

Abstract
Net, Elk-1, and Sap-1 are members of the ternary complex factor (TCF) subfamily of Ets transcription factors. They form ternary complexes with serum response factor (SRF) on serum response elements of immediate early genes such as c-fos and egr-1 and mediate responses to growth factors and mitogen-activated protein kinase signaling. Although the TCFs have been extensively studied as intermediates in signaling cascades, surprisingly little is known about their different target genes and physiological functions. We report that Net homozygous mutant mouse embryonic fibroblasts have a defect in cell migration. This defect results at least in part from increased expression of plasminogen activator inhibitor type 1 (PAI-1), a serine protease inhibitor (serpin) that controls extracellular proteolysis and cell matrix adhesion. The defect in cell migration can be reverted by the addition of a PAI-1 blocking antibody. Net represses PAI-1 promoter activity and binds to a specific region of the promoter containing Ets binding sites in the absence of SRF. We conclude that Net is a negative regulator of PAI-1 expression and is thereby involved in cell migration.
The three ternary complex factors (TCFs) Net (also called Elk-3, SAP-2, and ERP), Elk-1, and Sap-1 have become paradigms in the understanding of signal transduction and transcription regulation, and there is great current interest in establishing their physiological functions (for reviews, see references 8, 20, 48-50, 54, 55, 60, and 67). The three TCFs have a number of common features, including their four similar regions, A to D. The N-terminal A domain is the Ets DNA binding domain. The B domain interacts with the serum response factor (SRF) and is required for ternary complex formation. The C-terminal C domain is a transcription activation domain that is turned on by phosphorylation by mitogen-activated protein (MAP) kinases. The D domain is a docking site for MAP kinases. The TCFs bind to Ets binding sites (EBSs), composed of a purine-rich central core sequence, 5′-GGA(A/T)-3′. They form complexes with SRF dimers over serum response elements (SREs) located in immediate early gene promoters. SREs are formed by a combination of Ets and SRF (CArG box) binding sites (for a review, see reference 56). SRF is thought to act as an anchor factor that recruits the TCFs to certain SREs (c-fos and egr-1), but the TCFs appear to adopt this role and recruit SRF to others (pip92 and nur77) (28).
As well as similarities, the TCFs also have distinct features. Net can strongly repress transcription, in contrast to Elk-1 and Sap-1. Net has two autonomous inhibitory domains, the net inhibitory domain (NID) (34) and the C-terminal binding protein interaction domain (designated the CID) (15). The CID interacts with the corepressor E1A C-terminal binding protein, which represses transcription through histone deacetylase activity. The NID interacts with the sumoylation E2 and E3 enzymes Ubc9 and PIAS1, and NID sumoylation on lysine 162 increases repression by Net (62). Interestingly, sumoylation of Elk-1 leads to recruitment of the histone deacetylase HDAC2 and transcriptional repression (66).
Mouse mutant models suggest that the TCFs have redundant functions with some distinct features. Sap-1 null mice have a defect in positive selection of thymocytes (14), whereas Elk-1-deficient mice have mildly impaired neuronal gene activation (10). Homozygous Net mutant mice, which express a deletion mutant of Net that lacks the DNA binding domain, have defects in the vasculature (2, 68). Interestingly, the mutant mice also have a defect in wound healing (68). Angiogenesis and wound healing involve both cell migration and extracellular matrix remodeling, raising the possibility that Net plays a role in these processes.
Cell migration and pericellular proteolysis are also regulated by plasminogen activator inhibitor 1 (PAI-1). PAI-1 is a serpin (serine proteases inhibitor) that inhibits urokinase and tissue plasminogen activators (uPA and tPA) and thereby the conversion of inactive plasminogen to plasmin. It also binds to the extracellular matrix (ECM) component vitronectin and thereby modulates cell-ECM interactions (for a review, see reference 4). We show in the manuscript that Net represses PAI-1 activity and is thus involved in cell migration.
MATERIALS AND METHODS
Cell culture.
Mouse embryonic fibroblasts (MEFs) were isolated from wild-type and Net homozygous mutant 13.5-day-old embryos (2) and used between passages 3 and 5. Net mutant MEFs express a deleted form of Net that lacks the DNA binding domain. They were grown in Dulbecco's modified Eagle's medium (DMEM), 10% fetal calf serum (FCS), and 40-μg/ml gentamicin. In all of our experiments, the MEFs were derived from embryos coming from the same litters. The mutant mice were on a homogenous 129sv background, resulting from 9 to 11 backcrosses. Mouse skin endothelial (SEND) cells were grown in MEM, 10% FCS, and 40-μg/ml gentamicin.
Antibodies.
Uses and dilutions were as follows: sheep anti-mouse PAI-1 (American Diagnostica, Inc.) for Western blotting (WB; 1/200) and immunocytochemistry (ICC; 1/100); PAI-1-neutralizing monoclonal antibody (IMA-33H1F7; Innovative Research, Inc.) (6); rabbit anti-mouse Net no. 1996 (23) for WB (1/2,000) and immunoprecipitations (IPs; 10 μl per IP); rabbit anti-mouse Elk-1 no. 512 for WB (1/2,000); mouse anti-mouse Sap-1a 1A8 for WB (1/2,000); mouse anti-mouse TATA binding protein (TBP) (kind gift of L. Tora) for WB (1/2,000); mouse antibromodeoxyuridine (anti-BrdU; Sigma) for ICC (1/500); mouse antivinculin (Sigma) for ICC (1/100); rabbit anti-SRF G-20 (Santa Cruz Biotechnology) for WB (1/1,000) and for immunoprecipitations (10 μl per IP); rabbit anti-clathrin heavy-chain H-300 (Santa Cruz Biotechnology) for immunoprecipitations (10 μl per IP).
Plasmids.
The following plasmids were used: p601D-anti-Net and pTL2-Net (23) and pCMV LacZ (IGBMC core facilities). PAI-1-luficerase reporters were KpnI/BglII PCR fragments, amplified by Expand High Fidelity PCR (Roche Diagnostics) from mouse genomic DNA in the corresponding sites of pGL3-Basic (Promega). The 5′ primers were pP(−1001)L (5′-CGTTGGTACCCTAATCGGAGCACTCAGGAG), pP(−764)L (5′-CGTTGGTACCTCTGTGGTAACCTCTGTTCT), pP(−519)L (5′-CGTTGGTACCAGAACCCAGACAATCACAGG), and pP(−319)L (5′-CGTTGGTACCGGAATTCCAAACACCAGGCT). The 3′ primer was 5′-CGTTAGATCTAAAGGTGCCTTGTGATTGGC. The recombinants were verified by DNA sequencing.
Site-directed mutagenesis.
Base substitutions were introduced with the QuickChange Multi Site Directed Mutagenesis kit (Stratagene) to produce pP(−1001 and 3EBS mut)L. Primers (mutated nucleotides are underlined) were mut EBS 1 and 2 (5′-AGGCCAACTTTTTCTGA×ATGTAGGCCCAGG and 5′-CCTGGGCCTACATTCAGAAAAAGTTGGCCT) and mut EBS 3 (5′-AGACCAAGGCTCGATTAAGGGAATTCCAAA and 5′-TTTGGAATTCCCTTAATCGAGCCTTGGTCT). The recombinants were verified by sequencing.
Down-regulation by siRNA and antisense RNA.
Net small interfering RNA (siRNA) (5′-CAGGUUUGUGACCAAUAAA-3′; Dharmacon, Inc.) corresponds to the coding region (549 to 567) relative to the first nucleotide of the start codon. SRF siRNA (5′-AGAUGGAGUUCAUCGACAA-3′; Dharmacon, Inc.) corresponds to the coding region (428 to 446) relative to the first nucleotide of the start codon. GL2 luciferase control siRNA (21). p601D-anti-Net produces antisense net RNA (23). siRNA duplexes (final concentration, 20 nM) or increasing amounts of p601D-anti-Net (1 or 2 μg made up to 3 μg with pBSK) were transfected into SEND cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer's guidelines. Cells were harvested 48 h after transfection.
In vitro wound healing and time-lapse microscopy.
Wild-type and mutant MEFs were grown to confluence in 12-well tissue culture plates with flat bottoms and low-evaporation lids (Becton Dickinson). A wound was created by scraping the monolayer with 200-μl disposable plastic pipette tips. The plates were placed in a chamber fixed to a robotized platform of an inverted microscope (Leica DMRIB) and maintained at 37°C with 5% CO2. Images (magnification, ×40; Hoffman contrast) were collected every 5 min for 30 h with a Cool Snap FX camera using Metamorph software (Universal Imaging). Where indicated, 40 μM aphidicolin (Sigma) or various concentrations of PAI-1-neutralizing monoclonal antibody (0.04 to 8 ng/μl; Innovative Research, Inc.) were added. Individual cell movement was analyzed using NSURFX and TIMT programs (IGBMC Microscopy and Imaging Centre) to select individual cells from the time-lapse images and to measure the distance covered by each cell.
Adhesion and Boyden chamber migration assays.
Cell migration assays were performed according to the manufacturer's instructions (QCM VN/FN Quantitative Cell Migration assay; Chemicon International, Inc.). MEFs or SEND cells (80% confluent) were detached with 0.25% trypsin-EDTA, counted, and reseeded at 2.5 × 105cells/250 μl on Boyden chambers precoated with vitronectin (VN), fibronectin (FN), or bovine serum albumin (BSA). After 8 h at 37°C and 5% CO2, the cells that had passed through the matrix barrier were stained, and the eluent optical density (OD) was measured at 570 nm. Cell adhesion assays were performed according to the manufacturer's instructions (CytoMatrix cell adhesion strips coated with VN, FN, or BSA; Chemicon International, Inc.). After detachment with 5 mM EDTA in phosphate-buffered saline (PBS), MEFs were plated in DMEM at 2 × 104 cells/well on 96-well FN-, VN-, or BSA-coated CytoMatrix cell adhesion strips. The cells were incubated for 1 h at 37°C, rinsed in PBS, stained with 0.2% crystal violet in 10% ethanol for 5 min, and finally rinsed three times with PBS. The eluent OD was measured at 570 nm.
Immunofluorescence.
Wild-type and mutant MEFs were grown on coverslips, subjected to in vitro wound healing (as described above), and after 4 h or 10 h fixed with 3.7% formaldehyde in PBS for 10 min at room temperature (RT), permeabilized with 0.1% Triton X-100 for 5 min, washed three times with PBS, and blocked with 3% BSA in PBS for 40 min. The cells were incubated with a sheep anti-mouse PAI-1 (American Diagnostica, Inc.) or a mouse antivinculin (Sigma) (1/100 in PBS-0.5% BSA) for 2 h at 37°C, washed in PBS, and incubated for 1 h at 37°C with Cy3-conjugated anti-sheep antibody (Beckman Coulter). For actin cytoskeleton staining, Texas red-conjugated phalloidin (1/40 in PBS; Molecular Probes) was added for 20 min at RT. Cells were washed three times, stained for 1 min at RT with Hoechst (Sigma), washed three times, covered with coverslips using mounting solution (5% propylgallate in PBS diluted in glycerol), and observed with a fluorescent microscope.
BrdU incorporation.
BrdU (10 μM) was added to the medium 1 h before wounding. The cells were fixed with 3.7% formaldehyde, incubated for 10 min in 0.1% Triton X-100 and 2 M HCl at RT, washed, and treated with anti-BrdU (Sigma), Cy3-conjugated anti-mouse antibody (Beckman Coulter) and Hoechst stain (Sigma), as described in “Immunofluorescence,” above.
uPA activity assay.
Confluent plates of wild-type and Net mutant MEFs were scraped, and cell pellets were combined with assay buffer (uPA Activity Assay kit; Chemicon International, Inc., Temecula, CA). After a 2-h incubation with urokinase colorimetric substrate at 37°C, absorbance was read at 405 nm (Bio-Rad model 550 microplate reader). Absorbances were converted to units of activity using standard curves generated with pure uPA enzyme provided with the kit. Activity is expressed per milligram of total sample protein, as determined by the Bradford protein assay.
PAI-1 analysis during wound healing by real-time reverse transcription-PCR (RT-PCR) and immunoblotting.
Wild-type and mutant MEFs were grown to confluence in 10-cm plates and scraped with a 200-μl pipette tip to form a criss-cross of wounds 1 cm apart. At 0, 2 h, 4 h, 8 h, and 12 h after wounding, the cell culture medium was sampled, and total RNA or cell lysates were prepared as described below.
Real-time quantitative RT-PCR.
Total RNA was prepared using RNAsolv (Omega Bio-Tek). Real-time quantitative RT-PCR was performed with the LightCycler system (Roche Diagnostics) and the SYBR Green I (Roche Diagnostics) protocol. Primers for PAI-1 and the internal control 28S RNA were designed using the Oligo 4.0 program. The reaction mixtures, containing 500 ng of RNA and 1× master mixture (0.5 μM primers, 4 mM MgCl2, 200 μM deoxynucleoside triphosphates, 1× PCR buffer, 1-U/μl Superscript reverse transcriptase [Invitrogen], Taq polymerase [Promega], anti-Taq antibody diluted 1/200 [Taqstart antibody; Clontech], 4.3% glycerol, 0.15-mg/ml BSA, and 0.25× SYBR green I), were reverse transcribed for 10 min at 55°C, denatured for 30 s at 95°C, and cycled 40 times, each time for 2 s at 95°C, 10 s at 60°C, and 15 s at 72°C. Amplification specificity was verified by melting-curve analysis, and the data were quantified with LightCycler software. Genomic DNA contamination controls were performed by repeating the procedure on the same samples without reverse transcriptase; no amplification was observed. Oligonucleotides were as follows: PAI-1 (192 bp), 5′-CTCCGAGAATCCCACACAG and 5′-ACTTTGAATCCCATAGCATC; uPA (141 bp), 5′-CTGAAAATGTCCGTCGTAAA and 5′-TCCTCCAGAATCGCCCTTG; 28S (239 bp), 5′-GGCGGCCAAGCGTTCATAGC and 5′-GCCAAGCACATACACCAAAT.
Western blots.
Cells were harvested in lysis buffer (0.4 M KCl, 20 mM Tris-HCl [pH 7.5], 20% glycerol, 5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 1× complete protease inhibitor cocktail [Roche Diagnostics]) and snap frozen-thawed three times. A total of 50 μg of protein lysates in 1× Laemmli buffer was fractionated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and revealed with the indicated antibodies and the enhanced chemiluminescence kit (Amersham). To detect secreted PAI-1, 50 μl of cell culture medium (0.5% FCS) was mixed with 50 μl of 2× Laemmli buffer and analyzed by Western blotting as described above.
ChIP analysis.
Chromatin immunoprecipitation (ChIP) assay kits (Upstate Biotechnology) were used. Wild-type or Net mutant MEFs were cross linked in 1% formaldehyde for 10 min. Cells were washed in PBS, and 106 cells were resuspended in 200 μl ChIP-SDS lysis buffer and incubated for 10 min on ice. Following sonication to shear the chromatin, the cellular debris was pelleted. Supernatants were recovered, diluted with 1.8 ml ChIP dilution buffer (40 μl was removed to serve as a 2% input sample), precleared with 80 μl protein G for 30 min at 4°C with rotation, centrifuged to remove the protein G, incubated with antibodies (10 μl of rabbit anti-mouse Net [no. 1995], 10 μl of rabbit anti-SRF G-20 [Santa Cruz Biotechnology], or 10 μl of rabbit anti-clathrin heavy-chain H-300 [Santa Cruz Biotechnology]) for 18 h at 4°C with rotation, followed by 60 μl protein G for 1 h at 4°C with rotation. After being washed, the samples were resuspended in 30 μl distilled H2O, amplified by PCR (94°C for 5 min; 30 cycles, each consisting of 94°C for 30 s, 61°C for 30 s, and 72°C for 30 s; and finally a 7-min extension period at 72°C), resolved on 2% agarose gels, and visualized with ethidium bromide. Oligonucleotides were as follows: PAI-1 promoter distal region (−2019; −1540), 5′-GCTGGTTGCCTTGGTATCTG and 5′-TCTGACCCACCTCCCTCCGC; PAI-1 promoter proximal region (−715; −367), 5′-CATCAGCACCCCACCCAGTA and 5′-GACAGCCATCACAGAGAAGC. The c-fos promoter, SRE region (−338; −153) was 5′-CGTCAATCCCTCCCTCCTTT and 5′-CCGTCTTGGCATACATCTTT.
cDNA microarrays.
Atlas mouse cDNA expression arrays (588 genes; Clontech) were probed with poly(A)+ RNA that was purified from passage 3 subconfluent wild-type and Net mutant MEFs. Total RNA was isolated with RNASolv (Omega Bio-Tek) and treated with DNase I (0.1 units/μg RNA) for 30 min at 37°C. A total of 45 μg was incubated with biotinylated oligo(dT) and streptavidin-coated magnetic beads (Atlas Pure Total RNA Labeling system). RNA on the beads was converted to 32P-labeled first-strand cDNA probes with Moloney murine leukemia virus and the Atlas array gene-specific CDS primer mixture. Probes were purified by column chromatography (NucleoSpin extraction column; Macherey-Nagel), and specific activity was measured by scintillation counting. The cDNA expression arrays were hybridized for 16 h at 71°C using ExpressHyb (Clontech), washed, and autoradiographed with Kodak Biomax or exposed with Phosphorimager screens and scanned with a Typhoon 8600 (Amersham Biosciences). The array images were processed with Atlas Image 1.5 software (Clontech).
Luciferase assay.
Passage 3 wild-type and Net mutant MEFs were transfected in triplicate by the calcium phosphate method (34) in six-well clusters with 4 μg of DNA per well containing 1 μg of the PAI-1 reporters, 1 μg pCMV-LacZ, and 2 μg of pBSK. After 16 h, the cells were washed twice with DMEM, incubated in DMEM containing 0.5% FCS for 24 h, and scraped. Luciferase assays were performed with the Luciferase Assay system (Promega) according to the manufacturer's instructions and analyzed with a luminometer (EG&G Berthold). Luciferase activities were corrected for transfection efficiency using β-galactosidase activity as an internal control.
RESULTS
Impaired mobility of Net mutant MEFs.
Time-lapse microscopy was used to study the filling of wounds created by scraping monolayers of Net mutant and wild-type MEFs isolated from embryos from the same litters. Net mutant MEFs, which express a truncated form of Net (absence of DNA binding domain), filled the wounds more slowly than wild-type cells (Fig. 1A). They were slower throughout the time course of the experiment, finally closing the wound about 4 h later than the wild-type cells (Fig. 1Ba). Similar results were obtained with three experiments for each of three different pairs of mutant and wild-type mice.
FIG. 1.

Decreased migration of Net mutant MEFs during in vitro wound healing. (A) Time-lapse microscopy. Wild-type and Net mutant MEFs were grown to confluence and scrape wounded (wound edges are delimited by the white dashed lines). The plates were placed in a chamber fixed to a robotized platform of an inverted microscope. Images (magnification, ×40; Hoffman contrast) were collected every 5 min for 30 h. Time after wounding is indicated above the panels (0, 6 h, 12 h, and 18 h). Representative pictures of triplicates of two pairs of wild-type and Net mutant MEFs are shown. (B) Kinetics of wound healing. Wound areas are the average of triplicates with each of two pairs of wild-type and Net mutant MEFs. (C) BrdU incorporation during wound healing. Aphidicolin (40 μM) was added where indicated (panels b in B and C). (D) Actin and focal adhesion staining of migrating MEFs. Wild-type and Net mutant MEFs grown on coverslips were scrape wounded and stained after 10 h for the actin cytoskeleton (phalloidin staining) (top) or focal adhesions (vimentin immunocytostaining) (bottom). Representative pictures of migrating cells are shown. Mut, Net mutant.
To study the relative contributions of cell migration and division to wound filling, we first observed DNA-synthesizing cells at the wound edge by BrdU staining. A small percentage (about 5%) of the cells were BrdU labeled up to 12 h after wounding, increasing to about 20% after 24 h (Fig. 1Ca). There was no significant difference between wild-type and mutant cells. To focus on cell migration, we repeated the experiment in the presence of 40 μM aphidicolin (51), which efficiently inhibited DNA replication (Fig. 1Cb). The inhibitor decreased wound closure by both types of cells, the mutant cells remaining less efficient than the wild type (Fig. 1Bb). To follow migration in the absence of the inhibitor, we measured the distance covered by individual cells during an 8.5-h period. Wild-type MEFs migrated 711 (±55 μm [standard deviation]) compared to 530 (±72 μm) for the mutant, which is approximately a 25% decrease (eight MEFs each; P < 0.0005). These results show that Net mutant MEFs are defective in cell migration.
Contribution of the actin cytoskeleton, focal adhesion, and extracellular matrix to altered migration.
The defect in cell motility could result from alterations in the actin cytoskeleton, focal adhesions, and interactions with the ECM. Ten hours postwounding, the cells at the edge of the wound were examined by immunocytochemistry (Fig. 1D). There were no detectable differences in the orientation and distribution of actin filaments or the number and typical arrowhead shape of focal adhesions. Whether BSA, vitronectin, or fibronectin was used for coating, the proportion of cells that migrated through the porous membranes of Boyden chambers was less for Net mutant MEFs (about 20% less; P < 0.05%) (Fig. 2A). Adherence to the same substrates was similar for wild-type and mutant MEFs (Fig. 2B).
FIG. 2.
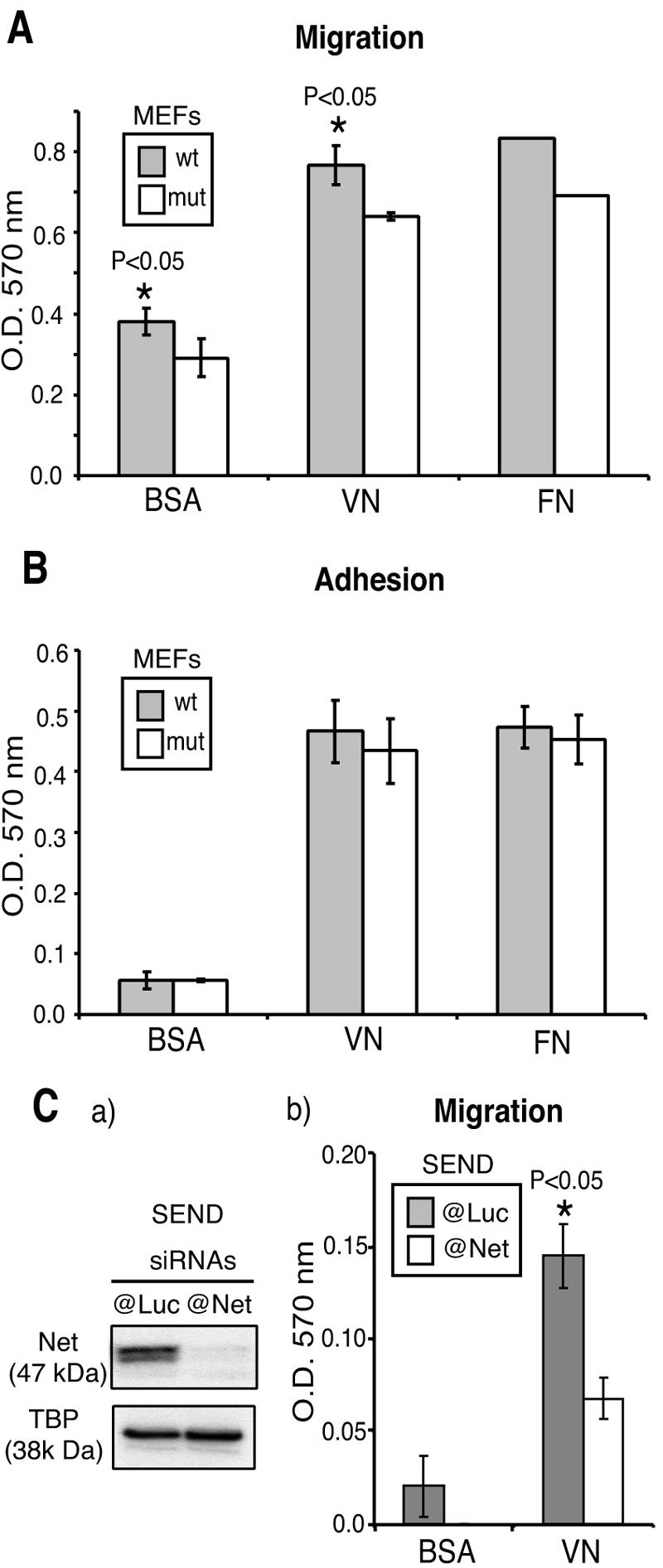
Migration and adhesion of Net mutant and wild-type MEFs on ECM substrates. (A) Boyden chamber migration assay. MEFs that had migrated through porous membranes coated with BSA, VN, or FN were quantified 8 h after seeding of 2.5 × 105 cells. (B) Adhesion assay. MEFs that had adhered to BSA-, VN-, or FN-coated wells were quantified 1 h after seeding of 2 × 104 cells. Two pairs of wild-type and Net mutant MEFs were used (A and B). The cells that had passed through the matrix barrier (A) or adhered to the substrate (B) were stained, and the eluent OD was measured at 570 nm. Mut, Net mutant. (C) Boyden chamber migration assays. Mouse SEND cells were transfected with 20 nM siRNA targeting Net (@Net) or control GL2-Luciferase (@Luc). At 48 h posttransfection, cells were harvested for Western blotting (a) or Boyden chamber migration assays (b). SEND cells that had migrated through porous membranes coated with BSA or VN were quantified 8 h after seeding of 2.5 × 105 cells per chamber.
Since the mutant MEFs express a truncated Net protein, we also studied the effect of down-regulating Net by RNA interference in SEND cells. Net siRNA specifically decreased Net protein expression (Fig. 2Ca, compare the luciferase siRNA and TBP loading controls) and significantly inhibited migration in Boyden chambers coated with BSA or vitronectin (Fig. 2Cb). These results indicate that the migration defect of Net mutant cells is not due to alterations of actin cytoskeleton, focal adhesions, attachment to the ECM, or expression of an abnormal truncated protein.
Net inhibits PAI-1 expression.
To identify Net target genes that could account for the defect in migration, we compared the gene expression profiles of wild-type and Net mutant MEFs using cDNA expression arrays (Atlas macroarrays; 588 genes). Of the 18 genes that were found to have an altered expression, the majority were up-regulated in Net mutant MEFS (15/18), suggesting that Net functions mainly as a repressor under the conditions analyzed. Only one of these genes is known to be directly involved in cell migration, PAI-1 (Fig. 3A). In contrast, there were no detectable differences in the expression levels of the PAI-1 substrates, uPA and tPA (Fig. 3A) or in several other genes involved in cell migration (matrix metalloproteinase 1 [MMP1], MMP2, MMP11, and MMP14) (data not shown). The difference in PAI-1 expression was confirmed at the RNA level by real-time quantitative RT-PCR (Fig. 3B) and at the protein level by Western blotting (Fig. 3C). We further investigated the expression of genes that were selected by the macroarrays or that are functionally connected with PAI-1 (uPA) and Net (the other TCFs and SRF). We confirmed the differential expression of 10 genes (including PAI-1) by real-time quantitative RT-PCR of independent RNA samples (Table 1). A major function of PAI-1 is to inhibit the protease uPA by direct binding. uPA expression at the mRNA level was not altered (Fig. 3A and B), whereas uPA activity was decreased in Net mutant cells (about 1.8-fold less; P < 0.05) (Fig. 3D). Elk-1, Sap-1a, and SRF were found to be expressed at similar levels in wild-type and mutant MEFs, indicating that altered characteristics of the mutant cells are not due to changes in the expression of functionally connected or related proteins.
FIG. 3.

PAI-1 is overexpressed in Net mutant MEFs. (A) Atlas 588 gene arrays were hybridized with probes derived from subconfluent growing MEFs. The spots for PAI-1, uPA, tPA, and the housekeeping gene HGPRT are shown. Mut, Net mutant. (B) PAI-1 and uPA mRNA quantified by real-time quantitative RT-PCR. The values were normalized to 28S rRNA. (C) Net, mutant Net (Net δ), Elk, Sap-1a, SRF, and PAI-1 proteins detected by immunoblotting; n.s., nonspecific band. (D) Chromogenic uPA activity assay performed on wild-type and Net mutant MEF cell extracts. (E) Down-regulation of endogenous Net by antisense RNA (a) or siRNA (b) increases PAI-1 expression in mouse SEND cells. (a) Cells were transfected with pBSK (lane 1) or increasing amounts of p601D-anti-Net that produces antisense (AS) net RNA (lane 2, 1 μg; lane 3, 2 μg), and immunoblotted for Net, PAI-1, and TBP. (b) Cells were not transfected (NT; lanes 1 and 4) or transfected with 20 nM siRNAs targeting GL2-luciferase (lanes 3 and 5), Net (lane 2), and SRF (lane 6). Protein extracts were harvested 48 h posttransfection and immunoblotted for Net, SRF, Elk-1, PAI-1, and TBP.
TABLE 1.
List of genes differentially expressed between wild-type (wt) and Net mutant (mut) MEFsa
| Ratio mutant/wt | Gene(s) expressed in subconfluent growing MEFsb | Accession no. |
|---|---|---|
| 5.0 | Cytokine receptor common gamma subunit precursor (gamma-C), interleukin-2 receptor gamma subunit (IL-2R gamma, IL2RG) | L20048 |
| 3.1 | T-cell death-associated protein (TDAG51) | U44088 |
| 2.9 | CD14 monocyte differentiation antigen precursor, lipopolysaccharide receptor, myeloid cell-specific leucine-rich glycoprotein | M34510 |
| 2.8 | Cyclin-dependent kinase inhibitor 1 (CDKN1A), melanoma differentiation-associated protein, CDK-interacting protein 1, WAF1 | U09507 |
| 2.6 | Cellular tumor antigen p53 (TRP53, TP53) | K01700 |
| 2.1 | Transforming growth factor beta 1 (TGF-β1, TGFB1) | M13177 |
| 2.1 | PAI-1 | M33960 |
| 0.27 | Delta-like protein precursor, preadipocyte factor 1, adipocyte differentiation inhibitor protein, SCP-1 | L12721 |
| 0.2 | Insulin-like growth factor binding protein 2 precursor (IGF-binding protein 2, IGFBP2, IBP2) | X81580 |
| 0.38 | IGFBP6 | X81584 |
Genes were obtained by cDNA array analysis and subsequent confirmation by quantitative RT-PCR.
The first seven genes were overexpressed in mutant MEFs; the last three genes were underexpressed in mutant MEFs.
To confirm that Net inhibits PAI-1 expression, we down-regulated Net expression by antisense Net RNA (Fig. 3Ea) and RNA interference (Fig. 3Eb) in mouse SEND cells. Progressively decreasing Net levels with increasing quantities of the antisense Net expression vector led to a concomitant increase in PAI-1, with no effect on the TBP control (Fig. 3Ea). We have previously shown that antisense Net is specific, since the level of the closely related protein Elk-1 is not affected (2, 23). In addition, diminishing Net levels with anti-Net siRNA markedly increased PAI-1 levels (Fig. 3Eb, lanes 1 and 2). Anti-Net siRNA did not affect Elk-1 or TBP expression (lanes 1 and 2). Control anti-luciferase siRNA did not significantly affect Net, PAI-1, Elk-1, or TBP levels (Fig. 3Eb, lanes 1 and 3 to 5). Since SRF is required for Net regulation of some genes (e.g., c-fos), we examined SRF regulation of PAI-1. Strongly decreasing SRF levels with anti-SRF siRNA did not affect expression of PAI-1 or the control TBP (lane 6) but as expected inhibited induction of Egr-1 (data not shown). These results provide strong evidence that Net inhibits PAI-1 expression and indicate that PAI-1 expression is not regulated by SRF.
PAI-1 expression during wound healing.
Since altered PAI-1 expression could contribute to differences in cell migration, we investigated its expression during wound healing at the levels of both RNA (Fig. 4A) and protein (Fig. 4B) and by immunocytochemistry (Fig. 4C). PAI-1 mRNA levels increased after wounding and were always higher in the Net mutant cells (1.7 to 3.1 fold) (Fig. 4A). Cell-associated and -secreted PAI-1 protein levels also increased, with higher levels in the mutant cells (Fig. 4B). There was a gradient of PAI-1 expression extending from the wound edge that was clearly visible after 4 h in both the wild-type and mutant cells, but the overall level of PAI-1 was higher in the mutant cells (Fig. 4C). Increased PAI-1 expression, restricted to the motile cell cohort, has already been described both in vitro (40) and in vivo (11). Net appears to be a repressor of PAI-1 under different conditions: PAI-1 expression is always higher in the mutant MEFs, in cycling subconfluent cells (Fig. 3A to C), in confluent cells at the time of wounding (Fig. 4A and B, time zero), and between 2 to 8 h after wounding (Fig. 4A to C).
FIG. 4.

PAI-1 expression after in vitro scrape wounding of wild-type and mutant MEFs. Wild-type and mutant MEFs were grown to confluence in 10-cm plates and scraped to form a criss-cross of wounds 1 cm apart. At 0, 2 h, 4 h, 8 h and 12 h after wounding, the cell culture medium was sampled and total RNA or cell lysates were prepared. (A) PAI-1 mRNA induction after wounding quantified by real-time quantitative RT-PCR (values normalized by 28S rRNA). Mut, Net mutant. (B) PAI-1 in cell extracts and secreted in the culture medium was detected by Western blotting. TBP was used as an internal control. n.s., nonspecific band. (C) Immunocytochemistry of PAI-1 protein expression at 0 and 4 h after wounding. The white dashed lines delimit the wound edges. Nuclei are stained blue (Hoechst), and PAI-1 is stained red.
PAI-1-blocking antibodies stimulate migration of Net mutant cells.
To study the contribution of PAI-1 to altered migration, we blocked PAI-1 activity by adding to the medium a neutralizing monoclonal antibody that has been shown to convert the inhibitory active conformation of PAI-1 to the noninhibitory substrate conformation that is proteolytically cleaved (6, 17). Low concentrations of neutralizing antibody did not significantly modify wound closure after 24 h (0.04 ng/μl and 0.1 ng/μl) (Fig. 5A, 5Bb, and 5Cb). Higher concentrations decreased wound closure by wild-type MEFs, whereas they accelerated closure by Net mutant cells (0.4 and 0.8 ng/μl) (Fig. 5Ba, 5Bb, 5Ca, and 5Cb). The highest dose (8 ng/μl) delayed migration of both wild-type and mutant cells. Similar results were obtained with a polyclonal antibody raised against full-length recombinant PAI-1 (data not shown). This dual effect was consistent with the published properties of PAI-1, that defined levels are required for optimum migration. Our results show that elevated expression of PAI-1 could account at least in part for the decreased migration of Net mutant MEFs.
FIG.5.

Effect of anti-PAI-1 blocking antibody on in vitro wound healing. Wild-type and Net mutant MEFs were grown to confluence and scrape wounded. The plates were placed in a chamber fixed to a robotized platform of an inverted microscope and examined by time-lapse video microscopy. The graphs show the kinetics of wound healing. Wound areas are the averages of duplicates with each of two pairs of wild-type and Net mutant MEFs. Wound areas of wild-type (wt; blue curve) and Net mutant (mut; red curve) fibroblasts in the absence of antibody (A) or in the presence of different concentrations of PAI-1-neutralizing monoclonal antibody in the medium of wild-type (Ba) or mutant (Ca) MEFs are shown. The histograms represent wound areas at 24 h postwounding with different concentrations of PAI-1-neutralizing monoclonal antibody for wild-type (Bb) and Net mutant MEFs (Cb). (*, P < 0.05; **, P < 0.01).
SRF-independent binding of Net to the PAI-1 promoter in vivo.
To establish whether regulation is direct, Net recruitment to the PAI-1 promoter in vivo was investigated by ChIP assays of wild-type and Net mutant MEFs (Fig. 6). The SRE region of the c-fos promoter was used as a positive control (59). Formaldehyde cross-linked protein-DNA complexes were immunoprecipitated with antibodies that targeted Net, SRF, and clathrin (a negative control protein that does not bind to DNA). Net was detected on the SRE-containing region (−338 to −153) of the c-fos promoter and the proximal region (−715 to −367) of the PAI-1 promoter (Fig. 6, lane 3) but not on the distal regions of the PAI-1 (−2019; −1540) and c-fos promoters (data not shown). SRF was only detected on the c-fos SRE region (lanes 4 and 8). The negative control (lanes 2 and 6) did not give specific products. The mutated form of Net that interacts with the antibody used was not detected on the c-fos or PAI-1 promoters (lane 7). These results suggest that Net directly regulates the PAI-1 gene in the absence of SRF.
FIG. 6.
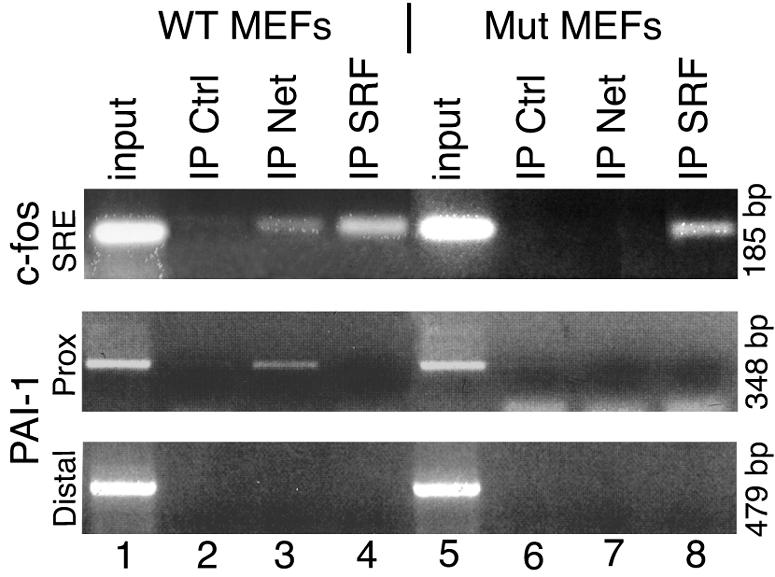
Endogenous Net binds to the PAI-1 promoter in vivo. Chromatin immunoprecipitation assays were performed with wild-type (WT, lanes 1 to 4) and Net mutant (Mut, lanes 5 to 8) MEFs. Formaldehyde cross-linked and sonicated cell lysates were used for clathrin (Ctrl, control; lanes 2 and 6), Net (lanes 3 and 7) and SRF (lanes 4 and 8) IPs. The Net antibody recognizes both wild-type (lane 3) and mutated (Net δ; lane 7) forms of Net. Coprecipitated DNA fragments were analyzed by PCR with specific primers for the c-fos SRE (positive control, promoter region −338 to −153), and PAI-1 proximal (−715 to −367) and distal (−2019 to −1540) promoter regions. PCR products were resolved in a 2% agarose gel.
Mapping of the Net-responsive elements of the PAI-1 promoter.
The Net-responsive elements of the PAI-1 promoter were delimited by transfection of promoter-luciferase reporters in wild-type and Net mutant MEFs (Fig. 7A). As expected, a reporter containing 1,001 bp of the upstream region, including the Net-interacting region determined by ChIP analysis, was more active in the mutant cells [pP(−1001)Luc]. 5′ deletion mutants, lacking sequences up to −764 and −519, retained higher activity in the mutant cells, whereas a further deletion to −319 abolished this activity. There are three EBSs in the Net-responsive region by Transfac analysis (Fig. 7B) (42). Disruption of the three EBSs by point mutation [pP(−1001, 3EBSmut)Luc] abolished Net-mediated repression. These results show that the EBS in the proximal region (−519 to −319) of the PAI-1 promoter mediates repression by Net.
FIG. 7.

Mapping of Net-responsive elements in the PAI-1 promoter. (A) Schematic representation of PAI-1 promoter 5′ deletion and point mutants linked to luciferase coding sequence (Luc) and relative luciferase activities of these constructs in wild-type (wt) and Net mutant (mut) MEFs. Each point was repeated in triplicate in two independent experiments (**, P < 0.005; *, P < 0.05). Luciferase activities were corrected for β-galactosidase activity expressed from the pCMV-LacZ internal control. For each construct, the values are relative to wild-type MEFs. (B) Sequence of the PAI-1 (−519; −319) promoter region and localization of the three putative EBSs mutated in pP(−1001, 3EBS mut)Luc.
DISCUSSION
We have shown that Net has a role in cell migration, using both in vitro wound healing and Boyden chamber assays. We showed that cell proliferation does not contribute significantly to healing within the time frame of the experiment, as wound closure by mutant cells was still observed in the presence of an inhibitor of DNA synthesis (aphidicolin). Direct observation of individual cells showed that the mutant cells move more slowly. Perturbation of the cytoskeleton, which can affect movement (44, 46), probably does not account for the differences, since Net mutant MEFs cells appear morphologically indistinguishable from the wild-type cells and neither the actin cytoskeleton nor focal adhesions are significantly altered. We provide evidence that the difference in motility is due, at least in part, to increased expression of PAI-1. Our cDNA array analysis showed that Net mutant cells have additional changes in gene expression (Table 1). Only PAI-1 is directly linked with cell migration. However, we cannot exclude that other deregulated genes could contribute to some extent to the phenotype of Net mutant cells.
Down-regulation of Net, by truncation of the DNA binding domain or by siRNA or anti-sense RNA, alleviates Net-mediated repression of PAI-1. It is unlikely that the truncated form of Net has a trans-dominant effect, since heterozygous animals and fibroblasts are identical to their wild-type counterparts in terms of both phenotype and expression of PAI-1 (2; data not shown). We used both fibroblasts and skin endothelial cells in our experiments. Endothelial cells express high levels of Net (1, 2; K. Alitalo, personal communication), which facilitates the study of endogenous Net. Moreover, endothelial cells are widely used to study PAI-1 (for reviews, see references 45 and 53). Fibroblasts and endothelial cell are implicated in wound healing in vivo (for a review, see reference 35). Dermal fibroblasts and endothelial cells in the neighborhood of wounds proliferate and migrate into the provisional matrix of wound clots. Fibroblasts synthesize extracellular matrix components that contribute to both the formation of granulation tissue and to keratinocytes migration at the wound bed (for a review, see references 31 and 35). We previously showed that wound healing and vascularization of skin wounds is retarded in Net mutant mice (68). Reduced migration of Net mutant cells could help to account for this phenotype.
PAI-1 has several activities that affect cell migration (for a review, see reference 53). PAI-1 controls turnover of the extracellular matrix. Furthermore, it binds to the N terminus of the ECM component VN and inhibits VN interactions with the cell surface proteins integrin αvβ3 (52) and the uPA receptor (26). The major consequence of PAI-1 deregulation in Net mutant cells is apparently extracellular proteolysis, since Net mutant cells display the same migration defect on VN as on FN or BSA, and they adhere as efficiently as wild-type cells to VN, FN, and BSA. This would agree with other studies that have shown that regulation of extracellular proteolysis by PAI-1 is important for cell migration (37, 63) and angiogenesis (3, 19).
Precise quantities of PAI-1 are required in various physiological processes, and over- or underexpression can be inhibitory. PAI-1 overexpression by adenovirus delivery inhibits many processes: smooth muscle cell migration in wound healing (9), HT-1080 cell migration (39) and cell invasion through Matrigel (38), glioma cell invasion and motility (24), metastasis in human and murine uveal melanomas (33), and migration and invasion in breast and gynecological cancer cells (63). Increasing PAI-1 expression with tumor necrosis factor alpha decreases migration of primary trophoblasts, an effect that is overcome with a PAI-1-blocking antibody (5). Conversely, down-regulation of endogenous PAI-1 leads to decreased in vitro wound closure by human keratinocytes (29), wound repair by renal epithelial cells (41), and cell motility in two- and three-dimensional culture models (7, 40). The critical dependence on precise PAI-1 levels is also observed for microvessel outgrowth from PAI-1 null aorta rings. Addition of recombinant PAI-1 is proangiogenic at physiological concentrations, whereas abnormally low or high concentrations are antiangiogenic (19). The effects of PAI-1 blocking antibodies on wild-type and Net mutant fibroblasts also illustrate this dual effect: they stimulate migration of Net mutant cells that overexpress PAI-1 and decrease migration of wild-type fibroblasts that express physiological concentrations of PAI-1.
MAP kinase phosphorylation switches Net from a repressor to an activator (23). Interestingly, the extracellular signal-regulated kinase pathway has been shown to be activated at the wound edge in an in vitro wound healing assay (40). We investigated whether Net is phosphorylated after wounding. We detected specific and transient phosphorylation of Net at the wound edge by immunocytochemistry 30 min after wounding (data not shown), suggesting that Net could be activated by scrape injury. However, phosphorylation was transient and was no longer detected at further times after wounding (1 h, 3 h, and 6 h; data not shown), indicating that Net acts predominantly as a repressor in the time scale of wound closure (about 24 h).
PAI-1 gene transcription is positively regulated by various transcription factors, including Sp1 (12, 36), Forkhead (61), and the hypoxia-inducible factor (27). Interestingly, an Sp1 and PAI-1 regulator, transforming growth factor β (16, 18), is overexpressed at the mRNA level in Net mutant cells (Table 1), raising the possibility that it may contribute to the increase of PAI-1. The mechanism of transforming growth factor β regulation by Net and its consequence on PAI-1 expression in these cells remains to be studied. Little is known about negative factors regulating PAI-1. Net represses PAI-1 gene expression and promoter activity and binds to the proximal promoter region in vivo. TCFs are especially known as regulators of immediate early genes, such as c-fos, egr-1, or junB, in association with SRF. We have identified a new target gene for a TCF, PAI-1, that is apparently SRF independent. SRF does not bind to the promoter region that recruits Net (−715 to −367), and there are no consensus SRF motifs for a further 300 bp upstream or downstream. However, we cannot exclude that SRF binds to a more distal site and interacts with Net through extended spatial flexibility in ternary complex formation (57). The three potential EBSs in the proximal region of the PAI-1 promoter may suffice for autonomous recruitment of Net, or there could be other interacting partners.
Several genes have been shown to be regulated by the TCFs in an SRF-independent manner. Elk-1 and Net interact with Pax5 on a composite element of the mb-1 promoter (22). Elk-1 binds to Ets motifs in the tumor necrosis factor alpha promoter and forms a multiprotein complex with Sp1, Egr-1, c-Jun, activating transcription factor 2, and CBP/p300 (58). The cis-acting element in the Cctq gene promoter is specifically recognized by the three TCFs in the absence of SRF (65). The 9E3/cCAF (30) and α(1, 3)-fucosyltransferase IV (64) genes have also been suggested to be SRF-independent targets. In our study, the evidence for SRF independence of PAI-1 expression includes several more recent tools, including siRNA and ChIPs.
PAI-1 is up-regulated in response to different inducers, including anisomycin (32), UV (47), heat shock (43), hypoxia (27), and tumorigenesis (for a review, see reference 13). It remains to be seen whether loss of Net repression contributes to PAI-1 induction in these conditions. PAI-1 has many physiological roles, in obesity, inflammation and atherosclerosis (for a review, see reference 25), and the identification of factors that regulate PAI-1 expression is consequently of great interest. Net repression of PAI-1 could be relevant for these pathologies.
Acknowledgments
We thank Jean-Luc Vonesch, Marcel Boeglin, and Didier Hentsch from the IGBMC Microscopy and Imaging Centre for their support with time lapse microscopy and image analysis; Jens Frauenfeld for his contribution to Net silencing with antisense RNA; Christine Wasylyk for her contribution to the cDNA array and Q-RT-PCR analysis; the Wasylyk laboratory members for support and encouragement; and the IGBMC core facilities.
G.B. is the recipient of a Ligue contre le Cancer fellowship. C.G. is the recipient of postgraduate fellowship from the Ministère de la Recherche et Technologie. Our laboratory is supported by the Ligue Nationale Française contre le Cancer (Equipe labelisée), the Ligue Régionale (Haut-Rhin) contre le Cancer, the Ligue Régionale (Bas-Rhin) contre le Cancer, the Association pour la Recherche sur le Cancer, BioAvenir (Aventis, Rhone-Poulenc), the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, and the Hôpital Universitaire de Strasbourg.
REFERENCES
- 1.Ayadi, A., M. Suelves, P. Dolle, and B. Wasylyk. 2001. Net, an Ets ternary complex transcription factor, is expressed in sites of vasculogenesis, angiogenesis, and chondrogenesis during mouse development. Mech. Dev. 102:205-208. [DOI] [PubMed] [Google Scholar]
- 2.Ayadi, A., H. Zheng, P. Sobieszczuk, G. Buchwalter, P. Moerman, K. Alitalo, and B. Wasylyk. 2001. Net-targeted mutant mice develop a vascular phenotype and up-regulate egr-1. EMBO J. 20:5139-5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajou, K., C. Maillard, M. Jost, R. H. Lijnen, A. Gils, P. Declerck, P. Carmeliet, J. M. Foidart, and A. Noel. 2004. Host-derived plasminogen activator inhibitor-1 (PAI-1) concentration is critical for in vivo tumoral angiogenesis and growth. Oncogene 23:6986-6990. [DOI] [PubMed] [Google Scholar]
- 4.Bass, R., and V. Ellis. 2002. Cellular mechanisms regulating non-haemostatic plasmin generation. Biochem. Soc. Trans. 30:189-194. [DOI] [PubMed] [Google Scholar]
- 5.Bauer, S., J. Pollheimer, J. Hartmann, P. Husslein, J. D. Aplin, and M. Knofler. 2004. Tumor necrosis factor-alpha inhibits trophoblast migration through elevation of plasminogen activator inhibitor-1 in first-trimester villous explant cultures. J. Clin. Endocrinol. Metab. 89:812-822. [DOI] [PubMed] [Google Scholar]
- 6.Bijnens, A. P., A. Gils, I. Knockaert, J. M. Stassen, and P. J. Declerck. 2000. Importance of the hinge region between alpha-helix F and the main part of serpins, based upon identification of the epitope of plasminogen activator inhibitor type 1 neutralizing antibodies. J. Biol. Chem. 275:6375-6380. [DOI] [PubMed] [Google Scholar]
- 7.Brooks, T. D., J. Slomp, P. H. Quax, A. C. De Bart, M. T. Spencer, J. H. Verheijen, and P. A. Charlton. 2000. Antibodies to PAI-1 alter the invasive and migratory properties of human tumour cells in vitro. Clin. Exp. Metastasis 18:445-453. [DOI] [PubMed] [Google Scholar]
- 8.Buchwalter, G., C. Gross, and B. Wasylyk. 2004. Ets ternary complex transcription factors. Gene 324:1-14. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet, P., L. Moons, R. Lijnen, S. Janssens, F. Lupu, D. Collen, and R. D. Gerard. 1997. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation: a gene targeting and gene transfer study in mice. Circulation 96:3180-3191. [DOI] [PubMed] [Google Scholar]
- 10.Cesari, F., S. Brecht, K. Vintersten, L. G. Vuong, M. Hofmann, K. Klingel, J. J. Schnorr, S. Arsenian, H. Schild, T. Herdegen, F. F. Wiebel, and A. Nordheim. 2004. Mice deficient for the ets transcription factor Elk-1 show normal immune responses and mildly impaired neuronal gene activation. Mol. Cell. Biol. 24:294-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan, J. C., D. A. Duszczyszyn, F. J. Castellino, and V. A. Ploplis. 2001. Accelerated skin wound healing in plasminogen activator inhibitor-1-deficient mice. Am. J. Pathol. 159:1681-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Y. Q., M. Su, R. R. Walia, Q. Hao, J. W. Covington, and D. E. Vaughan. 1998. Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J. Biol. Chem. 273:8225-8231. [DOI] [PubMed] [Google Scholar]
- 13.Chorostowska-Wynimko, J., E. Skrzypczak-Jankun, and J. Jankun. 2004. Plasminogen activator inhibitor type-1: its structure, biological activity and role in tumorigenesis (review). Int. J. Mol. Med. 13:759-766. [PubMed] [Google Scholar]
- 14.Costello, P. S., R. H. Nicolas, Y. Watanabe, I. Rosewell, and R. Treisman. 2004. Ternary complex factor SAP-1 is required for Erk-mediated thymocyte positive selection. Nat. Immunol. 5:289-298. [DOI] [PubMed] [Google Scholar]
- 15.Criqui-Filipe, P., C. Ducret, S. M. Maira, and B. Wasylyk. 1999. Net, a negative Ras-switchable TCF, contains a second inhibition domain, the CID, that mediates repression through interactions with CtBP and de-acetylation. EMBO J. 18:3392-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datta, P. K., M. C. Blake, and H. L. Moses. 2000. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-beta-induced physical and functional interactions between smads and Sp1. J. Biol. Chem. 275:40014-40019. [DOI] [PubMed] [Google Scholar]
- 17.Debrock, S., and P. J. Declerck. 1997. Neutralization of plasminogen activator inhibitor-1 inhibitory properties: identification of two different mechanisms. Biochim. Biophys. Acta 1337:257-266. [DOI] [PubMed] [Google Scholar]
- 18.Dennler, S., S. Itoh, D. Vivien, P. ten Dijke, S. Huet, and J. M. Gauthier. 1998. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devy, L., S. Blacher, C. Grignet-Debrus, K. Bajou, V. Masson, R. D. Gerard, A. Gils, G. Carmeliet, P. Carmeliet, P. J. Declerck, A. Noel, and J. M. Foidart. 2002. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 16:147-154. [DOI] [PubMed] [Google Scholar]
- 20.Dittmer, J., and A. Nordheim. 1998. Ets transcription factors and human disease. Biochim. Biophys. Acta 1377:F1-11. [DOI] [PubMed] [Google Scholar]
- 21.Elbashir, S. M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494-498. [DOI] [PubMed] [Google Scholar]
- 22.Fitzsimmons, D., W. Hodsdon, W. Wheat, S. M. Maira, B. Wasylyk, and J. Hagman. 1996. Pax-5 (BSAP) recruits Ets proto-oncogene family proteins to form functional ternary complexes on a B-cell-specific promoter. Genes Dev. 10:2198-2211. [DOI] [PubMed] [Google Scholar]
- 23.Giovane, A., A. Pintzas, S. M. Maira, P. Sobieszczuk, and B. Wasylyk. 1994. Net, a new ets transcription factor that is activated by Ras. Genes Dev. 8:1502-1513. [DOI] [PubMed] [Google Scholar]
- 24.Hjortland, G. O., K. Bjornland, S. Pettersen, S. S. Garman-Vik, E. Emilsen, J. M. Nesland, O. Fodstad, and O. Engebraaten. 2003. Modulation of glioma cell invasion and motility by adenoviral gene transfer of PAI-1. Clin. Exp. Metastasis 20:301-309. [DOI] [PubMed] [Google Scholar]
- 25.Juhan-Vague, I., M. C. Alessi, A. Mavri, and P. E. Morange. 2003. Plasminogen activator inhibitor-1, inflammation, obesity, insulin resistance and vascular risk. J. Thromb. Haemost. 1:1575-1579. [DOI] [PubMed] [Google Scholar]
- 26.Kanse, S. M., C. Kost, O. G. Wilhelm, P. A. Andreasen, and K. T. Preissner. 1996. The urokinase receptor is a major vitronectin-binding protein on endothelial cells. Exp. Cell Res. 224:344-353. [DOI] [PubMed] [Google Scholar]
- 27.Kietzmann, T., U. Roth, and K. Jungermann. 1999. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood 94:4177-4185. [PubMed] [Google Scholar]
- 28.Latinkic, B. V., M. Zeremski, and L. F. Lau. 1996. Elk-1 can recruit SRF to form a ternary complex upon the serum response element. Nucleic Acids Res. 24:1345-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, F., J. Goncalves, K. Faughnan, M. G. Steiner, I. Pagan-Charry, D. Esposito, B. Chin, K. M. Providence, P. J. Higgins, and L. Staiano-Coico. 2000. Targeted inhibition of wound-induced PAI-1 expression alters migration and differentiation in human epidermal keratinocytes. Exp. Cell Res. 258:245-253. [DOI] [PubMed] [Google Scholar]
- 30.Li, Q. J., S. Vaingankar, F. M. Sladek, and M. Martins-Green. 2000. Novel nuclear target for thrombin: activation of the Elk1 transcription factor leads to chemokine gene expression. Blood 96:3696-3706. [PubMed] [Google Scholar]
- 31.Lorena, D., K. Uchio, A. M. Costa, and A. Desmouliere. 2002. Normal scarring: importance of myofibroblasts. Wound Repair Regen. 10:86-92. [DOI] [PubMed] [Google Scholar]
- 32.Lund, L. R. 1996. Expression of urokinase-type plasminogen activator, its receptor and type-1 plasminogen activator inhibitor is differently regulated by inhibitors of protein synthesis in human cancer cell lines. FEBS Lett. 383:139-144. [DOI] [PubMed] [Google Scholar]
- 33.Ma, D., R. D. Gerard, X. Y. Li, H. Alizadeh, and J. Y. Niederkorn. 1997. Inhibition of metastasis of intraocular melanomas by adenovirus-mediated gene transfer of plasminogen activator inhibitor type 1 (PAI-1) in an athymic mouse model. Blood 90:2738-2746. [PubMed] [Google Scholar]
- 34.Maira, S. M., J. M. Wurtz, and B. Wasylyk. 1996. Net (ERP/SAP2) one of the Ras-inducible TCFs, has a novel inhibitory domain with resemblance to the helix-loop-helix motif. EMBO J. 15:5849-5865. [PMC free article] [PubMed] [Google Scholar]
- 35.Martin, P. 1997. Wound healing—aiming for perfect skin regeneration. Science 276:75-81. [DOI] [PubMed] [Google Scholar]
- 36.Motojima, M., T. Ando, and T. Yoshioka. 2000. Sp1-like activity mediates angiotensin-II-induced plasminogen-activator inhibitor type-1 (PAI-1) gene expression in mesangial cells. Biochem. J. 349:435-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmieri, D., J. W. Lee, R. L. Juliano, and F. C. Church. 2002. Plasminogen activator inhibitor-1 and -3 increase cell adhesion and motility of MDA-MB-435 breast cancer cells. J. Biol. Chem. 277:40950-40957. [DOI] [PubMed] [Google Scholar]
- 38.Praus, M., D. Collen, and R. D. Gerard. 2002. Both u-PA inhibition and vitronectin binding by plasminogen activator inhibitor 1 regulate HT1080 fibrosarcoma cell metastasis. Int. J. Cancer 102:584-591. [DOI] [PubMed] [Google Scholar]
- 39.Praus, M., K. Wauterickx, D. Collen, and R. D. Gerard. 1999. Reduction of tumor cell migration and metastasis by adenoviral gene transfer of plasminogen activator inhibitors. Gene Ther. 6:227-236. [DOI] [PubMed] [Google Scholar]
- 40.Providence, K. M., and P. J. Higgins. 2004. PAI-1 expression is required for epithelial cell migration in two distinct phases of in vitro wound repair. J. Cell Physiol. 200:297-308. [DOI] [PubMed] [Google Scholar]
- 41.Providence, K. M., S. M. Kutz, L. Staiano-Coico, and P. J. Higgins. 2000. PAI-1 gene expression is regionally induced in wounded epithelial cell monolayers and required for injury repair. J. Cell Physiol. 182:269-280. [DOI] [PubMed] [Google Scholar]
- 42.Quandt, K., K. Frech, H. Karas, E. Wingender, and T. Werner. 1995. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23:4878-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roca, C., L. Primo, D. Valdembri, A. Cividalli, P. Declerck, P. Carmeliet, P. Gabriele, and F. Bussolino. 2003. Hyperthermia inhibits angiogenesis by a plasminogen activator inhibitor 1-dependent mechanism. Cancer Res. 63:1500-1507. [PubMed] [Google Scholar]
- 44.Samarakoon, R., and P. J. Higgins. 2002. MEK/ERK pathway mediates cell-shape-dependent plasminogen activator inhibitor type 1 gene expression upon drug-induced disruption of the microfilament and microtubule networks. J. Cell Sci. 115:3093-3103. [DOI] [PubMed] [Google Scholar]
- 45.Schleef, R. R., and D. J. Loskutoff. 1988. Fibrinolytic system of vascular endothelial cells. Role of plasminogen activator inhibitors. Haemostasis 18:328-341. [DOI] [PubMed] [Google Scholar]
- 46.Schratt, G., U. Philippar, J. Berger, H. Schwarz, O. Heidenreich, and A. Nordheim. 2002. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J. Cell Biol. 156:737-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seite, S., A. Colige, C. Deroanne, C. Lambert, P. Piquemal-Vivenot, C. Montastier, A. Fourtanier, C. Lapiere, and B. Nusgens. 2004. Changes in matrix gene and protein expressions after single or repeated exposure to one minimal erythemal dose of solar-simulated radiation in human skin in vivo. Photochem. Photobiol. 79:265-271. [DOI] [PubMed] [Google Scholar]
- 48.Sharrocks, A. D. 2002. Complexities in ETS-domain transcription factor function and regulation: lessons from the TCF (ternary complex factor) subfamily. The Colworth Medal Lecture. Biochem. Soc. Trans. 30:1-9. [DOI] [PubMed] [Google Scholar]
- 49.Sharrocks, A. D. 2001. The ETS-domain transcription factor family. Nat Rev. Mol. Cell Biol. 2:827-837. [DOI] [PubMed] [Google Scholar]
- 50.Shaw, P. E., and J. Saxton. 2003. Ternary complex factors: prime nuclear targets for mitogen-activated protein kinases. Int. J. Biochem. Cell Biol. 35:1210-1226. [DOI] [PubMed] [Google Scholar]
- 51.Spadari, S., F. Focher, C. Kuenzle, E. J. Corey, A. G. Myers, N. Hardt, A. Rebuzzini, G. Ciarrocchi, and G. Pedrali-Noy. 1985. In vivo distribution and activity of aphidicolin on dividing and quiescent cells. Antiviral Res. 5:93-101. [DOI] [PubMed] [Google Scholar]
- 52.Stefansson, S., and D. A. Lawrence. 1996. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature 383:441-443. [DOI] [PubMed] [Google Scholar]
- 53.Stefansson, S., G. A. McMahon, E. Petitclerc, and D. A. Lawrence. 2003. Plasminogen activator inhibitor-1 in tumor growth, angiogenesis and vascular remodeling. Curr. Pharm. Des. 9:1545-1564. [DOI] [PubMed] [Google Scholar]
- 54.Treisman, R. 1995. Journey to the surface of the cell: Fos regulation and the SRE. EMBO J. 14:4905-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treisman, R. 1996. Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol. 8:205-215. [DOI] [PubMed] [Google Scholar]
- 56.Treisman, R. 1992. The serum response element. Trends Biochem. Sci. 17:423-426. [DOI] [PubMed] [Google Scholar]
- 57.Treisman, R., R. Marais, and J. Wynne. 1992. Spatial flexibility in ternary complexes between SRF and its accessory proteins. EMBO J. 11:4631-4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai, E. Y., J. V. Falvo, A. V. Tsytsykova, A. K. Barczak, A. M. Reimold, L. H. Glimcher, M. J. Fenton, D. C. Gordon, I. F. Dunn, and A. E. Goldfeld. 2000. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol. Cell. Biol. 20:6084-6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Riggelen, J., G. Buchwalter, U. Soto, J. De-Castro Arce, H. Z. Hausen, B. Wasylyk, and F. Rosl. 2005. Loss of net as repressor leads to constitutive increased c-fos transcription in cervical cancer cells. J. Biol. Chem. 280:3286-3294. [DOI] [PubMed] [Google Scholar]
- 60.Verger, A., and M. Duterque-Coquillaud. 2002. When Ets transcription factors meet their partners. Bioessays 24:362-370. [DOI] [PubMed] [Google Scholar]
- 61.Vulin, A. I., and F. M. Stanley. 2002. A Forkhead/winged helix-related transcription factor mediates insulin-increased plasminogen activator inhibitor-1 gene transcription. J. Biol. Chem. 277:20169-20176. [DOI] [PubMed] [Google Scholar]
- 62.Wasylyk, C., P. Criqui-Filipe, and B. Wasylyk. 2005. Sumoylation of the net inhibitory domain (NID) is stimulated by PIAS1 and has a negative effect on the transcriptional activity of Net. Oncogene 24:820-828. [DOI] [PubMed] [Google Scholar]
- 63.Whitley, B. R., D. Palmieri, C. D. Twerdi, and F. C. Church. 2004. Expression of active plasminogen activator inhibitor-1 reduces cell migration and invasion in breast and gynecological cancer cells. Exp. Cell Res. 296:151-162. [DOI] [PubMed] [Google Scholar]
- 64.Withers, D. A., and S. I. Hakomori. 2000. Human α(1,3)-fucosyltransferase IV (FUTIV) gene expression is regulated by Elk-1 in the U937 cell line. J. Biol. Chem. 275:40588-40593. [DOI] [PubMed] [Google Scholar]
- 65.Yamazaki, Y., H. Kubota, M. Nozaki, and K. Nagata. 2003. Transcriptional regulation of the cytosolic chaperonin theta subunit gene, Cctq, by Ets domain transcription factors Elk-1, Sap-1a, and Net in the absence of serum response factor. J. Biol. Chem. 278:30642-30651. [DOI] [PubMed] [Google Scholar]
- 66.Yang, S. H., and A. D. Sharrocks. 2004. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 13:611-617. [DOI] [PubMed] [Google Scholar]
- 67.Yang, S. H., A. D. Sharrocks, and A. J. Whitmarsh. 2003. Transcriptional regulation by the MAP kinase signaling cascades. Gene 320:3-21. [DOI] [PubMed] [Google Scholar]
- 68.Zheng, H., C. Wasylyk, A. Ayadi, J. Abecassis, J. A. Schalken, H. Rogatsch, N. Wernert, S. M. Maira, M. C. Multon, and B. Wasylyk. 2003. The transcription factor Net regulates the angiogenic switch. Genes Dev. 17:2283-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]