

Abstract
Melatonin has been experimentally implicated in skin functions such as hair growth cycling, fur pigmentation, and melanoma control, and melatonin receptors are expressed in several skin cells including normal and malignant keratinocytes, melanocytes, and fibroblasts. Melatonin is also able to suppress ultraviolet (UV)-induced damage to skin cells and shows strong antioxidant activity in UV exposed cells. Moreover, we recently uncovered expression in the skin of the biochemical machinery involved in the sequential transformation of l-tryptophan to serotonin and melatonin. Existence of the biosynthetic pathway was confirmed by detection of the corresponding genes and proteins with actual demonstration of enzymatic activities for tryptophan hydroxylase, serotonin N-acetyl-transferase, and hydroxyindole-O-methyltransferase in extracts from skin and skin cells. Initial evidence for in vivo synthesis of melatonin and its metabolism was obtained in hamster skin organ culture and in one melanoma line. Therefore, we propose that melatonin (synthesized locally or delivered topically) could counteract or buffer external (environmental) or internal stresses to preserve the biological integrity of the organ and to maintain its homeostasis. Furthermore, melatonin could have a role in protection against solar radiation or even in the management of skin diseases.
Keywords: Skin, melatonin, serotonin, N-acetylserotonin, ultraviolet radiation
Introduction
The strategic location of the skin as barrier between environment and internal milieu renders it critically important for the preservation of body homeostasis. By being constantly subjected to the actions of solar, thermal, and mechanical energy, and chemical and biological agents, the skin has developed unique properties to deal with these stressors (1–4). Thus, the skin is endowed with regional, local, and focal capabilities to recognize, discriminate, and to integrate specific signals within a highly heterogeneous environment (2–4), and to integrate them into a stress-response neuroendocrine system (2,5–7).
A known neuroendocrine mediator is melatonin [molecule discovered by Lerner (8,9)], with pleiotropic bioactivities such as hormonal, neurotransmitter, immunomodulator, and biological modifier actions, which are mediated through interactions with high-affinity membrane-bound or nuclear receptors (10–14). Moreover, melatonin itself can function as a free-radical scavenger and broad-spectrum antioxidant, or as activator of pathways protective against oxidative stress or metabolic modulator (15–17). Thus, melatonin exhibits a number of properties that could be extremely useful for a stress-response system of the skin (5).
Melatonin connections to the skin are well recognized since the initial identification of its actions producing lightening of skin pigmentation in frogs (9,18). It has been experimentally implicated in hair growth cycling, fur pigmentation, and melanoma growth control (5,19–22). Because melatonin receptors are expressed in skin cells, these have the potential to mediate phenotypic actions on cellular proliferation and differentiation. In addition, its chemical activity suggests that melatonin could also have a protective role against UV-induced pathology. Both biosynthetic and biodegradative pathways for melatonin have initially been characterized in whole human and rodent skin and in the major cutaneous cellular populations (5). Therefore, we have proposed that the expression of a cutaneous melatoninergic system would serve to counteract or buffer external (environmental) or internal stresses to preserve the biological integrity of the organ and maintain its homeostasis.
Cutaneous Pathway for Melatonin Synthesis
Using molecular, biochemical, and chemical techniques, we have uncovered the full expression in the skin of the biochemical machinery involved in the sequential transformation of l-tryptophan to serotonin and melatonin (Fig. 1) (5). This included detection of the genes and proteins for tryptophan hydroxylase (TPH), serotonin N-acetyl-transferase (NAS), and hydroxyindole-O-methyltransferase (HIOMT) in the whole skin and skin cells, and actual demonstration of the corresponding enzymatic activities (23–28).
Fig. 1.
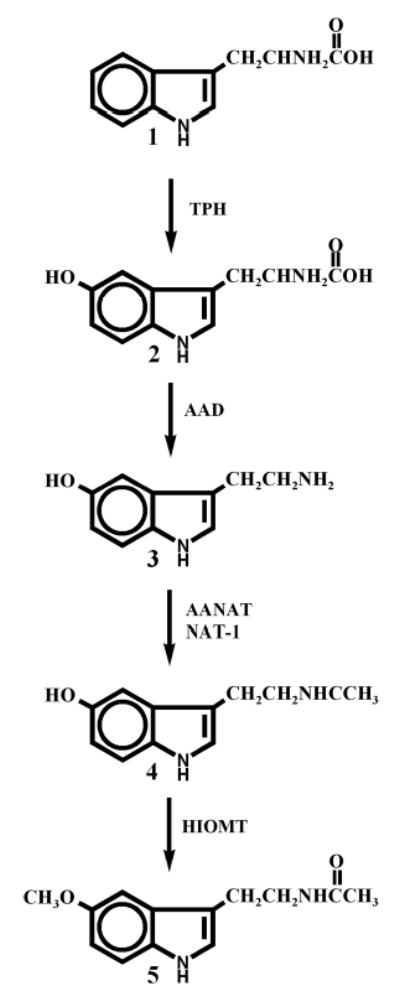
Proposed pathway on melatonin synthesis in the skin. 1, tryptophan; 2,5-hydroxytryptophan; 3, serotonin(5-hydroxytryptamine); 4, N-acetylserotonin (N-acetyl-5-hydroxytryptamine); 5, melatonin.
The skin also expresses epidermal equivalents of another bioamine-based system, the catecholaminergic systems (29, 30). This also includes a 6-tetrahydrobiopterin (6BH4) generating system (30,31), which acts as a co-factor for phenylalanine, tyrosine, and tryptophan hydroxylases. Expression and activity of aromatic amino acid decarboxylase (AAD) has also been demonstrated in human skin cells (29). Thus, the same rate-limiting factors in their biosynthesis are shared by catecholaminergic and serotonin/melatoninergic systems, 6BH4 and AAD, are expressed in the skin. Because central melatonin production is positively regulated by activation of adrenergic receptors with following activation of adenylate cyclase, in analogy, the cutaneous melatoninergic system could also be regulated by locally produced catecholamines. These putative bidirectional interactions between local catecholaminergic and melatoninergic pathways represent a current challenge in skin biology research.
Acetylation of serotonin to N-acetylserotonin (NAS), a limiting step in melatonin formation (32), may be catalyzed in the skin by either arylalkylamine N-acetyltransferase (AANAT) and/or arylamine N-acetyltransferase (NAT), most likely NAT-1 (5,25). NAS can in fact be produced in the C57BL/6 mouse strain (25), which represents natural AANAT “knockdown” (33), through an alternate pathway (5,25). NAS produced in the skin may be released into the circulation (5), and could be transformed into melatonin after delivery to organs expressing HIOMT (34). It remains to be tested whether in the C57BL/6 mice HIOMT-producing organs also have the enzymatic ability to produce NAS, similar to the skin.
Experimental data in organ culture of hamster skin (35) and in cultured melanoma cells (28) indicate that melatonin can be synthesized in vivo in skin cells. In human scalp, melatonin immunoreactivity has been localized in different cellular compartments using immunocytochemistry (5).
Cutaneous Pathways for Melatonin Degradation
The melatonin metabolites 5-methoxytryptamine (5MTT) and 5-methoxytryptophol (5MTOL) have been detected in cultured mammalian skin and skin cells (23,28,35) indicating similarity in degradative pathways with melatonin metabolism in frog skin and retina (36,37), including expression of monoaminoxidase (MAO) activity in mammalian skin (5,26). Recent experimental evidence indicates that cutaneous degradation of melatonin may also include pathways known to be operative in the liver and kidney (Fisher et al., manuscript in preparation). Based on the above, together with the known mechanism for melatonin degradation or transformation in peripheral organs, we propose that in the skin melatonin may be metabolized through alternative metabolic pathways (Fig. 2). Pathway activities and nature of the final product would be linked to the spatial distribution of melatonin in the skin, to the specific cell type and subcellular compartment.
Fig. 2.
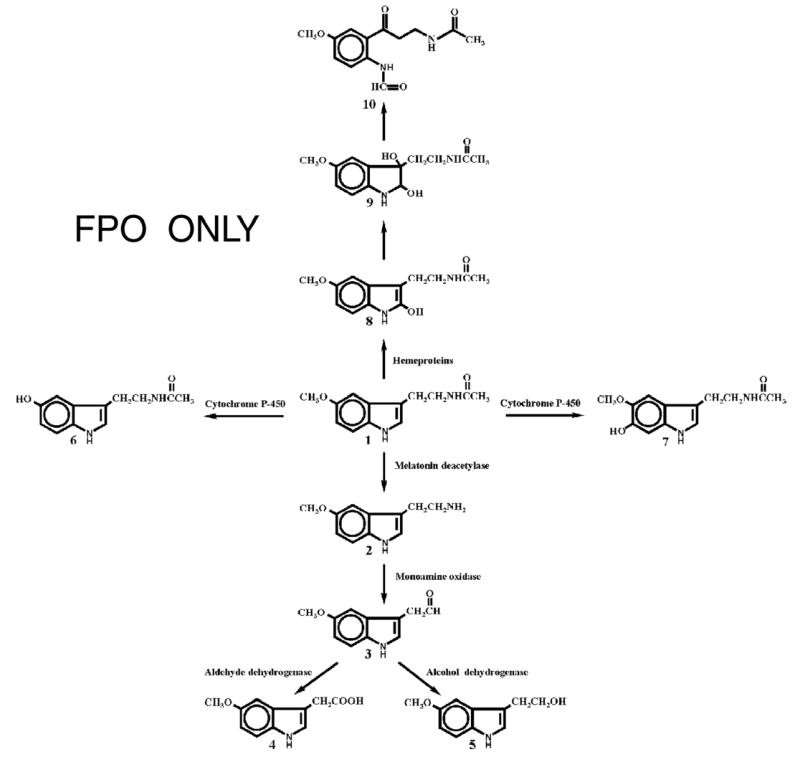
Proposed model of melatonin metabolism in the skin. 1, melatonin; 2, 5-methoxytryptamine; 3, 5-methoxyindoleacetaldehyde; 4, 5-methoxyindole acetic acid; 5, 5-methoxytryptophol; 6, N-acetylserotonin; 7, 6-hydroxymelatonin; 8, 2-hydroxymelatonin; 9, 2,3-dihydroxymelatonin; 10, N1-acetyl-N2-formyl-5-methoxykynuramine.
Expression of Melatonin Receptors in the Skin
Membrane-Bound Melatonin Receptors
Phenotypic effects of melatonin can be mediated through interaction with the G protein–coupled membrane bound MT1 (MTNRa) and MT2 (MTNRb) receptors (11) or with nuclear receptors of RZR/ROR subfamily of orphan receptors (14,38,39). Quinone reductase II (NQO2) has also been proposed as a melatonin receptor type 3 (MT3) (40,41); however, an alternative explanation could be the function as a co-factor or regulator of the enzyme NQO2. Expression of membrane-bound cell surface MT receptors in the skin is variable, depending on the species. For example, skin from the C57BL/6 mouse predominantly or exclusively expresses MT2 (19), while human skin expresses both receptors, although with a bias toward MT1 (the predominant form found in both whole skin and cultured cells) (42). Immunocytochemical studies on the human skin showed cell-type- and compartment-dependent expression of MT1 and MT2 proteins (Table 1) (5). This pattern suggests that selectivity for melatonin action could be achieved by spatial compartmentalization and specificity of signal transduction pathways.
Table 1.
Localization of MT1 and MT2 Immunoreactivities in Human Scalp (5)
| MT1 | MT2 | |
|---|---|---|
| Epidermis | ++ (Stratum Granulosum) | − |
| + (Stratum Spinosum) | ||
| Hair follicle | + (Upper outer root sheath) | |
| + (Inner root sheath) | + (Inner root sheath) | |
| Eccrine glands | +++ | +++ |
| Blood vessels | +++ (Endothelium) | ++ (Endothelium) |
(−) = negative; (+) = weak; (++) = moderate; (+++) = strong.
Expression of the MT1 and MT2 genes was found to be modified by environmental factors, and defined by genetic background (Fig. 3). For example, exposure to UVB (100 mJ/cm2)-induced expression of MT1 in normal neonatal epidermal melanocytes, but it downregulated MT1 expression in two melanoma lines (Fig. 1A, C). Expression of MT1 was also dependent on the donor, e.g., in newly obtained samples of dermal fibroblasts it was below the limits of detectability for the sequence of primers used (Fig. 1A, lanes 3 and 4). Analysis of MT2 gene expression showed this to be induced/upregulated or modified by UVB in adult normal epidermal or immortalized (HaCaT) keratinocytes, in normal epidermal melanocytes, or in dermal fibroblasts (Fig. 3C). Of great interest is the UVB-induced expression of the MT2b isoform, previously cloned by us (42). This was detected in neonatal melanocytes, normal keratinocytes, and dermal fibroblasts (Fig. 3B, lanes 2, 4, 6, and 8) indicating that UVB modifies alternative splicing of the MT2 gene, and consistent with the UVB-induced switch from MT2 to MT2b gene expression in dermal fibroblasts. Because we have previously described environmental regulation of alternative splicing, in the G protein–coupled cell surface receptor gene (CRH-R1), we analyzed in detail the DNA sequence of the MT2b isoform (Fig. 4). Thus, alternative splicing of the MT2 gene generating MT2b isoforms results in a DNA sequence containing two open reading frames (orf) that encode the putative proteins MT2b1 and MT2b2 (Fig. 4). If translated, MT2b1 would generate a truncated protein of 79 aa containing N-terminal and the first transmebrane sequence followed by 8 aa of MT2 with the sequence gehhs added because of frame shift and addition of a stop codon (Fig. 4). The putative MT2b2 protein has 247 aa, lacking the TM 1–3 domains of MT2. It starts with the mrclfppqpglpfpf sequence followed consecutively by the predicted transmembrane domain (TMX) and the MT2 sequence. Thus, MT2b has only five TM domains (TMX and TM4–TM7 of MT2). The biological significance of these putative protein products of alternative splicing of the MT2 gene will be tested, following a protocol similar as that described for CRH-R1 isoforms (43,44).
Fig. 3.

Expression of genes for membrane-bound melatonin receptors in human skin cells. (A) Nested RT-PCR for MT1 receptor. (B) Nested RT-PCR for MT2 receptor; 100 kbp DNA ladder (M), neonatal melanocytes (1), neonatal melanocytes after UVB treatment (2), immortalized keratinocytes (HaCaT) (3), immortalized keratinocytes (HaCaT) after UVB treatment (4), adult epidermal keratinocytes (5), adult epidermal keratinocytes after UVB treatment (6), adult dermal fibroblasts (7), adult dermal fibroblasts after UVB treatment (8), immortalized melanocytes (PIG-1) (9). (C) Nested RT-PCR for MT1 receptor in melanoma cells; 100 kbp DNA ladder (M), SKMEL188 (1), SKMEL188 after UVB treatment (2), WM164 (3), WM164 after UVB treatment (4), WM98 (5), WM98 after UV treatment (6). Sequences of primers and conditions for nested RT-PCR were as described (23).
Fig. 4.

Predicted amino acid sequence for two new human iosforms of MT2 (MTNRb) receptor. Sequence of human MT2 (MTNRb) (gene accession #NP_005950.1) is shown in comparison to the predicted sequences of two newly discovered open reading frames (orf) detected after alternative splicing of MT2 mRNA. Alternative splicing was confirmed by sequencing the 209 bp PCR fragment of MT2 cDNA (gene accession #AY114100) (for gene structure cf. ref. 23). Both the stop codon for MT2b1 and the start codon for MT2b2 are coded by sequences generated after alternative splicing. There is a gap, 72 bp long, between the stop codon for MT2B1 and the start codon for MT2b2 (not shown). Splicing is marked with an arrow, and with A for MT2 or B for the splicing variant MT2b. Transmembrane regions (TM) are marked with # and with numbers 1–7 listed above the sequences. TMX is an alternative transmebrane fragment for MT2b2. MT2b2 has only five TM helices and lacks TM 1–3 helices, a portion partially substituted by a unique transmembrane helix (TMX), causing a shift of two amino acids to C-terminus in TM 4 helices (not show).
Nuclear Melatonin Receptors
Nuclear receptor RORα (retinoid-related orphan receptor α) is a member of the RZR/ROR subfamily that contains at least four splicing variants: RORα1, RORα2, RORα3, RZRα (RORα4) (38,39,45). We suggest renaming the last isoform RZRα to RORα4 for the sake of consistency, although its sequence was described independently (46). The sequence for that fourth isoform of RORα–RORαd has been deposited in Genebank (NM_134262), noting that it only has single nucleotide substitution when compared to RZRα. We used a unique set of primers developed by others to test for expression of the common fragment of RORαmRNA (47) and then to identify each isoform (45). All of the tested skin cells expressed at least one of three RORα isoforms; RORα3 was consistently absent (Table 2). Moreover, gene expression was modified by UVB, which down-regulated the expression of RZR1 (RORα4) in HaCaT keratinocytes and upregulated it in normal neonatal melanocytes (Table 2, Fig. 5). The modifying effect of UVB on expression of genes coding nuclear melatonin receptors (Table 2) suggests that these receptors can be subject to environmental regulation in vivo. RORα1 and RORα2 expression was detected only in dermal fibroblasts (both isoforms) and in immortalized PIG-1 melanocyte line (RORα2 only) being below detectability levels in normal epidermal melanocytes and keratinocytes and in HaCaT cells (Table 2, Fig. 5). It is probable that RORα gene possesses more splicing variants, because the mRNA for this gene contains 23 exons within a genomic region that spans 732,840 bp (according to NCBI Evidence Viewer Homo sapiens RORA entry), and for the possible explanation for the additional DNA fragments of unexpected length amplified with RORα1 and RORα2 primers (Fig. 5B, C).
Table 2.
Expression of Genes Coding RORa/RZR Receptors and NQO2 in Human Skin Cells
| Genes or isoforms |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RORα |
RORα1 |
RORα2 |
RORα3 |
RZR1 (RORα4) |
NQO2 |
|||||||
| Cell line | −UVB | +UVB | −UVB | +UVB | −UVB | +UVB | −UVB | +UVB | −UVB | +UVB | −UVB | +UVB |
| Adult epidermal keratinocytes | + | + | − | − | − | − | − | − | + | + | + | + |
| HaCaT keratinocytes | + | − | − | − | − | + | − | − | + | +↓ | + | + |
| Neonatal melanocytes | + | +↑ | − | − | − | − | − | − | + | +↑ | + | + |
| Immortalized melanocytes (PIG–1) | + | ND | − | ND | + | ND | − | ND | − | ND | + | ND |
| Adult dermal fibroblasts | + | + | + | +* | − | +* | − | − | + | + | + | + |
Possible splicing variant; + present; − absent; ↑ stimulation; ↓ inhibition; ND: not done; UVB ultraviolet radiation (100 mJ/cm2).
Fig. 5.

Expression of genes coding for nuclear melatonin receptors and quinone reductase 2 (NQO2) in skin cells. RT-PCR for nuclear receptor izoforms: RORα receptor (common fragment for all isoforms, A), RORα1 (B), RORα2 (C), RORα3 (D), RZR1 (RORα4) (E), and NQO2 (F). 100 kB DNA ladder (M), neonatal melanocytes (1), neonatal melanocytes after UVB treatment (2), immortalized keratinocytes (HaCaT) (3), immortalized keratinocytes (HaCaT) after UVB treatment (4), adult epidermal keratinocytes (5), adult epidermal keratinocytes after UVB treatment (6), adult dermal fibroblasts (7), adult dermal fibroblasts after UVB treatment (8), immortalized melanocytes (PIG-1) (9). The RT-PCR primers conditions for common RORα fragment and RORα1–4 isoforms were as described (common fragment, ref. 47; isoforms, ref. 45). RT-PCR to amplify NQO2 was preformed as described (49). UVB was used at dose of 100 mJ/cm2 as described previously (94).
NQO2 is a cytosolic flavoprotein that possesses a melatonin binding site (previously proposed MT3) (41,48); it is involved in cellular resistance to oxidative stress and in cellular detoxification (49,50). Thus, melatonin could be involved in the regulation of NQO2 function and, consequently, in its antioxidant activity. Molecular testing for the NQO2 gene, using primers and conditions described in the literature (49), revealed that expression of this gene in skin cells is ubiquitous (Table 2, Fig. 6).
Fig. 6.

Effect of UVB (35 mJ/cm2) on HaCaT keratinocytes with or without prior addition of 10−3 M melatonin for 30 min and collected 20 h after irradiation. RNA was extracted, cDNA produced, and microarray analysis performed as described (73). Differences between control (UVB-irradiated cells) and cells irradiated with UVB and treated with melatonin are presented as mean of the respective ratios (ratio 1 represents no difference) ± SEM, followed by analysis with Student’s t test (n = 4, *p < 0.05, **p < 0.005).
Phenotypic Effects of Melatonin
The major compartments of the skin epidermis, dermis, and adnexa are targets for melatonin regulation (5). More specifically, melatonin has been implicated in hair growth cycling (20), cutaneous pigmentation (3), and skin physiology and pathology (5). Because those actions have been discussed in recent reviews, the description below will focus on areas not been discussed in detail in those papers.
The field of hair growth has been the subject of intensive research in Australia and New Zealand, countries with a prospering wool industry, where testing in fur-producing animals revealed that melatonin stimulates significantly hair growth. Thus, animals fed with a melatonin-supplemented diet showed an increased rate of hair growth in springtime, the period of change from the winter coat to summer fur (51). The results were independently confirmed in other experimental models (52,53). The fur growth may be mediated by melatonin binding sites/receptors, because these were identified in the rodent skin (19,54,55). Studies with a human hair organ culture model showed that melatonin had bimodal effects on hair shaft elongation consisting of stimulation (at 30 μM), and inhibition (in millimolar range) (56). A clinical study in women suffering from androgenetic or diffuse alopecia provided evidence for a positive effect of melatonin in human hair growth (57), suggesting that melatonin could be a potential target for hair growth regulation in humans (58).
Melatonin can also reduce collagen deposition observed in the skin of rats after pinealectomy (59,60), suggesting that this accumulation could also be modulated by the local supply of melatonin. Further evidence for a role of melatonin in dermis metabolism is indicated by a decrease in glycosaminoglycan content seen after pre-treatment with melatonin of rats exposed to acute stress (61). Indeed, melatonin treatment increased levels of uronic acid and hexosamines, and moderately affected collagen turnover in stressed animals. Melatonin can also modulate monocyte and fibroblast proliferation and alter cytokine levels, suggestive of an effect on angiogenesis. Indeed, recent data obtained in rats suggest that melatonin may enhance both angiogenesis and wound healing (62).
Moreover, receptors for melatonin are expressed in keratinocytes, melanocytes, and fibroblasts, and these mediate many of its phenotypic actions (42).
Melatonin as Protector Against Skin Damage
Because of its broad antioxidant and radical scavenger properties (16), melatonin may act as a protective agent against UV radiation (UVR)-induced damage in the skin (reviewed in refs. 5 and 63). Clinical studies indicated that melatonin is able to prevent sun damage only when it is administered before UVR and it is present in relevant concentrations directly at the irradiation site (64–67). These observation have strong experimental support from in vitro studies. Thus, melatonin increases cell viability in UV-irradiated fibroblasts by counteracting the formation of polyamine levels (68), and the accumulation of malondialdehyde while decreasing apoptosis cells (69). Melatonin has also been shown to protect against UV light–induced damage in human leukocytes. In this cell model, melatonin significantly suppresses the formation of reactive oxygen species leading consecutively to an increased rate of cell survival when applied to the cells prior UV irradiation (70,71). In fact, melatonin exhibited stronger radical-scavenging properties than vitamin C and Trolox (72). In human epidermal keratinocytes, melatonin revealed a strong protective effect against UV-induced reduction of cell viability, but only at high doses (10−3 M). This effect was found to be receptor independent, and was further investigated in colony-forming assays, which showed higher numbers of colonies at 14 d post-UVR when subjected to pre- and post-UV treatment with melatonin- as compared with non-melatonin–treated colony plates. In that setting the antiapoptotic properties of melatonin were also confirmed (Fischer et al., in preparation). The interactions between intracellular pathways stimulated by UVB and by melatonin may be complex because using microarray analysis we found that melatonin attenuates the expression of several genes known to be stimulated by UVB (73) (Fig. 6).
Given the antioxidant effects of melatonin, it could have a role in skin biology (58,63), where its short plasma half-life and low molecular weight suggest that it could be useful as a constituent of sun-protective creams. In this regard the photo-stability of melatonin has been a limiting factor, although liquid chromatography–mass spectrometric analysis has shown that the melatonin metabolites 6-hydroxymelatonin and N1-acetyl-N2-formyl-5-methoxykynurenamine may retain significant antioxidant activity (74). Moreover, melatonin does not appear to be a skin sensitizer (75).
The radioprotective effects of melatonin may include X-ray-induced skin damage, recently found in an albino rat model (76), where pretreatment with melatonin protected against degenerative cellular change. Other studies have also shown that melatonin administration can reverse oxidative injury in other tissues and organs, as assessed with biochemical and histopathologic techniques. Thus, melatonin could be useful as a damage-limiting supplement in patients undergoing radiotherapy (77,78). The mechanisms of action could include inhibition of lipid peroxidation, shown in human fibroblasts (79), direct radical-scavenging properties, and stimulation of antioxidative enzymes in human skin fibroblasts (80) and other organ systems (16,17).
It has be reported that intraperitoneal administration of melatonin at physiological levels to pinealectomized rats undergoing skin surgery can reduce malondialdehyde and nitric oxide levels in the skin, while glutathione, glutathione-peroxidase, and superoxide dismutase are increased, as compared with non-treated pinealectomized animals (81). The effect of melatonin treatment on oxidant damage caused by thermal injury and burns has been examined in rats (82). Treatment of animals with melatonin resulted with post-injury glutathione levels to be increased significantly and lipid peroxidation to be significantly decreased in skin homogenates. However, application of melatonin to incisional full-thickness skin wounds in Wister–Albino rats with 20% surface burns failed to modify glutathione peroxidase levels, although superoxide dismutase and catalase activities were elevated as compared group with the control group (83).
Finally, antioxidants such as melatonin are important for the defense against oxidative stress and, consequently, may prevent carcinogenesis (84). Melatonin is a strong radical scavenger directed especially against hydroxyl radicals, which are thought to be the most damaging effectors produced during UVR (5,63). Therefore, melatonin may able to prevent skin cancer development acting at the molecular, cellular, and clinical levels through its radical scavenging and direct protection of DNA (21,84). Melatonin has in fact been shown to suppress not only the initiation but also the promotion stages of skin carcinogenesis (85).
Melatonin as a Topically Applied Drug in Dermatology
In that context the pharmacodynamic properties of melatonin have been studied in humans. Topical application of 20 mg and 100 mg melatonin dissolved in 70% ethanol lead to a rise within the first hour after application, which was dose-dependent and lasted over 8 h. Thus, it appears that melatonin may build a depot in the stratum corneum from which it is continuously released into the dermis and blood vessels (65,86). Melatonin in cream preparations builds an even stronger depot in the stratum corneum, but with lower release into the blood over the 24 h observation period, as compared with melatonin in an alcohol solution (87). Thus, the skin may be an optimal organ not only for the treatment of local pathways with topical melatonin application, but also for the possibility of transdermal delivery to create stable plasma levels for systemic treatment with melatonin through constant release from the stratum corneum (58,88).
Oncostatic Properties of Melatonin in Skin Cancer
Melatonin has also been reported to exhibit tumorostatic properties in different tumor models that include melanomas and tumors of epithelial origin (22,89). For example, in breast cancer melatonin appears to be useful as adjuvant in the therapy of this malignancy. We have also performed initial testing of melatonin in the C1-4 squamous cell carcinoma line of cervical origin and found inhibition of cell viability by increasing concentrations (Fig. 7), representing an additional model for the testing of the oncostatic properties of melatonin.
Fig. 7.

Melatonin suppresses cell viability in squamous cell carcinoma cells. C1–4 cells were incubated for 24 h in serum free medium in the presence of graded concentrations of melatonin. Cell viability was measured by MTT test (23). Data are presented as means ± SEM (n = 16 combined from two experiments), and the statistical analysis was performed with ANOVA (+p < 0.01; #p < 0.001).
Severalclinicalstudieshavereportedpositiveresultswith melatonin in patients with metastatic malignant melanoma. In one of the studies melatonin was used as a monotherapy in 40 patients, at four different doses, from 5 to 700 mg/m²/d, with evaluation and followup at 5 wk. Partial response occurred in six patients and a stable disease resulted in a further six patients for an overall response rate of 30%. The median response duration in the partial responders was 33 wk. Among the response sites were central nervous system, subcutaneous tissue, and lung. Toxicity was generally low, and a dose–response relationship was found (90). In other studies melatonin was applied in combination with interleukin-2 (IL-2), because melatonin enhances the IL-2 modulated stimulation of the immune system against tumor progression. This was tested in 14 patients with untreatable metastatic melanoma using melatonin as a complementary drug to IL-2 and combined in addition with naltrexon; the therapy increased significantly Th1 lymphocyte number and suppressed Th2, which represent the most important favorable prognostic criteria in IL-2-mediated anticancer immunotherapy (91,92). Apart from this cooperative tumor-suppressing effect with IL-2, melatonin has been shown to suppress significantly the toxicity of chemotherapeutic drugs and to increase their anticancer cytotoxicity (93). In a clinical study of 13 patients with metastatic malignant melanoma, melatonin was used in combination with cisplatin and IL-2 as a second-line therapy after failure of the first-line therapy with dacarbazine and interferon alpha. The objective tumor response-rate (CR + PR) was 31% with stable disease occurring in five patients. Tolerance to this second-line therapy with melatonin was good and especially that the well-known critical neurotoxicity of dacarbazine was significantly reduced (92). Because melatonin blood levels are considerably suppressed in patients at the early stages of tumor development, treatment with melatonin might be useful in cancer patients at early tumor stages (21).
Concluding Remarks
Melatonin (either produced locally or delivered to the skin) may be a significant contributor to regulation of the local system that preserves the physical and functional integrity of the skin. Such a system, which would be activated by environmental stressors or internal dyshomeostatic stimuli, would act counteracting or buffering any damaging effects. In this setting melatonin could act through intra-, auto-, or paracrine mechanisms, to participate in regulation of skin functions at a highly compartmentalized level (5). Melatonin phenotypic effects would then be determined by control of its in situ availability (5). The reported differential expression of melatoninergic system in human and fury animal skin may then be related to the prevailing species stressor (solar radiation in humans vs hair cycling or chemical stimuli in rodents) (5).
An efficient epidermal biological barrier requires an efficient local immune system, highly organized differentiation of epidermal keratinocytes, activation of hair follicles (important in fury animals), activation of fibroblasts activity (builds and regulates structure of the dermis), and activation of the pigmentary system (important in social communication and protection against solar radiation). All these actions clearly reside within the domain of the cutaneous melatoninergic system (5), in addition to melatonin protective effect against solar radiation and possible antimutagenic and anticarcinogenic actions.
Acknowledgments
This work was supported in part by grants from Vitiligo Foundation, NIH #AR047079, Center of Genomic and Bioinformatics, University of Tennessee to AS; University of Tennessee Cancer Center Pilot Grant to AS and TWF, “German Academy of Natural Scientists Leopoldina” with Ref-No. BMBF-LPD 9901/8-113 to TWF. Generous support of ASATONA AG is also acknowledged.
References
- 1.Slominski A, Pawelek J. Clin Dermatol. 1998;16:503–515. doi: 10.1016/s0738-081x(98)00023-6. [DOI] [PubMed] [Google Scholar]
- 2.Slominski A, Wortsman J. Endocr Rev. 2000;21:457–487. doi: 10.1210/edrv.21.5.0410. [DOI] [PubMed] [Google Scholar]
- 3.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Physiol Rev. 2004;84:1155–1228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- 4.Slominski A, Wortsman J. Minerva Endocrinol. 2003;28:135–143. [PubMed] [Google Scholar]
- 5.Slominski A, Wortsman J, Tobin DJ. FASEB J. 2005;19:176–194. doi: 10.1096/fj.04-2079rev. [DOI] [PubMed] [Google Scholar]
- 6.Slominski A, Wortsman J, Luger T, Paus R, Solomon S. Physiol Rev. 2000;80:979–1020. doi: 10.1152/physrev.2000.80.3.979. [DOI] [PubMed] [Google Scholar]
- 7.Slominski A, Wortsman J, Pisarchik A, et al. FASEB J. 2001;15:1678–1693. doi: 10.1096/fj.00-0850rev. [DOI] [PubMed] [Google Scholar]
- 8.Lerner AB, Case JD, Takahashi Y. J Biol Chem. 1960;235:1992–1997. [PubMed] [Google Scholar]
- 9.Lerner AB, Case J, Takahashi Y. J Am Chem Soc. 1958;80:2587. [Google Scholar]
- 10.Yu, H. S. and Reiter, R. J. (1993). Melatonin biosynthesis, physiological effects, and clinical implications. CRC Press, Boca Raton, FL.
- 11.Dubocovich ML, Rivera-Bermudez MA, Gerdin MJ, Masana MI. Front Biosci. 2003;8:d1093–d1108. doi: 10.2741/1089. [DOI] [PubMed] [Google Scholar]
- 12.Carlberg C. Ann NY Acad Sci. 2000;917:387–396. doi: 10.1111/j.1749-6632.2000.tb05403.x. [DOI] [PubMed] [Google Scholar]
- 13.Reiter RJ. Best Pract Res Clin Endocrinol Metab. 2003;17:273–285. doi: 10.1016/s1521-690x(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 14.Wiesenberg I, Missbach M, Carlberg C. Restor Neurol Neurosci. 1998;12:143–150. [PubMed] [Google Scholar]
- 15.Reiter RJ, Tan DX, Manchester LC, El Sawi MR. Ann NY Acad Sci. 2002;959:238–250. doi: 10.1111/j.1749-6632.2002.tb02096.x. [DOI] [PubMed] [Google Scholar]
- 16.Tan DX, Reiter RJ, Manchester LC, et al. Curr Top Med Chem. 2002;2:181–197. doi: 10.2174/1568026023394443. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez C, Mayo JC, Sainz RM, et al. J Pineal Res. 2004;36:1–9. doi: 10.1046/j.1600-079x.2003.00092.x. [DOI] [PubMed] [Google Scholar]
- 18.Lerner AB, Case JD. J Invest Dermatol. 1959;32:211–221. [PubMed] [Google Scholar]
- 19.Slominski A, Pisarchik A, Wortsman J. Biochim Biophys Acta. 2004;1680:67–70. doi: 10.1016/j.bbaexp.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Slominski A, Wortsman J, Plonka PM, Schallreuter KU, Paus R, Tobin DJ. J Invest Dermatol. 2005;124:13–21. doi: 10.1111/j.0022-202X.2004.23528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartsch C, Bartsch H, Karasek M. Neuroendocrinol Lett. 2002;23(Suppl 1):30–38. [PubMed] [Google Scholar]
- 22.Gupta, D., Atanasio, A., and Reiter, R. J. (1988). Brain research promotion. Oxford, UK.
- 23.Slominski A, Pisarchik A, Semak I, et al. FASEB J. 2002;16:896–898. doi: 10.1096/fj.01-0952fje. [DOI] [PubMed] [Google Scholar]
- 24.Slominski A, Pisarchik A, Johansson O, et al. Biochim Biophys Acta. 2003;1639:80–86. doi: 10.1016/s0925-4439(03)00124-8. [DOI] [PubMed] [Google Scholar]
- 25.Slominski A, Pisarchik A, Semak I, Sweatman T, Wortsman J. Eur J Biochem. 2003;270:3335–3344. doi: 10.1046/j.1432-1033.2003.03708.x. [DOI] [PubMed] [Google Scholar]
- 26.Semak I, Korik E, Naumova M, Wortsman J, Slominski A. Arch Biochem Biophys. 2004;421:61–66. doi: 10.1016/j.abb.2003.08.036. [DOI] [PubMed] [Google Scholar]
- 27.Slominski A, Pisarchik A, Semak I, Sweatman T, Szczesniewski A, Wortsman J. J Invest Dermatol. 2002;119:934–942. doi: 10.1046/j.1523-1747.2002.00156.x. [DOI] [PubMed] [Google Scholar]
- 28.Slominski A, Semak I, Pisarchik A, Sweatman T, Szczesniewski A, Worstman J. FEBS Lett. 2002;511:102–106. doi: 10.1016/s0014-5793(01)03319-1. [DOI] [PubMed] [Google Scholar]
- 29.Gillbro JM, Marles LK, Hibberts NA, Schallreuter KU. J Invest Dermatol. 2004;123:346–353. doi: 10.1111/j.0022-202X.2004.23210.x. [DOI] [PubMed] [Google Scholar]
- 30.Schallreuter KU, Lemke KR, Pittelkow MR, Wood JM, Korner C, Malik R. J Invest Derm. 1995;104:953–957. doi: 10.1111/1523-1747.ep12606218. [DOI] [PubMed] [Google Scholar]
- 31.Schallreuter KU, Schulz-Douglas V, Bunz A, Beazley WD, Korner C. J Invest Dermatol. 1997;109:31–35. doi: 10.1111/1523-1747.ep12276418. [DOI] [PubMed] [Google Scholar]
- 32.Coon SL, Roseboom PH, Baler R, et al. Science. 1995;270:1681–1683. doi: 10.1126/science.270.5242.1681. [DOI] [PubMed] [Google Scholar]
- 33.Roseboom PH, Namboodiri MA, Zimonjic DB, et al. Brain Res Mol Brain Res. 1998;63:189–197. doi: 10.1016/s0169-328x(98)00273-3. [DOI] [PubMed] [Google Scholar]
- 34.Conti A, Conconi S, Hertens E, et al. J Pineal Res. 2000;28:193–202. doi: 10.1034/j.1600-079x.2000.280401.x. [DOI] [PubMed] [Google Scholar]
- 35.Slominski A, Baker J, Rosano T, et al. J Biol Chem. 1996;271:12281–12286. doi: 10.1074/jbc.271.21.12281. [DOI] [PubMed] [Google Scholar]
- 36.Cahill GM, Besharse JC. Proc Natl Acad Sci USA. 1989;86:1098–1102. doi: 10.1073/pnas.86.3.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grace MS, Cahill GM, Besharse JC. Brain Res. 1991;559:56–63. doi: 10.1016/0006-8993(91)90286-5. [DOI] [PubMed] [Google Scholar]
- 38.Becker-Andre M, Wiesenberg I, Schaeren-Wiemers N, et al. J Biol Chem. 1994;269:28531–28534. [PubMed] [Google Scholar]
- 39.Carlberg C, Hooft van Huijsduijnen R, Staple JK, DeLamarter JF, Becker-Andre M. Mol Endocrinol. 1994;8:757–770. doi: 10.1210/mend.8.6.7935491. [DOI] [PubMed] [Google Scholar]
- 40.Nosjean O, Nicolas JP, Klupsch F, Delagrange P, Canet E, Boutin JA. Biochem Pharmacol. 2001;61:1369–1379. doi: 10.1016/s0006-2952(01)00615-3. [DOI] [PubMed] [Google Scholar]
- 41.Nosjean O, Ferro M, Coge F, et al. J Biol Chem. 2000;275:31311–31317. doi: 10.1074/jbc.M005141200. [DOI] [PubMed] [Google Scholar]
- 42.Slominski A, Pisarchik A, Zbytek B, Tobin DJ, Kauser S, Wortsman J. J Cell Physiol. 2003;196:144–153. doi: 10.1002/jcp.10287. [DOI] [PubMed] [Google Scholar]
- 43.Slominski A, Pisarchik A, Tobin DJ, Mazurkiewicz JE, Wortsman J. Endocrinology. 2004;145:941–950. doi: 10.1210/en.2003-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pisarchik A, Slominski A. Eur J Biochem. 2004;271:2821–2830. doi: 10.1111/j.1432-1033.2004.04216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozo D, Garcia-Maurino S, Guerrero JM, Calvo JR. J Pineal Res. 2004;37:48–54. doi: 10.1111/j.1600-079X.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- 46.Becker-Andre M, Andre E, DeLamarter JF. Biochem Biophys Res Commun. 1993;194:1371–1379. doi: 10.1006/bbrc.1993.1976. [DOI] [PubMed] [Google Scholar]
- 47.Carrillo-Vico A, Garcia-Perganeda A, Naji L, Calvo JR, Romero MP, Guerrero JM. Cell Mol Life Sci. 2003;60:2272–2278. doi: 10.1007/s00018-003-3207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mailliet F, Ferry G, Vella F, Thiam K, Delagrange P, Boutin JA. FEBS Lett. 2004;578:116–120. doi: 10.1016/j.febslet.2004.10.083. [DOI] [PubMed] [Google Scholar]
- 49.Strassburg A, Strassburg CP, Manns MP, Tukey RH. Mol Pharmacol. 2002;61:320–325. doi: 10.1124/mol.61.2.320. [DOI] [PubMed] [Google Scholar]
- 50.Buryanovskyy L, Fu Y, Boyd M, et al. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welch RAS, Gurnsey MP, Betteridge K, Mitchell RJ. Proc N Z Soc Anim Prod. 1990;50:335–338. [Google Scholar]
- 52.Nixon AJ, Choy VJ, Parry AL, Pearson AJ. J Exp Zool. 1993;267:47–56. doi: 10.1002/jez.1402670108. [DOI] [PubMed] [Google Scholar]
- 53.Ibraheem M, Galbraith H, Scaife J, Ewen S. J Anat. 1994;185(Pt 1):135–142. [PMC free article] [PubMed] [Google Scholar]
- 54.Slominski A, Chassalevris N, Mazurkiewicz J, Maurer M, Paus R. Exp Dermatol. 1994;3:45–50. doi: 10.1111/j.1600-0625.1994.tb00265.x. [DOI] [PubMed] [Google Scholar]
- 55.Kobayashi H, Paus R. Exp Dermatol. 2005;14:157. [Google Scholar]
- 56.Fischer TW, Fischer A, Knöll B, Hipler UC, Elsner P. Arch Derm Res. 2000;292:147. [Google Scholar]
- 57.Fischer TW, Burmeister G, Schmidt HW, Elsner P. Br J Dermatol. 2004;150:341–345. doi: 10.1111/j.1365-2133.2004.05685.x. [DOI] [PubMed] [Google Scholar]
- 58.Fischer T, Wigger-Alberti W, Elsner P. Hautarzt. 1999;50:5–11. doi: 10.1007/s001050050857. [DOI] [PubMed] [Google Scholar]
- 59.Drobnik J, Dabrowski R. Cytobios. 1999;100:49–55. [PubMed] [Google Scholar]
- 60.Drobnik J, Dabrowski R. Cytobios. 1996;85:51–58. [PubMed] [Google Scholar]
- 61.Pertsov SS, Abramov YV, Volodina TV, Rebrov LB. Bull Exp Biol Med. 2004;137:327–330. doi: 10.1023/b:bebm.0000035120.75368.73. [DOI] [PubMed] [Google Scholar]
- 62.Soybir G, Topuzlu C, Odabas O, Dolay K, Bilir A, Koksoy F. Surg Today. 2003;33:896–901. doi: 10.1007/s00595-003-2621-3. [DOI] [PubMed] [Google Scholar]
- 63.Fischer TW, Elsner P. Curr Probl Dermatol. 2001;29:165–174. doi: 10.1159/000060665. [DOI] [PubMed] [Google Scholar]
- 64.Bangha E, Elsner P, Kistler GS. Arch Dermatol Res. 1996;288:522–526. doi: 10.1007/BF02505248. [DOI] [PubMed] [Google Scholar]
- 65.Bangha E, Elsner P, Kistler GS. Dermatology. 1997;195:248–252. doi: 10.1159/000245953. [DOI] [PubMed] [Google Scholar]
- 66.Dreher F, Denig N, Gabard B, Schwindt DA, Maibach HI. Dermatology. 1999;198:52–55. doi: 10.1159/000018064. [DOI] [PubMed] [Google Scholar]
- 67.Dreher F, Gabard B, Schwindt DA, Maibach HI. Br J Dermatol. 1998;139:332–339. doi: 10.1046/j.1365-2133.1998.02447.x. [DOI] [PubMed] [Google Scholar]
- 68.Lee KS, Lee WS, Suh SI, et al. Exp Mol Med. 2003;35:263–268. doi: 10.1038/emm.2003.35. [DOI] [PubMed] [Google Scholar]
- 69.Ryoo YW, Suh SI, Mun KC, Kim BC, Lee KS. J Dermatol Sci. 2001;27:162–169. doi: 10.1016/s0923-1811(01)00133-5. [DOI] [PubMed] [Google Scholar]
- 70.Fischer TW, Scholz G, Knoll B, Hipler UC, Elsner P. J Pineal Res. 2001;31:39–45. doi: 10.1034/j.1600-079x.2001.310106.x. [DOI] [PubMed] [Google Scholar]
- 71.Fischer TW, Scholz G, Knoll B, Hipler UC, Elsner P. J Pineal Res. 2004;37:107–112. doi: 10.1111/j.1600-079X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 72.Fischer TW, Scholz G, Knoll B, Hipler UC, Elsner P. Skin Pharmacol Appl Skin Physiol. 2002;15:367–373. doi: 10.1159/000064543. [DOI] [PubMed] [Google Scholar]
- 73.Pisarchik A, Wortsman J, Slominski A. Gene. 2004;341:199–207. doi: 10.1016/j.gene.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 74.Maharaj DS, Anoopkumar-Dukie S, Glass BD, et al. J Pineal Res. 2002;32:257–261. doi: 10.1034/j.1600-079x.2002.01866.x. [DOI] [PubMed] [Google Scholar]
- 75.Kanikkannan N, Jackson T, Shaik MS, Singh M. Eur J Pharm Sci. 2001;14:217–220. doi: 10.1016/s0928-0987(01)00176-2. [DOI] [PubMed] [Google Scholar]
- 76.Hussein MR, Abu-Dief EE, Abd El-Reheem MH, Abd-Elrahman A. Int J Exp Pathol. 2005;86:45–55. doi: 10.1111/j.0959-9673.2005.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sener G, Atasoy BM, Ersoy Y, Arbak S, Sengoz M, Yegen BC. J Pineal Res. 2004;37:241–246. doi: 10.1111/j.1600-079X.2004.00161.x. [DOI] [PubMed] [Google Scholar]
- 78.Vasin MV, Ushakov IB, Kovtun V, Komarova SN, Semenova LA, Galkin AA. Radiat Biol Radioecol. 2004;44:68–71. [PubMed] [Google Scholar]
- 79.Kim BC, Shon BS, Ryoo YW, Kim SP, Lee KS. J Dermatol Sci. 2001;26:194–200. doi: 10.1016/s0923-1811(01)00088-3. [DOI] [PubMed] [Google Scholar]
- 80.Kilanczyk E, Bryszewska M. Cell Mol Biol Lett. 2003;8:333–336. [PubMed] [Google Scholar]
- 81.Gurlek A, Aydogan H, Parlakpinar H, et al. J Pineal Res. 2004;36:58–63. doi: 10.1046/j.1600-079x.2003.00099.x. [DOI] [PubMed] [Google Scholar]
- 82.Tunali T, Sener G, Yarat A, Emekli N. Life Sci. 2005;76:1259–1265. doi: 10.1016/j.lfs.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 83.Basak PY, Agalar F, Gultekin F, Eroglu E, Altuntas I, Agalar C. Ulus Travma Derg. 2003;9:96–101. [PubMed] [Google Scholar]
- 84.Karbownik M. Neuroendocrinol Lett. 2002;23(Suppl 1):39–44. [PubMed] [Google Scholar]
- 85.Kumar CA, Das UN. Med Sci Monit. 2000;6:471–475. [PubMed] [Google Scholar]
- 86.Bangha E, Lauth D, Kistler GS, Elsner P. Skin Pharmacol. 1997;10:298–302. doi: 10.1159/000211518. [DOI] [PubMed] [Google Scholar]
- 87.Fischer TW, Greif C, Fluhr JW, Wigger-Alberti W, Elsner P. Skin Pharmacol Physiol. 2004;17:190–194. doi: 10.1159/000078822. [DOI] [PubMed] [Google Scholar]
- 88.Oh HJ, Oh YK, Kim CK. Int J Pharm. 2001;212:63–71. doi: 10.1016/s0378-5173(00)00598-6. [DOI] [PubMed] [Google Scholar]
- 89.Cos S, Sanchez-Barcelo EJ. Front Neuroendocrinol. 2000;21:133–170. doi: 10.1006/frne.1999.0194. [DOI] [PubMed] [Google Scholar]
- 90.Gonzalez R, Sanchez A, Ferguson JA, et al. Melanoma Res. 1990;1:237–243. doi: 10.1097/00008390-199111000-00003. [DOI] [PubMed] [Google Scholar]
- 91.Lissoni P, Malugani F, Malysheva O, et al. Neuroendocrinol Lett. 2002;23:341–344. [PubMed] [Google Scholar]
- 92.Lissoni P, Vaghi M, Ardizzoia A, et al. In Vivo. 2002;16:93–96. [PubMed] [Google Scholar]
- 93.Reiter RJ, Tan DX, Sainz RM, Mayo JC, Lopez-Burillo S. J Pharm Pharmacol. 2002;54:1299–1321. doi: 10.1211/002235702760345374. [DOI] [PubMed] [Google Scholar]
- 94.Pisarchik A, Slominski AT. FASEB J. 2001;15:2754–2756. doi: 10.1096/fj.01-0487fje. [DOI] [PubMed] [Google Scholar]