can confer the Ski− phenotype, and it is notable that different mutations of the same protein can either stimulate or repress virus replication. In all cases of the single mutants, the Mak− phenotype also correlated with frameshifting defects (maintenance of frame [Mof−]) (Table 3). As shown by the example in the lane labeled rpl3-Mak− of Fig. 2B, loss of the killer phenotype strains was a consequence of the inability of these cells to support propagation of M1 dsRNA satellite virus. Loss of the killer phenotype could be due to indirect effects, e.g., defects in the translation or processing of the M1-encoded killer toxin, or 60S subunit biogenesis defects, rather than changes affecting −1 PRF, which should also affect propagation of L-A, the helper virus of M1. Figure 2B also shows that expression of these rpl3 alleles either eliminated or dramatically reduced L-A dsRNA copy numbers, supporting the hypothesis that the loss of killer is directly due to changes in −1 PRF on the L-A mRNA as opposed to, e.g., general 60S ribosomal subunit biogenesis defects (37).
FIG. 2.

Killer virus maintenance profiles of representative rpl3 mutants. A. Killer phenotypes of wild-type RPL3 and rpl3 alleles that promoted the Ski− and Mak− phenotypes. JD1090 cells expressing pRPL3 were used as the wild-type example, mutant L17S was used as the rpl3-Ski− example, and W255C was used as the rpl3-Mak− example. B. Analysis of viral dsRNAs. The colonies from panel A were used as sources of RNA, and dsRNAs isolated from JD19 (ski3) were included for comparison. The L-A and M1 dsRNAs are responsible for the killer phenotype. L-BC is a dsRNA related to L-A but does not affect killer phenotype. Chromosomal DNA is also indicated.
Decreased peptidyltransfer rates correlate with increased −1 PRF and killer virus loss.
Changes in PRF efficiency have profound negative impacts on virus propagation by changing the ratio of structural to enzymatic proteins available for virus particle assembly (12). Programmed −1 ribosomal frameshifting is kinetically driven, and it has been hypothesized that decreased rates of peptidyltransfer would allow ribosomes paused at the slippery site more time to shift, thus promoting increased rates of −1 PRF (23). This prediction has been borne out by both genetic and pharmacological approaches (11, 31, 33). Programmed −1 ribosomal frameshifting can also be influenced by changes in other biophysical parameters of the ribosome that promote pausing, e.g., by altered affinities for tRNAs (31, 32) and changes in rates of accommodation of aa-tRNAs (28). Thus, mutants that alter −1 PRF can be used to probe the relationship between ribosome structure and function.
The peptidyltransferase activities of ribosomes purified from two mutants and wild-type strains were characterized. The two mutants were selected to represent one from each phenotypic category related to killer virus maintenance: P18S is a superkiller, and W255C cannot maintain killer because of increased −1 PRF. To evaluate peptidyltransferase activity rates, complex C, i.e., a ternary complex composed of ribosomes, Ac-Phe-tRNA, and poly(U), was first isolated. This approach served to separate process steps related to the binding of the donor tRNA to the ribosome from peptide bond formation, ensuring that each ribosome's peptidyltransferase center performs only one round of catalysis according to the following scheme:
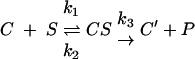 |
where C is complex C, S is substrate (puromycin), and P is product Ac-Phe-puromycin, quantitated by extraction in ethyl acetate. A time course series of single-round peptidyltransferase reactions using a fixed concentration of puromycin (2 mM) is shown in Fig. 3. In Fig. 3A the results are plotted as percentage of bound donor (Ac-Phe-tRNA) reacted (x). The reactions ran to 69% to 92% depletion of substrate with reduced velocity for mutant ribosomes compared to wild type. Figure 3B shows the corrected values of x′ (x′ = x/Aα) fitted to a first-order time plot. Instead of the initial rate, an apparent first-order rate constant (kobs) was used to evaluate the entire course of reaction. The peptidyltransferase reaction follows the rate law: ln[Co/(Co − P)] = ln[100/(100 − x′)] = kobst, where Co is the concentration of complex C at time zero and P is the concentration of the product formed in the time interval t. The slopes of the first-order time plots, i.e., kobs, are independent of the initial concentration of complex C. At a concentration of puromycin 2 mM, apparent rate constants for wild-type, P18S, and W255C ribosomes were 0.27 min−1, 0.14 min−1, and 0.05 min−1, respectively. Thus, there is a good correlation between the defects in virus propagation defects caused by increased −1 PRF and decreased peptidyltransferase activities. However, P18S retained the superkiller phenotype despite the near-twofold decrease in peptidyltransfer activity. As discussed below, multiple factors aside from frameshifting can influence killer virus maintenance.
FIG. 3.

Characterization of peptidyltransferase activities and affinities for aa-tRNAs by wild-type and two mutant ribosomes. Ribosomes isolated from yeast cells expressing the wild-type or P18S or W255C mutant forms of Rpl3 were used for the assays. A. Time course assay of the percent bound donor reacted. Single-round peptidyltransfer assays were performed using isolated complex C [120 pmol ribosomes plus 0.4 mg/ml poly(U) and 100 pmol Ac-[14C]Phe-tRNA]. Reactions were initiated by adding puromycin and terminated at the indicated time intervals by addition of NaOH. Reaction products were extracted with ethyl acetate, and radioactivity was determined by scintillation counting as described in Materials and Methods. B. Determination of Kobs by first-order time plot. The percentage of bound Ac-[14C]Phe-tRNA converted to Ac-[14C]Phe-puromycin (x) was corrected with the complex C stability factor and “extent factor” α, as previously described (15, 54). The value of x′ was obtained for various time intervals and fitted into the integrated form of a first-order reaction, such as Kobs · t = ln[100/(100 − x′)], which represents a straight line. The slope of this line gives the value of Kobs, the apparent rate constant of the entire course of the reaction at 2 mM puromycin. C. aa-tRNA binding studies. aa-tRNA binding to the A-site of the ribosome was carried out in reaction mixtures containing 12 to 25 pmol of ribosomes primed with 0.4 μg/ml of poly(U). Reaction mixtures were preincubated with uncharged tRNAs (4:1 tRNA-ribosomes) at 30°C for 15 min to ensure full occupation of P-sites by uncharged tRNA, after which increasing amounts of (4 to 264 pmol) of [14C]Phe-tRNA were added. Incubations continued for 15 min at 30°C to allow formation of [14C]Phe-tRNA-80S-poly(U) complexes. Aliquots were applied onto nitrocellulose membranes, filters were washed with binding buffer, and radioactivity was measured by scintillation counting. The data were plotted onto a double reciprocal Scatchard plot, and Ka values were determined as the slopes of the linear regression trend lines.
Correlation of anisomycin resistance and increased ribosome affinity for aa-tRNA.
Anisomycin increases the dissociation rate of aa-tRNAs from ribosomes (7, 20) by competing for the binding site of the aa-tRNA 3′ end in the peptidyltransferase center (21). Thus, mutants resistant to this drug might be expected to have increased affinities for aa-tRNAs. To test this model, aa-tRNA binding kinetics to the A-site were determined for the ribosomes purified from the same three strains, taking care to block the nonspecific binding to ribosomal P- and E-sites by preincubating ribosomes with uncharged tRNAs. The results of these experiments are represented in the form of a Scatchard plot in Fig. 3C. This analysis reveals that the Kas of wild-type, P18S, and W255C ribosomes for aa-tRNAs are 1.1 × 106 M−1, 4.3 × 106 M−1, and 7.3 × 106 M−1, respectively. Thus, as predicted, the anisomycin-resistant strains have increased A-site affinities for aa-tRNA.
The Ski− phenotype is caused by enhanced translation of cap−/poly(A)− mRNAs.
Mutants of the SKI2, -3, -4, -6, -7, and -8 genes derepress L-A and M1 dsRNA copy numbers (reviewed in reference 62), but ski1/xrn1 mutants do not (57). In contrast to the rpl3 alleles that promoted the Mak− phenotype, those promoting the Ski− phenotype resulted in increased copy numbers of both L-A and M1 dsRNAs compared to wild type, though not to as great an extent as ski2 or ski3 mutants. An example appears in Fig. 2B. The L-A and M1 mRNAs lack 5′ 7mGppp caps and poly(A) tails (6, 56). Previous studies have shown that expression of uncapped, poly(A)− mRNAs is derepressed in ski2, ski3, ski4, ski6, ski7, and ski8 mutants but not in those of xrn1/ski1 (2, 3, 30, 65). To examine this issue, lacZ expression plasmids in which transcription was driven from either RNA polymerase I (RDN1-lacZ) or RNA polymerase II (PGK1-lacZ) promoters were employed. Wild-type cells and rpl3 mutants that conferred the strongest Ski− phenotypes were chosen for comparison, and ski1, ski2, and ski3 strains were used as positive controls. Examination of the ratios of β-galactosidase expressed from these two different constructs revealed increased levels of translational derepression of the RNA polymerase I transcript in the strong Ski− rpl3 alleles compared to the wild-type and ski1 control (Table 4). As observed for viral dsRNA copy numbers, the extent of derepression tended to be less than that observed for the ski2 and ski3 mutants. The observation that translation of this transcript was not derepressed by deletion of xrn1/ski1 suggests that increased relative expression of β-galactosidase by this class of rpl3 alleles is due to a diminished ability to repress translation of cap−, poly(A)− mRNAs as opposed to stabilization of this reporter mRNA.
TABLE 4.
Relative abilities of rpl3 mutants to translate uncapped poly(A)− mRNAsa
| Strain | Killer | β-Galactosidase activity
|
(Pol I)/(Pol II)b | Fold WTc | |
|---|---|---|---|---|---|
| RDN1-lacZ | PGK1-lacZ | ||||
| JD1090 | K+ | 181 ± 8 | 3,875 ± 116 | 4.7 | 1.0 |
| JD1123 (xrn1/ski1) | K++ | 85 ± 6 | 1,778 ± 77 | 4.8 | 1.0 |
| JD2 (ski2) | K++ | 31 ± 1 | 122 ± 5 | 25.4 | 5.4 |
| JD19 (ski3) | K++ | 1,328 ± 74 | 2,793 ± 32 | 47.5 | 10.1 |
| rpl3 allele (in JD1090) | |||||
| L17S | K++ | 332 ± 12 | 2,240 ± 85 | 14.8 | 3.2 |
| T54A | K++ | 161 ± 5 | 1,792 ± 85 | 8.9 | 1.9 |
| T344S | K++ | 116 ± 3 | 1,592 ± 72 | 7.3 | 1.6 |
| F365Y | K++ | 179 ± 6 | 1,774 ± 67 | 10.1 | 2.1 |
| K369S | K++ | 223 ± 4 | 1,929 ± 65 | 11.6 | 2.5 |
| K384T | K++ | 130 ± 8 | 1,770 ± 64 | 7.3 | 1.6 |
Cells were transformed with lacZ reporter plasmids in which transcription was driven from either RNA polymerase I (RDN1-lacZ) or RNA polymerase II (PGK1-lacZ) promoters. The RDN1-lacZ mRNA product is uncapped and does not contain a poly(A) tail, while the PGK1-lacZ mRNA has both 7mGppp caps and poly(A) tails. β-Galactosidase activities were normalized for total protein, experiments were repeated in triplicate on three separate occasions, and means and standard deviations were calculated.
Ratios obtained by dividing the β-galactosidase activities generated from the RDN1-lacZ reporter by those generated from the PGK1-lacZ reporter were multiplied by 100%.
(Pol I)/(Pol II) ratio of mutant divided by the wild-type ratio.
That many of the L3 mutants can promote Ski− phenotypes similar to the ski2/3/8 class of mutants is truly novel. In contrast to Xrn1p/Ski1p, which degrades uncapped mRNAs, the other Ski proteins are involved in functions associated with the presence or absence of poly(A) tails (reviewed in reference 38). Two nonexclusive models have been proposed for how these Ski proteins function. In the first, they act during translation initiation at the subunit joining step by helping ribosomes discriminate whether or not to initiate on poly(A)− versus poly(A)+ mRNAs (50). In the second model, the involvement in translation by this class of proteins is indirect: they act in the process of “nonstop mRNA decay” by recruiting the exosome to ribosomes stalled at the 3′ ends of transcripts lacking termination codons (17, 60). By this model, L3 might participate in the interaction between the ribosome and the Ski complex, and the Ski− L3 mutants may disrupt this binding site. To test this hypothesis, cells expressing the wild-type gene and three rpl3 alleles that conferred the Ski− phenotype (L17S, P18S, and Y283T) were transformed with plasmids expressing either FLAG-tagged Ski7p or myc-tagged Ski2p. Wild-type RPL3 cells transformed with empty vectors were used as controls. Cycloheximide-stalled ribosomes were isolated by ultracentrifugation from logarithmically growing cells, and proteins were separated by sodium dodecyl sulfate-PAGE, transferred to PVDF membranes, and probed using anti-FLAG or antihemagglutinin antibodies. Control blots were probed using an anti-L3 antibody. The results of these experiments demonstrated there were no apparent quantitative differences in the abilities of mutant ribosomes to associate with either FLAG-Ski7p (Fig. 4) or myc-Ski2p (data not shown) compared to wild type. These data lend further support to the hypothesis that the Ski− phenotype in these mutants is due to enhanced translation of cap−, poly(A)− mRNAs as opposed to stabilization of this reporter mRNA.
FIG. 4.

The superkiller rpl3 mutants do not confer defects on the ability of mutant ribosomes to recruit the Ski-complex to mRNAs. JD1090 cells expressing pRPL3 (WT) or the L17S, P18S, or Y283C alleles were transformed with pAJ1203 (FLAG-tagged SKI7). WT-c represents wild-type cells transformed with a vector control. Cycloheximide-arrested ribosomes were purified by ultracentrifugation from logarithmically growing cells, separated through sodium dodecyl sulfate-PAGE, transferred to PVDF membranes, and probed using anti-FLAG or anti-L3 antiserum. Locations of the Ski7-FLAG and L3 are indicated. Anti-FLAG-reactive nonspecific bands are indicated by an asterisk.
DISCUSSION
Although accumulating evidence indicates that rRNA is the main, and perhaps only, catalytic component of the ribosome, mutational analysis has long ascribed significant functional relevance to several ribosomal proteins (10, 18, 36, 53). Phenotypic expression of RPL3 mutants (effects on translational accuracy, resistance to PTC specific translation inhibitors and PAP protein) and the structural features indicates that L3 belongs to this class of ribosomal proteins. The questions are whether the functional effects of the mutations characterized in this study are due to changes in rRNA structure conferred by the mutant proteins, or might L3 itself be directly involved in catalysis and interaction with anisomycin? Examination of the X-ray crystal structure of the H. marismortui large subunit suggests that the latter possibility is unlikely, as no amino acid residues of L3 are close enough to the PTC catalytic center to be directly involved in its activity, nor are any positioned to directly interact with anisomycin (1, 21).
Examination of the structure of L3 reveals that it contains two globular domains and two extensions (29). The globular domains are located on the solvent side of the large subunit near helices 94 and 96 and are positioned flanking the sarcin/ricin loop (SRL), which comprises an important site of interaction between the ribosome and the elongation factors eEF1 and eEF2. One of the extensions is at the N terminus of the protein (residues 1 to 22 in H. marismortui and 1 to 24 in S. cerevisiae), and the other is internal to the protein (206 to 260 in H. marismortui and 217 to 278 in S. cerevisiae) and reaches deep into the core of the large subunit. W255 is located on the tip of the latter extension (the tryptophan finger). Figure 5A maps the amino acids of the original yeast mutants to the proposed structure of S. cerevisiae L3 (19, 51). In Fig. 5B, the single amino acid substitutions listed in Table 3 are color coded according to phenotype. Examination of these figures reveals that the mutations identified in this study are located throughout the structure of the protein.
FIG. 5.

Linking L3 structure to function. A to C. Modeling of the L3 mutants. A. Locations of all mutant amino acids from the original screen mapped onto the structure of yeast L3, based on reference 52. B. Locations of all single mutants that result in phenotypes mapped onto the yeast L3 molecular structure. Blue, K− and increased −1 PRF; red, anisomycin resistant; black, superkiller only; yellow, effects on killer (either K− or superkiller) and anisomycin resistant. C. Wider view showing L3 in context from the PTC to the SRL. The SRL is an important site of interaction between the ribosome and the translation elongation factors eEF1 and eEF2. A- and P-loops, sites of interaction with the 3′ ends of the aa- and peptidyl-tRNAs, respectively, are labeled A and P. Helices of the 25S rRNA that are involved in forming the continuum between the PTC, L3, and the SRL are numbered. D to F. Model to explain anisomycin resistance of the rpl3 alleles. D. Tip of the L3 finger and the PTC with anisomycin. The central adenosine in the PTC (equivalent to E. coli A2451) is indicated in red. Coordinates were derived from reference 21. E. Cartoon of image in panel D, with wild-type L3. The peptidyl-tRNA 3′ end is shown in blue, and aa-tRNA is in magenta. F. Mutant forms of L3 destabilize the A-site proximal region of the PTC, allowing room for both anisomycin and the acceptor stem.
The rRNA-protein contacts have been mapped in the high-resolution structure of the H. marismortui 50S subunit (29), and a lower-resolution structure of the yeast ribosomal proteins threaded into the H. marismortui 23S rRNA is also available (52). We have used these structures to visualize the interrelationships between L3, the PTC, the SRL, and other related 25S rRNA sequences (Fig. 5C). Strikingly, the pattern of rpl3 mutants identified in this study mirrored the interactions observed between L3 and 23S rRNA in the X-ray crystal structure of the H. marismortui 50S subunit (29). Specifically, most of the rpl3 mutations were located in close proximity to helices 61, 72, 73, 90, 92, 94, 96, 100, and 101, while the majority of the H. marismortui L3 contacts mapped to the same helices. This analysis suggests that these mutations may affect the contacts between L3 and 25S rRNA. Although we cannot conclude that these genetic studies have identified amino acids of S. cerevisiae L3 that are directly involved in interactions with rRNA bases, this map provides evidence for involvement of L3 with specific functional regions of 25S rRNA, specifically with the A-site proximal region of the PTC and with the environment of the SRL.
The L3 mutations identified in this study can be divided into three separate classes according to their topological features. The first two classes represent mutations in regions likely to be involved in interactions with rRNA bases, i.e., (i) substitutions of amino acids in the extensions and (ii) mutations in the globular domains. With regard to the class 1 mutants, those at the tip of the tryptophan finger (e.g., W255C, H256Q, and P257T) displayed the most pronounced phenotypes: they were all anisomycin resistant and two could not maintain the killer virus. W255 makes the closest approach of any amino acid in the ribosome to the PTC active center, extending to the A-site proximal side of the PTC, where it can make numerous stacking interactions with rRNA (Fig. 5C). The cluster of mutations in the N-terminal extension of L3 can potentially make numerous contacts with helices 90, 94, and 96. Considering the class 2 mutants, the globular region of H. marismortui L3 contains two tightly packed globular domains which interact with helices 94 and 96, and it was proposed that these interactions work together to stabilize the tertiary structure of domain VI, which contains the SRL and with which L3 makes extensive contacts (29). Most class 2 mutants identified in this study are positioned within H-bonding distances to helices 94 and 96. Also noteworthy are the substitutions of basic residues in the C-terminal tail that extend toward helix 96. Although the precise topology of these residues in the yeast ribosome is not yet clear, it was proposed that basic residues in extension regions of ribosomal proteins neutralize the negatively charged rRNA backbone, which is likely critical for proper folding of rRNA (29). The third class of mutations represents substitutions of amino acids far away from rRNA bases. In light of the above considerations, we believe these mutations may promote conformational changes in L3 that affect the positions of amino acids involved in rRNA interactions. For instance, the I282T mutation may affect the orientation and/or flexibility of the middle extension, thus promoting phenotypes similar to but weaker than mutations in the tryptophan finger. It is also conceivable that the mutations generated in this study could affect L3 by altering potential protein modification sites. Analyses of the mutations by using the PROSITE protein signatures database (http://us.expasy.org/prosite/) revealed several substitutions which could potentially inactivate or create new phosphorylation sites on the solvent-accessible face of L3 (e.g., K384T) (data not shown).
Models to explain observed phenotypes of the mutants.
Combining the structural and genetic analyses enables us to propose the following models of how the structure of L3 works to determine its function. Biochemical and structural studies designed to test the models proposed here are currently under way.
Resistance to anisomycin.
As shown in Fig. 3C, there is a correlation between anisomycin resistance and increased ribosomal Ka for aa-tRNA. Examination of the cocrystal structure of the H. marismortui large subunit and anisomycin reveals that the aromatic ring of anisomycin approaches the active site crevice from the opposite direction as the amino acid side chain that extends from the A-site substrate (aa-tRNA) (21). A cartoon of this is shown in Fig. 5D. Anisomycin makes a number of specific contacts with the A-site of the peptidyltransferase center, the most interesting of which is the ability of its p-methoxyphenyl group to occupy the hydrophobic crevice that normally accepts the amino acid side chains of A-site-bound aa-tRNAs. This allows anisomycin to compete with the amino acid for access to the peptidyltransferase center and is consistent with reports that anisomycin interferes with the binding of A-site substrates (reviewed in reference 40). This is cartooned in Fig. 5E. As diagrammed in Fig. 5F, we hypothesize that conformational changes in the PTC induced by the L3 mutants may increase the size of this crevice by disrupting interactions with neighboring bases, thus providing enough space for the A-site substrate to access the PTC active site in the presence of anisomycin. This may also help to deepen the binding pocket for the aa-tRNA, resulting in the observed increase in affinity. This mechanism is supported by observation that the original anisomycin-resistant tcm1 mutation (W255C) did not prevent binding of anisomycin to ribosomes (25, 26). Interestingly, specific mutations in the 23S rRNAs of many organisms have also been shown to confer resistance to anisomycin, and these mapped to bases that normally stabilize the structure of the active site crevice (21). An alternative explanation has been offered that such mutations make the PTC smaller, thus excluding anisomycin (44, 48).
Increased −1 ribosomal frameshifting.
An mRNA pseudoknot-induced ribosomal pause is integral to the mechanism of −1 PRF, and decreased rates of peptidyltransfer promote increased −1 PRF efficiencies, presumably by increasing the amount of time for paused ribosomes to shift. We propose that the L3 mutations that affect −1 PRF induce conformational changes in the PTC region, which in turn affect the proper positioning of A- and P-site substrates, resulting in altered peptidyltransferase activities. As demonstrated by the P18S mutant, however, it must be noted that there is not a total correspondence between decreased peptidyltransferase activity and killer virus maintenance: other considerations, e.g., the ability of ribosomes to discriminate between cap+/poly(A)+ and cap−/poly(A)− mRNAs, must be factored into the final outcome. Two additional, nonexclusive mechanisms could also be operative. First, that the mutants may promote changes in the interactions between the aa-tRNA 3′ ends and/or their misalignment with the PTC. Such changes could be transduced down the body of the aa-tRNA to the decoding center, destabilizing the codon-anticodon interactions and thus helping to facilitate frameshifting. A second possibility arises from the observation that stronger interactions between the Hirsch suppressor tRNA and the ribosome serve to increase the rate of EF-Tu activation and GTPase hydrolysis and aa-tRNA accommodation, resulting in increased rates of nonsense suppression (8, 9). The 9-Å model of −1 PRF predicts that increased rates of accommodation would promote increased −1 PRF frequencies (42), and increased affinities for aa-tRNAs have been demonstrated for ribosomes containing the mak8-1, W255C, P257T, and I282T forms of L3 (41). Thus, it is possible that accommodation rates could be enhanced by a subset of the rpl3 mutants, resulting in increased −1 PRF efficiencies.
Induction of the superkiller phenotype.
As noted above, the Ski− phenotype could have resulted from an intrinsic defect in ribosomes ability to discriminate between mRNAs having or lacking caps and/or polyA tails, or they could be due to a defect in the abilities of mutant ribosomes to recruit the Ski-complex. The immunoblot assay data (Fig. 4 and not shown) demonstrating that the mutant ribosomes were not defective in their abilities to interact with the Ski complex show that the Ski− phenotype can be induced independently of this interaction. Thus, though these findings do not further our understanding of the Ski complex per se, for this class of mutants, they do suggest that ability of the translational complex to discriminate between mRNAs with or without 5′ cap structures and/or poly(A) tails can be an intrinsic feature of the ribosome itself.
Does L3 function as a transducer of information between the PTC and the SRL?
A striking feature of Fig. 5B is that there is no correlation between any specific phenotypes and the physical location of mutations within the protein. However, as shown in Fig. 5C, their localization close to the bases in helices 73, 90, 94, and 96 suggests that L3 may be involved in a functional continuum between the PTC and the SRL. We suggest that L3 may act to help communicate the tRNA occupancy status of the PTC to the SRL. What is particularly striking is the potential for the tip of the L3 “finger” to move into and out of the PTC from the A-site side in response to conformational changes in other parts of protein. We hypothesize that when the A-site is occupied by aa-tRNA in the A/A state, the W255 tip of L3 is excluded from the PTC. This movement would be transduced down the tryptophan finger to the globular domains of L3 and over to the SRL (and also possibly indirectly to helix 89, which interacts with domain V of eEF-2), with the result that the SRL is no longer able to interact with eEF-1. After peptidyltransfer, the A-site would be semioccupied (peptidyl-tRNA is in the A/P hybrid state), the tip of L3 could move into this space, and this movement would be transduced to reposition the SRL (and helix 89?), so that it could then interact with eEF-2. After translocation and release of eEF-2, the A-site would be wholly unoccupied, repositioning the SRL to be able to interact with eEF-1. Accommodation of new aa-tRNA into the A-site would complete the cycle. By this mechanism, the proper coordination between these two functional centers of the large subunit might be ensured. Precedence for such an allosteric model of intermolecular communication can be found in the mechanism by which a cognate codon-anticodon interaction at the ribosomal decoding center is transduced through the body of the aa-tRNA to activate the GTPase activity of EF-Tu (reviewed in reference 43). The anisomycin resistance phenotype of mutations in globular domains positioned far away from the actual drug binding site provides additional support for this allosteric model.
Acknowledgments
We thank Arlen Johnson, Reed Wickner, and Jonathan Warner for plasmids, strains, and antibodies. We also thank Ewan Plant, Rasa Rakauskaite, Jennifer Baxter-Roshek, and John Russ for their insightful comments.
This work was supported by grants from the National Institutes of Health to J.D.D. (GM58859) and the National Science Foundation (MCB 0130531).
REFERENCES
- 1.Ban, N., P. Nissen, J. Hansen, P. B. Moore, and T. A. Steitz. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905-920. [DOI] [PubMed] [Google Scholar]
- 2.Benard, L., K. Carroll, R. C. P. Valle, D. C. Masison, and R. B. Wickner. 1999. The ski7 antiviral protein is an EF1-alpha homolog that blocks expression of non-poly(A) mRNA in Saccharomyces cerevisiae. J. Virol. 73:2893-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benard, L., K. Carroll, R. P. C. Valle, and R. B. Wickner. 1998. Ski6p is a homolog of RNA-processing enzymes that affects translation of non-poly(A) mRNAs and 60S ribosomal subunit biogenesis. Mol. Cell. Biol. 18:2688-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosling, J., S. M. Poulsen, B. Vester, and K. S. Long. 2003. Resistance to the peptidyl transferase inhibitor tiamulin caused by mutation of ribosomal protein l3. Antimicrob. Agents Chemother. 47:2892-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. T., X. Bai, and A. W. Johnson. 2000. The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6:449-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruenn, J., and B. Keitz. 1976. The 5′ ends of yeast killer factor RNAs are pppGp. Nucleic Acids Res. 3:2427-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrasco, L., M. Barbacid, and D. Vazquez. 1973. The tricodermin group of antibiotics, inhibitors of peptide bond formation by eukaryotic ribosomes. Biochim. Biophys. Acta 312:368-376. [DOI] [PubMed] [Google Scholar]
- 8.Cochella, L., and R. Green. 2005. An active role for tRNA in decoding beyond codon:anticodon pairing. Science 308:1178-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daviter, T., F. V. Murphy, and V. Ramakrishnan. 2005. A renewed focus on transfer RNA. Science 308:1123-1124. [DOI] [PubMed] [Google Scholar]
- 10.Deusser, E., G. Stoffler, and H. G. Wittmann. 1970. Ribosomal proteins. XVI. Altered S4 proteins in Escherichia coli revertants from streptomycin dependence to independence. Mol. Gen. Genet. 109:298-302. [DOI] [PubMed] [Google Scholar]
- 11.Dinman, J. D., M. J. Ruiz-Echevarria, K. Czaplinski, and S. W. Peltz. 1997. Peptidyl transferase inhibitors have antiviral properties by altering programmed −1 ribosomal frameshifting efficiencies: development of model systems. Proc. Natl. Acad. Sci. USA 94:6606-6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dinman, J. D., and R. B. Wickner. 1992. Ribosomal frameshifting efficiency and Gag/Gag-pol ratio are critical for yeast M1 double-stranded RNA virus propagation. J. Virol. 66:3669-3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinman, J. D., and R. B. Wickner. 1994. Translational maintenance of frame: mutants of Saccharomyces cerevisiae with altered −1 ribosomal frameshifting efficiencies. Genetics 136:75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dresios, J., I. L. Derkatch, S. W. Liebman, and D. Synetos. 2000. Yeast ribosomal protein L24 affects the kinetics of protein synthesis and ribosomal protein L39 improves translational accuracy, while mutants lacking both remain viable. Biochemistry 39:7236-7244. [DOI] [PubMed] [Google Scholar]
- 15.Dresios, J., P. Panopoulos, C. P. Frantziou, and D. Synetos. 2001. Yeast ribosomal protein deletion mutants possess altered peptidyltransferase activity and different sensitivity to cycloheximide. Biochemistry 40:8101-8108. [DOI] [PubMed] [Google Scholar]
- 16.Fried, H. M., and J. R. Warner. 1981. Cloning of yeast gene for trichodermin resistance and ribosomal protein L3. Proc. Natl. Acad. Sci. USA 78:238-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frischmeyer, P. A., A. van Hoof, K. O'Donnell, A. L. Guerrerio, R. Parker, and H. C. Dietz. 2002. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295:2258-2261. [DOI] [PubMed] [Google Scholar]
- 18.Funatsu, G., and H. G. Wittmann. 1972. Ribosomal proteins. 33. Location of amino-acid replacements in protein S12 isolated from Escherichia coli mutants resistant to streptomycin. J. Mol. Biol. 68:547-550. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Lorenzo, M. G., C. M. Spahn, R. K. Agrawal, R. A. Grassucci, P. Penczek, K. Chakraburtty, J. P. Ballesta, J. L. Lavandera, J. F. Garcia-Bustos, and J. Frank. 2000. Three-dimensional cryo-electron microscopy localization of EF2 in the Saccharomyces cerevisiae 80S ribosome at 17.5 Å resolution. EMBO J. 19:2710-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grollman, A. P. 1967. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J. Biol. Chem. 242:3226-3233. [PubMed] [Google Scholar]
- 21.Hansen, J. L., P. B. Moore, and T. A. Steitz. 2003. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 330:1061-1075. [DOI] [PubMed] [Google Scholar]
- 22.Harger, J. W., and J. D. Dinman. 2003. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. RNA 9:1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harger, J. W., A. Meskauskas, and J. D. Dinman. 2002. An “integrated model” of programmed ribosomal frameshifting and post-transcriptional surveillance. Trends Biochem. Sci. 27:448-454. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs, J. L., and J. D. Dinman. 2004. Systematic analysis of bicistronic reporter assay data. Nucleic Acids Res. 32:e160-e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jimenez, A., L. Sanchez, and D. Vazquez. 1975. Simultaneous ribosomal resistance to trichodermin and anisomycin in Saccharomyces cerevisiae mutants. Biochim. Biophys. Acta 383:427-434. [DOI] [PubMed] [Google Scholar]
- 26.Jimenez, A., and D. Vazquez. 1975. Quantitative binding of antibiotics to ribosomes from a yeast mutant altered on the peptidyl-transferase center. Eur. J. Biochem. 54:483-492. [DOI] [PubMed] [Google Scholar]
- 27.Kaneko, I., and R. H. Doi. 1966. Alteration of valyl-sRNA during sporulation of bacillus subtilis. Proc. Natl. Acad. Sci. USA 55:564-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinzy, T. G., J. W. Harger, A. Carr-Schmid, J. Kwon, M. Shastry, M. C. Justice, and J. D. Dinman. 2002. New targets for antivirals: the ribosomal A-site and the factors that interact with it. Virology 300:60-70. [DOI] [PubMed] [Google Scholar]
- 29.Klein, D. J., P. B. Moore, and T. A. Steitz. 2004. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 340:141-177. [DOI] [PubMed] [Google Scholar]
- 30.Masison, D. C., A. Blanc, J. C. Ribas, K. Carroll, N. Sonenberg, and R. B. Wickner. 1995. Decoying the cap− mRNA degradation system by a double-stranded RNA virus and poly(A)− mRNA surveillance by a yeast antiviral system. Mol. Cell. Biol. 15:2763-2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meskauskas, A., J. L. Baxter, E. A. Carr, J. Yasenchak, J. E. G. Gallagher, S. J. Baserga, and J. D. Dinman. 2003. Delayed rRNA processing results in significant ribosome biogenesis and functional defects. Mol. Cell. Biol. 23:1602-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meskauskas, A., and J. D. Dinman. 2001. Ribosomal protein L5 helps anchor peptidyl-tRNA to the P-site in Saccharomyces cerevisiae. RNA 7:1084-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meskauskas, A., J. W. Harger, K. L. M. Jacobs, and J. D. Dinman. 2003. Decreased peptidyltransferase activity correlates with increased programmed −1 ribosomal frameshifting and viral maintenance defects in the yeast Saccharomyces cerevisiae. RNA 9:982-992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muhlrad, D., R. Hunter, and R. Parker. 1992. A rapid method for localized mutagenesis of yeast genes. Yeast 8:79-82. [DOI] [PubMed] [Google Scholar]
- 35.Nowotny, V., and K. H. Nierhaus. 1982. Initiator proteins for the assembly of the 50S subunit from Escherichia coli ribosomes. Proc. Natl. Acad. Sci. USA 79:7238-7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Connor, M., S. T. Gregory, and A. E. Dahlberg. 2004. Multiple defects in translation associated with altered ribosomal protein L4. Nucleic Acids Res. 32:5750-5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohtake, Y., and R. B. Wickner. 1995. Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol. Cell. Biol. 15:2772-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parker, R., and H. Song. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11:121-127. [DOI] [PubMed] [Google Scholar]
- 39.Peltz, S. W., A. B. Hammell, Y. Cui, J. Yasenchak, L. Puljanowski, and J. D. Dinman. 1999. Ribosomal protein L3 mutants alter translational fidelity and promote rapid loss of the yeast Killer virus. Mol. Cell. Biol. 19:384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pestka, S. 1977. Inhibitors of protein synthesis, p. 467-553. In H. Weissbach and S. Pestka (ed.), Molecular mechanismns of protein biosynthesis. Academic Press, New York, N.Y.
- 41.Petrov, A., A. Meskauskas, and J. D. Dinman. 2004. Ribosomal protein L3: influence on ribosome structure and function. RNA Biol. 1:59-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plant, E. P., K. L. M. Jacobs, J. W. Harger, A. Meskauskas, J. L. Jacobs, J. L. Baxter, A. N. Petrov, and J. D. Dinman. 2003. The 9-angstrom solution: how mRNA pseudoknots promote efficient programmed −1 ribosomal frameshifting. RNA 9:168-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poole, E. S., M. E. Askarian-Amiri, L. L. Major, K. K. McCaughan, D. J. Scarlett, D. N. Wilson, and W. P. Tate. 2003. Molecular mimicry in the decoding of translational stop signals. Prog. Nucleic Acid Res. Mol. Biol. 74:83-121. [DOI] [PubMed] [Google Scholar]
- 44.Pringle, M., J. Poehlsgaard, B. Vester, and K. S. Long. 2004. Mutations in ribosomal protein L3 and 23S ribosomal RNA at the peptidyl transferase centre are associated with reduced susceptibility to tiamulin in Brachyspira spp. isolates. Mol. Microbiol. 54:1295-1306. [DOI] [PubMed] [Google Scholar]
- 45.Rose, M. D., F. Winston, and P. Hieter. 1990. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 46.Schilling-Bartetzko, S., F. Franceschi, H. Sternbach, and K. H. Nierhaus. 1992. Apparent association constants of transfer-RNAs for the ribosomal A-site, P-site, and E-site. J. Biol. Chem. 267:4693-4702. [PubMed] [Google Scholar]
- 47.Schindler, D., P. Grant, and J. Davies. 1974. Trichodermin resistance—mutation affecting eukaryotic ribosomes. Nature 248:535-536. [DOI] [PubMed] [Google Scholar]
- 48.Schlunzen, F., E. Pyetan, P. Fucini, A. Yonath, and J. M. Harms. 2004. Inhibition of peptide bond formation by pleuromutilins: the structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 54:1287-1294. [DOI] [PubMed] [Google Scholar]
- 49.Schulze, H., and K. H. Nierhaus. 1982. Minimal set of ribosomal components for reconstitution of the peptidyltransferase activity. EMBO J. 1:609-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Searfoss, A., T. E. Dever, and R. Wickner. 2001. Linking the 3′ poly(A) tail to the subunit joining step of translation initiation: relations of Pab1p, eukaryotic translation initiation factor 5B (Fun12p), and Ski2p-Slh1p. Mol. Cell. Biol. 21:4900-4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spahn, C. M., R. Beckmann, N. Eswar, P. A. Penczek, A. Sali, G. Blobel, and J. Frank. 2001. Structure of the 80S ribosome from Saccharomyces cerevisiae—tRNA-ribosome and subunit-subunit interactions. Cell 107:373-386. [DOI] [PubMed] [Google Scholar]
- 52.Spahn, C. M., M. G. Gomez-Lorenzo, R. A. Grassucci, R. Jorgensen, G. R. Andersen, R. Beckmann, P. A. Penczek, J. P. Ballesta, and J. Frank. 2004. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 23:1008-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stoffler, G., E. Deusser, H. G. Wittmann, and D. Apirion. 1971. Ribosomal proteins. XIX. Altered S5 ribosomal protein in an Escherichia coli revertant from streptomycin dependence to independence. Mol. Gen. Genet. 111:334-341. [DOI] [PubMed] [Google Scholar]
- 54.Synetos, D., and C. Coutsogeorgopoulos. 1987. Studies on the catalytic rate constant of ribosomal peptidyltransferase. Biochim. Biophys. Acta 923:275-285. [DOI] [PubMed] [Google Scholar]
- 55.Theocharis, D. A., and C. Coutsogeorgopoulos. 1989. Recovery of active ribosomal complexes from cellulose nitrate membranes. Anal. Biochem. 176:278-283. [DOI] [PubMed] [Google Scholar]
- 56.Thiele, D. J., E. M. Hannig, and M. J. Leibowitz. 1984. Multiple L double-stranded RNA species of Saccharomyces cerevisiae: evidence for separate encapsidation. Mol. Cell. Biol. 4:92-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toh-e, A., P. Guerry, and R. B. Wickner. 1978. Chromosomal superkiller mutants of Saccharomyces cerevisiae. J. Bacteriol. 136:1002-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Triana, F., K. H. Nierhaus, and K. Chakraburtty. 1994. Transfer RNA binding to 80S ribosomes from yeast: evidence for three sites. Biochem. Mol. Biol. Int. 33:909-915. [PubMed] [Google Scholar]
- 59.Triana-Alonso, F. J., C. M. Spahn, N. Burkhardt, B. Rohrdanz, and K. H. Nierhaus. 2000. Experimental prerequisites for determination of tRNA binding to ribosomes from Escherichia coli. Methods Enzymol. 317:261-276. [DOI] [PubMed] [Google Scholar]
- 60.van Hoof, A., P. A. Frischmeyer, H. C. Dietz, and R. Parker. 2002. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295:2262-2264. [DOI] [PubMed] [Google Scholar]
- 61.von der Haar, F. 1974. Affinity elution: principles and applications to purification of aminoacyl-tRNA synthetases. Methods Enzymol. 34:163-171. [DOI] [PubMed] [Google Scholar]
- 62.Wickner, R. B. 1991. Yeast RNA virology: the killer systems, p. 263-296. In J. R. Broach, E. W. Jones, and J. R. Pringle (ed.), The molecular and cellular biology of the yeast Saccharomyces: genome dynamics, proteins synthesis, and energetics, vol. 1. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- 63.Wickner, R. B., and M. J. Leibowitz. 1974. Chromosomal and non-chromosomal mutations affecting the “killer character” of Saccharomyces cerevisiae. Genetics 76:423-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wickner, R. B., S. Porter-Ridley, H. M. Fried, and S. G. Ball. 1982. Ribosomal protein L3 is involved in replication or maintenance of the killer double-stranded RNA genome of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 79:4706-4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Widner, W. R., and R. B. Wickner. 1993. Evidence that the SKI antiviral system of Saccharomyces cerevisiae acts by blocking expression of viral mRNA. Mol. Cell. Biol. 13:4331-4341. [DOI] [PMC free article] [PubMed] [Google Scholar]