

Abstract
Growth inhibition and oxidized guanine lesion formation were studied in a number of base excision repair (BER) deficient Escherichia coli (E. coli) following chromate exposure. The only BER deficient bacterial strain that demonstrated significant growth inhibition by chromate, in comparison to its matched wild-type cell line, was the Nei deficient (TK3D11). HPLC coupled with electrospray ionization mass spectrometry showed that the Nei deficient E. coli accumulated the further oxidized guanine lesion, spiroiminodihydantoin (Sp), in genomic DNA at levels that were ~20-fold greater than its wild-type counterpart. However, no accumulation of the putative intermediate of Sp, 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxodG), was observed in the Nei deficient strain. A MutM−/MutY− double deletion mutant that was deficient in BER enzymes for the recognition and repair of 8-oxodG demonstrated no sensitivity toward chromate nor was there an associated increase in Sp accumulation over that of its wild type. However, the MutM−/MutY− double deletion mutant did show ~20-fold accumulation of 8-oxodG upon chromate exposure over that of the wild type and the Nei deficient E. coli. These data demonstrate that the Nei BER enzyme is critical for the recognition and repair of the Sp lesion in bacterial cell lines and demonstrates the protective effect of a specific BER enzyme on DNA lesions formed by chromate. To our knowledge, these are the first studies to show the formation and biological significance of the Sp lesion in a cellular system. This study has significant mechanistic and toxicological implications for how chromate may serve as an initiator of carcinogenesis and suggests a role for specific repair enzymes that may ameliorate the carcinogenic potential of chromate.
Introduction
Further oxidation of the ubiquitous 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-oxodG)1 lesion in DNA has been a recent topic of considerable interest. An ever increasing number of oxidants including photosensitizing agents (1), peroxynitrite (2), carbonate radicals (3), and high valent metals such as Ir(IV) (4) and Cr(V) (5) have all been observed to specifically react at 8-oxodG sites within duplex DNA to form further oxidized lesions (Figure 1). Of particular interest are the lesions of spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh), which were first observed in the reaction of 8-oxodG with Ir(IV) by the Burrows group (4, 6–8). Recently, we have shown the formation of Sp directly from guanine in synthetic oligonucleotides during the reduction of chromate with the endogenous cellular reductant, ascorbate (9).
Figure 1.
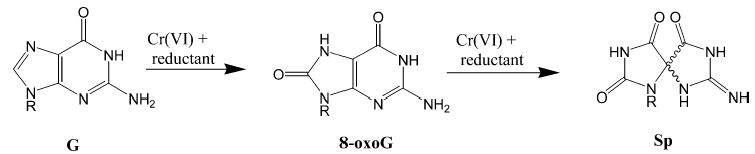
General reaction scheme for the formation of 8-oxoG and Sp from the oxidation of guanine (G) by Cr(VI).
Sp and Gh lesions show an enhanced polymerase arresting capability in comparison to the parent 8-oxodG lesion as well as significantly increasing levels of mutation in vitro and in cellular systems (10–13). Recognition and excision of the Sp and Gh lesions in DNA in vitro have been shown to occur through the bacterial Nei (endonuclease VIII) base excision repair (BER) enzyme (14) and through the mammalian NEIL (Nei-like) BER glycosylases (15). While the thermodynamics of 8-oxodG oxidation to the Sp and Gh lesions coupled with the demonstrated recognition of these lesions by endogenous BER enzymes argues for their formation in cellular DNA, the refractory nature of these lesions to detection has hitherto kept them from being observed in cellular systems.
On the basis of the in vitro studies, Nei deficient bacteria should show sensitivity, manifested as differential growth, toward the carcinogen chromate if lesions such as Sp are formed during the intracellular reduction of Cr(VI) to Cr(III). Furthermore, the Nei deficient cell lines should accumulate the Sp lesion within genomic DNA that correlates with the observed growth inhibition. This study analyzed a series of BER deficient Escherichia coli (E. coli) for differential growth inhibition toward chromate with respect to their wild-type counterparts. The MutM−, MutY−, and MutM−/MutY− double mutants all demonstrated similar growth curves following chromate treatment to their matched wild-type counterparts. Only the Nei deficient E. coli (TK3D11) demonstrated a significant difference in growth with increasing doses of chromate over that of its wild type. The genomic DNA of the Nei− and the MutM−/MutY−double mutant, with respect to their wild-type controls, was assayed for the formation of the Sp lesion and the putative intermediate of this lesion, 8-oxodG. This study focused solely on the Sp lesion since this lesion is formed preferentially over Gh at neutral pH (7). Using high-performance liquid chromatography coupled with electrospray ionization–mass spectrometry (HPLC-ESI-MS) and an isotopically labeled 18O-Sp internal standard, Sp arising within the genomic DNA of the different cell lines was identified and quantified.
Materials and Methods
Hazardous Materials
Caution: Chromium(VI) is a known human lung carcinogen, and Cr(V) is an intermediate metabolite of Cr(VI) reduction. Proper care should be taken in the handling of these materials.
Bacterial Strains
The strains WP2 and CM1322 were generous gifts from Dr. Bryn Bridges and Dr. Andy Timms of MRC (University of Sussex, United Kingdom). WP2 is trpE65-(oc), lon-11, sulA1 (16), CM1322 is as WP2 but mutY68::kanR, mutM::Tn10 [source P1(TT101) × CM1307] (17), CM1319 was as WP2 but mutM::TN10, CM1307 is as WP2 but mutY68::kanR (18). TK3D11 (ΔkdpFABC-gltA) 219, Δ(galK-bioD) 76, trkA405, trkD1, rha-4, thi-1 and its control strain CSR06 [thr-1, araC14, leuB6(Am), Δ(gpt-proA)62, lacY1, tsx-33, glnV44(AS), phr-1, galK2(Oc), LAM-, Rac-0, hisG4(Oc), rfbC1, mgl-51, rpsL31(strR), kdgK51, xylA5, mtl-1, arg E3(Oc), thi-1, uvrA6] were obtained from the E. coli Genetic Stock Center at Yale University (New Haven, CT). The ΔkdpFABC-gltA encompasses the five genes of the Nei operon (19), and TK3D11 is designated herein as Nei−in order to reflect the deletion of this operon.
Differential Growth of BER Deficient E. coli Following Chromate Treatment
Fourteen hour growth curves were conducted on the BER deficient strains of E. coli and their wild-type controls (WP2 for the MutM−/MutY− CM1322 and CSRO6 for the Nei− TK3D11). The E. coli strains were grown with either 0 (control), 100, or 250 μM Cr(VI) over a time course of 14 h in LB broth at 37 °C. Chromate, Cr(VI), as potassium dichromate (J. T. Baker Chem. Co., Phillipsburg, NJ) was added at an OD600 of 0.05, and measurements of cell density were taken every hour for 8 h and then every 2 h for the next 6 h.
Extraction of Genomic DNA for Mass Spectral and Electrochemical Analysis of DNA Lesions
Cultures of CSR06, TK3D11, WP2, and CM1322 were grown in 50 mL of LB broth at 37 °C until they reached an OD600 of ~0.5. Cell densities were normalized to ensure identical numbers of cells in each treatment. When an OD600 of 0.5 was reached, Cr(VI) was added to the cell/LB mixture to give concentrations of 0, 250, and 500 μM. Cells were allowed to grow for 3 h after the addition of Cr(VI) and prior to harvesting. Cells were harvested and lysed using the Qiagen bacterial lysis buffer (Qiagen Inc., Valencia, CA), and genomic DNA was extracted using phenol/ chloroform as previously described (20).
Enzymatic Digestion of Oxidized DNA
Phosphodiesterase I was purchased from Sigma-Aldrich (St. Louis, MO). Phosphodiesterase II was purchased from Worthington Biochemical Corporation (Lakewood, NJ). Calf intestinal phosphatase was purchased from New England BioLabs (Ipswich, MA). DNA was purified from E. coli. as described above. DNA samples were dissolved in Nanopure H2O and normalized to a final concentration of about 500 μg/mL (OD260 = 10) and a final volume of 100 μL. The DNA was denatured by heating to 95 °C for 10 min and flash cooled. Samples were hydrolyzed to the individual nucleotides by the addition of 10 units of phosphodiesterase II (unbuffered) and incubated for 3 h at 37 °C. Samples were further hydrolyzed by the addition of 10 units of phosphodiesterase I buffered in 100 mM Tris-HCl, 100 mM NaCl, and 15 mM MgCl2 (pH 8.9) and incubated for 3 h at 37 °C. The resulting nucleotides were hydrolyzed to their respective nucleosides by the addition of 10 units of calf intestinal phosphatase and incubated for 3 h at 37 °C. Following hydrolysis, samples were filtered using a Millipore YM-30 centrifugal filter, lyophilized to dryness, and dissolved in 100 μL of Nanopure H2O.
HPLC Quantification of Deoxyguanosine
Hydrolyzed DNA samples were analyzed using an Agilent 1100 series HPLC coupled with diode array detection. Separations were performed using a C18 reverse phase Microsorb MV 2.1 mm × 250 mm column, with a 0.5% aqueous acetonitrile/min gradient mobile phase and a flow rate of 0.4 mL/min. A calibration curve was generated from standards of dG ranging from 2 to 100 nmol. The amount of dG per DNA sample was calculated by comparing the peak area from 10 μL of injected enzymatic hydrolysate with the calibration curve.
HPLC-ESI-MS Analysis of Oxidized Duplex DNA
Hydrolyzed DNA samples were analyzed using a Waters 2790 HPLC coupled to a Micromass LCT mass spectrometer with electrospray ionization. Separations were performed using a C18 reverse phase Microsorb MV 2.1 mm × 250 mm column, with a 0.5% aqueous acetonitrile/min gradient mobile phase and a flow rate of 0.4 mL/min. Mass spectral analysis was performed using selected ion monitoring (SIM) with a cone voltage of 30 V, a capillary voltage of 3000 V, a desolvation temperature of 250 °C, a source temperature of 150 °C, and an aperture of 15. Sp was identified and quantified through the introduction of the stable isotopic 18O internal standard of 18O-Sp described below. A linear calibration curve was obtained using 1.0 nmol of 18O-Sp as the internal standard and variable amounts of 16O-Sp ranging from 0.01 to 1.0 nmol (slope = 1.0, r2 = 0.997). For the measurements of Sp in hydrolyzed DNA samples, the amount of 18O-Sp internal standard was chosen according to the limits of this standard curve. Levels of Sp in hydrolyzed DNA samples were expressed as the number of Sp per 106 dG.
Synthesis and Purification of 16O-Sp and 18O-Sp Nucleoside Standards
Na2IrCl6·6H2O, Ir(IV), was purchased from Acros Organics (Geel, Belgium). 8-Oxo-2′-deoxyguanosine was purchased from Sigma-Aldrich. Isotopically enriched water-18O (18O 95%) was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA). 18O-Sp was synthesized by oxidation of 8-oxodG in H218O using Ir(IV) as described previously (21). Briefly, a reaction volume containing 300 μL of 8-oxo-2′-deoxyguanosine (2.5 mM) was incubated for 1 h at 60 °C with Na2IrCl6 (5 mM) in 75 mM phosphate buffer (pH 7.4) followed by addition of 30 μL (200 mM; pH 4.8) of Na–Acetate buffer. Excess Ir(IV) was removed by microanion exchange chromatography, using a column containing 20 mg of DEAE Sephadex A-25 anion exchange resin. Samples were eluted with four 100 μL washes with Nanopure water. Samples were pooled and further purified by HPLC using a Zorbax C18 semipreparative 9.4 mm × 25 cm column with a gradient of 5%/min acetonitrile in water and a flow rate of 1.8 mL/min. Samples were collected and lyophilized to ×50 μL and quantified by UV–vis, ε230 = 4900 M−1cm−1 (4). The identity of 18O-Sp was confirmed by mass spectral analysis and MS-MS fragmentation as previously described (4, 9).
HPLC-ECD Analysis of Genomic DNA from E. coli
HPLC-ECD identification of 8-oxodG was performed using a previously established method (20). 8-OxodG and dG in enzymatically digested DNA samples were separated by HPLC with a 4.6 mm × 150 mm reverse phase YMC basic column and quantified using a CoulArray electrochemical detection system (ESA Inc., Chelmsford, MA). Nucleosides were eluted from the column using an isocratic mobile phase of 100 mM sodium acetate, pH 5.2, in 4% methanol. Potentials of the four coulometric analytical cells of the CoulArray system placed in series were as follows: 50, 125, 175, 200, 250, 380, 500, 700, 785, 850, 890, and 900 mV. Calibration curves were generated from a dG standard (0.1–2 μg) and an 8-oxodG standard (50–1000 pg). The amount of 8-oxodG was calculated by comparing the peak area from a 50 μL injection of enzymatic hydrolysate of the oxidized DNA sample with the calibration curve. Levels of 8-oxodG in the genomic DNA were expressed as the number of 8-oxodG per million dG.
Results
Growth Inhibition of BER Deficient E. coli Strains as a Function of Chromate Treatment
This study focused on a set of known BER deficient bacterial cells lines that included the MutM−, MutY−, the MutM−/ MutY− double mutant, and an Nei deficient E. coli cell line. The MutM− (CM1319), MutY− (CM1307), and the MutM−/MutY− double mutant (CM1322) showed no change in growth with respect to the wild-type strain (WP2) as Cr(VI) concentration was increased. A representative growth curve for the MutM−/MutY− (CM1322) double mutant vs its matched wild-type cell line is shown in Figure 2 (MutM− and MutY− not shown).
Figure 2.
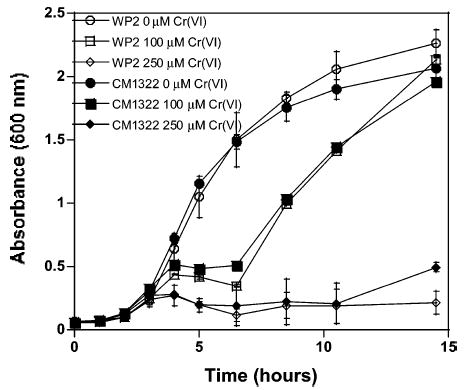
Growth inhibition of the MutM−/MutY− double mutant (CM1322) and its corresponding wild type (WP2) with increasing chromate treatment. Growth inhibition data for the wild type are represented by open symbols and the mutant with the identical closed symbol for each chromium concentration. The error bars represent the standard deviation of three replicates.
In contrast, the Nei deficient strain (TK3D11) showed a significant change in growth inhibition over that of its wild-type control (CSR06) following chromate treatment (Figure 3). Growth of the Nei deficient strain at the 8 h time point for the 100 μM and 250 μM Cr(VI) treatments was observed to be inhibited by 27 and 67% with respect to its wild-type control strain. To our knowledge, this is the first example of a BER deletion mutant to show differential growth sensitivity toward Cr(VI).
Figure 3.
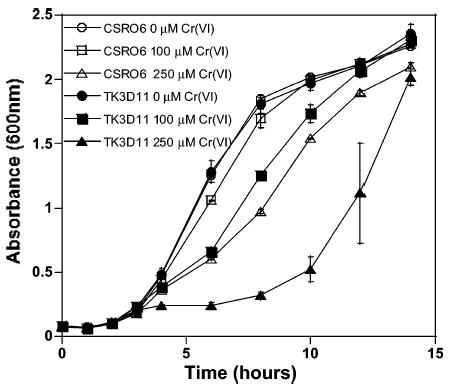
Growth inhibition of the Nei− mutant (TK3D11) and its corresponding wild type (CSR06) with increasing chromate treatment. Growth inhibition data for the wild type are represented by open symbols and the mutant with the identical closed symbol for each chromium concentration. The error bars represent the standard deviation of three replicates.
HPLC-ESI-MS Analysis for the Sp Lesion in Genomic DNA of BER Deficient E. coli
The ability of the Nei deficient E. coli (TK3D11) and the MutM−/MutY−double mutant (CM1322) to accumulate the Sp lesion in genomic DNA in response to chromate treatment was studied by HPLC-ESI-MS. An isotopically labeled 18O-Sp internal standard allowed the identification and quantification of 16O-Sp in digested DNA samples as we have described previously (9). A typical ion current profile for digested E. coli genomic DNA containing the 18O-Sp internal standard and 16O-Sp from oxidized DNA is shown in Figure 4. An exact mass calculation of the M + Na peak for 16O-Sp using the 18O-Sp internal standard was 322.0761 as compared to a calculated mass of 322.0764 to yield an 0.8 ppm mass accuracy. Accumulation of Sp in the genomic DNA was assessed in high-density cell cultures (OD600 = 0.5) of the Nei deficient (TK3D11), the MutM−/MutY− double mutant (CM1322), and their matched wild-type controls treated with 250 or 500 μM chromate. HPLC-ESI-MS analysis of the digested genomic DNA for Sp formation, using the 18O-Sp internal standard for quantification showed a dose-dependent accumulation of Sp with increasing chromate concentration only in the Nei deficient (chromate sensitive; TK3D11) E. coli (Figure 5). Accumulation of the Sp lesion in the Nei deficient E. coli was observed to be approximately 20-fold greater than that observed for its wild-type counterpart (CSR06). The MutM−/MutY− double mutant (CM1322), which is Nei proficient, and its wild-type counterpart (WP2) showed no significant accumulation of Sp with chromate treatment over that of control. This result correlated well with our previous studies on in vitro Sp formation in Cr(VI)-oxidized DNA (9) and suggests that the Nei BER enzyme is the primary glycosylase that recognizes Sp in bacterial DNA.
Figure 4.
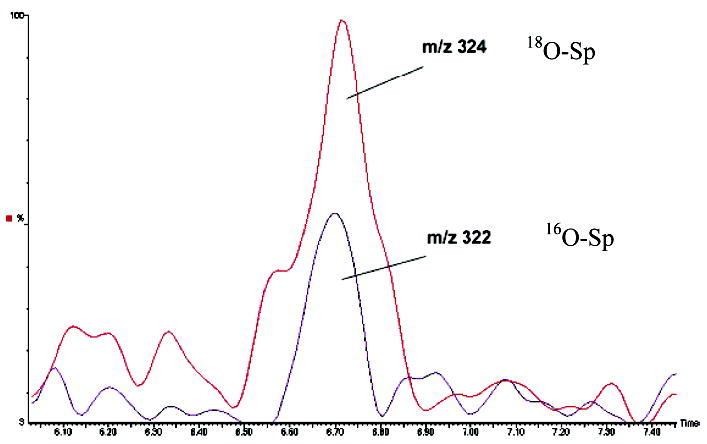
Typical HPLC-ESI-MS ion current profiles of a genomic DNA digest from Cr(VI)-treated TK3D11 E. coli using SIM. The peak at m/z 324 corresponds to the Na + 18O-Sp internal standard, and the m/z 322 peak with identical retention time corresponds to the Na + 16O-Sp species arising from Cr(VI) oxidation of genomic DNA.
Figure 5.
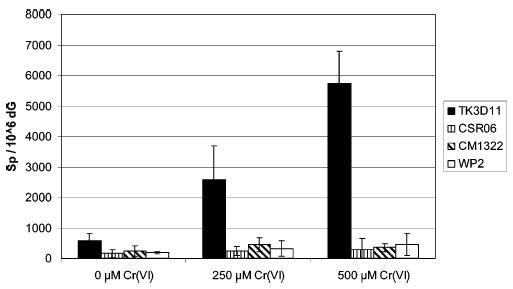
Formation of Sp in chromate-treated E. coli. The Nei deficient (TK3D11) E. coli shows a dose dependence for Sp formation with increasing chromate concentration. The data are normalized on a per million dG basis, and each data point is a minimum of n = 5 replicates.
The toxicity of chromate in these high-density cell cultures (OD600 = 0.5) was assessed by a plating assay as growth curves cannot be conducted on high-density cultures. At the 250 μM concentration of chromate, no overt toxicity was observed for any of the cell lines in the plating assay. At the 500 μM chromate treatment, the Nei deficient (TK3D11) cell line showed an 18% survival rate vs 40% for its matched wild type (CSRO6) in the plating assay. The MutM−/MutY− double mutant (CM1322) displayed a 35% survival in the plating assay with its matched wild type (WP2) showing 43% survival at the 500 μM chromate treatment.
HPLC-ECD Analysis for the 8-OxodG Lesion in the Genomic DNA of BER Deficient E. coli
Enzymatically digested genomic DNA from the different E. coli strains were also analyzed for 8-oxodG using HPLC coupled with electrochemical detection (HPLC-ECD). 8-OxodG is the putative intermediate of Sp formation and is primarily removed from genomic DNA by the MutM (Fpg) and MutY BER enzymes (22). Figure 6 shows that the MutM/MutY proficient bacterial cell lines, TK3D11, CSR06, and WP2, all gave typical levels of 8-oxodG (typical background levels for 8-oxodG have been established by the ESCODD to be 0.3–4.2 8-oxodG/106 dG; 23) even at the highest concentration of chromate used (500 μM). Not surprisingly, the MutM−/MutY− double mutant (CM1322) was the only cell line studied that showed accumulation of 8-oxodG in its genomic DNA following chromate treatment. However, the 8-oxodG accumulation in the MutM−/MutY− double mutant was modest with respect to the amount of Sp accumulated in the Nei deficient cell line and was not differentially toxic to the cell by either a plating assay or growth curves.
Figure 6.
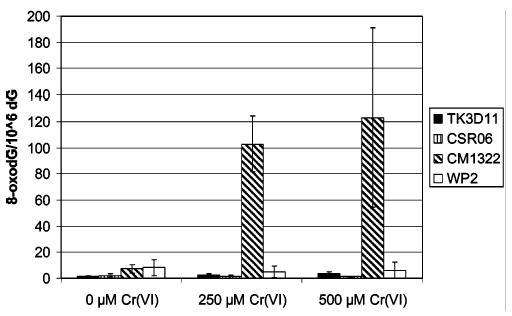
Formation of 8-oxodG in chromate-treated wild-type and BER deficient E. coli genomic DNA. Each data point is normalized on a per million dG basis and has a minimum of n = 3 replicates.
Discussion
Growth inhibition in repair deficient strains of bacteria by a toxicant that exceeds that observed for the wild-type bacterial strain implies both reaction with DNA and a role for the repair gene in ameliorating its genotoxic effects. The reaction of chromate with DNA in cellular systems is thought to produce a number of different adducts including those derived from an oxidative pathway (strand breaks, abasic sites, 8-oxodG; 24–26) and those derived from a metal-binding pathway (DNA interstrand cross-links and DNA–protein cross-links; 27, 28). Little is known about the exact nature of many of these adducts or their mechanism of repair. One recent study has shown that a human XP-A fibroblast cell line, deficient in nucleotide excision repair (NER), was sensitive to chromate (29). This chromate sensitivity in the XP-A fibroblasts was attributed to the formation of DNA cross-links arising from the metal-binding pathway.
Even though Cr(VI) is a known intracellular oxidant, no systematic analysis of the impact of Cr(VI) exposure on BER deficient bacterial strains had previously been undertaken. We studied the ability of BER deficient E. coli to grow in the presence of Cr(VI) since the formation of 8-oxodG has long been considered to be a relevant lesion induced by Cr(VI) in a variety of in vitro and cellular systems. If Cr(VI) exposure in E. coli generated 8-oxodG as a key genotoxic lesion, it would be expected that strains that are repair deficient in MutM (Fpg) and/ or MutY would exhibit significant growth inhibition. MutM, or Fpg, recognizes and excises 8-oxodG opposite cytosine while MutY recognizes and excises adenine opposite 8-oxodG (adenine is the base that is misincorporated for cytosine opposite an 8-oxodG upon DNA replication) (22). MutM recognizes Sp in duplex DNA when paired opposite cytosine, C, as a complementary base but shows little or no recognition of this lesion when paired opposite guanine, G, nor adenine, A (14). In fact, it has been recently shown that a functional MutY repair enzyme effectively eliminates MutM recognition of Sp opposite G (14). The BER enzyme, Nei, has shown the ability to recognize Sp in duplex DNA opposite C, G, and A with the greatest affinity for Sp opposite G (14). Nei is also the only BER enzyme known to recognize Sp opposite A. This finding is of considerable significance since recent cellular mutation studies have suggested that G→C transversion mutations, and to a lesser degree G→T transversion mutations, predominate when Sp is formed in duplex DNA (10–13). Interestingly enough, these transversion mutations are the primary mutations observed in the lung tumors of chromate-exposed workers and in shuttle vector replication assays in Cr(VI)-treated mammalian cells (30, 31). On the basis of these lesion specific mutation profiles, we propose that the formation of the Sp lesion is more consistent with known Cr(VI) mutation patterns than is the 8-oxodG lesion.
DNA repair studies using E. coli are generally considered significant to humans due to the homology and significant substrate overlap between the E. coli and mammalian BER enzymes. Recently, NEIL BER genes were found in both the human and the mouse genome (32, 33). These genes have been designated as NEIL1, NEIL2, and NEIL3. The mammalian NEIL genes are homologous to E. coli Nei and Fpg, and we have hypothesized that they play a fundamental role in repair of chromium-damaged DNA. We have recently established that the murine NEIL1 and NEIL2 BER enzymes have a high degree of affinity for the Sp lesion with little or no activity toward 8-oxodG (15).
The formation of Sp in DNA in vitro has been observed in a wide variety of oxidizing systems including reactions with chromium. The identification and quantification of the Sp lesion in cellular systems have been complicated by its refractory nature toward enzymatic digestion and the polarity of the resulting nucleoside that makes it difficult to separate by HPLC. The use of 18O-Sp as an internal standard in our digestion mixtures has allowed us to quantify the formation of 16O-Sp arising in genomic DNA extracted from E. coli as well as to identify lesion formation through characteristic mass shifts and retention times using HPLC-ESI-MS and selective ion monitoring (SIM). At the highest dose of chromate used in this study, 500 μM, an approximate 20-fold increase in Sp formation was observed for the Nei deficient E. coli over its matched wild-type control. In this study, the chromate was added to E. coli in the logarithmic growth phase. We hypothesize that these quickly replicating cells accumulated Sp opposite the misincorporated bases guanine and adenine, conditions under which Nei has been shown to play the major role in repair (14) (Figure 7). The failure of the Nei deficient cell line to accumulate even modest amounts of 8-oxodG, while accumulating large quantities of Sp, indicates that chromate oxidation of 8-oxodG effectively and efficiently out-competes the proficient MutM and MutY repair systems in this cell line. This is not unexpected since 8-oxodG is kinetically and thermodynamically prone to oxidation to Sp. Indeed, we have previously shown (5) that chromium preferentially oxidized 8-oxodG in DNA over any other nucleic acid base, including guanine. These results suggest, however, the additional possibility that chromate forms Sp within genomic DNA directly without going through an 8-oxodG intermediate. While there has not been an established mechanistic route for such an oxidative event with chromate, our current results cannot rule out this possibility.
Figure 7.
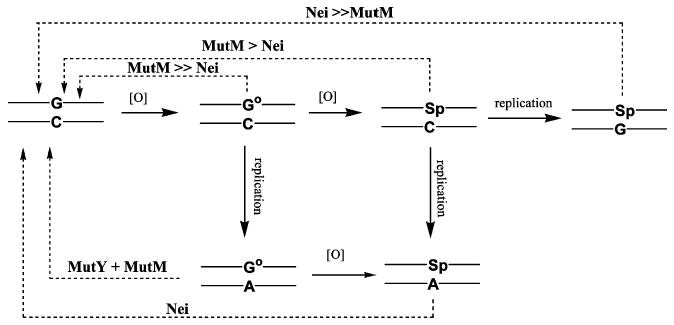
Schematic representation of 8-oxodG (G°) and Sp formation and lesion repair by E. coli BER enzymes in genomic DNA. The recognition specific glycosylase initiating each repair enzyme sequence is indicated above each pathway.
Lesion formation in the MutM−/MutY− double mutant, which has a functional Nei BER gene, can remove the Sp lesion from the genomic DNA pool, resulting in no accumulation of Sp and thus no sensitivity toward chromate. However, the lack of the MutM and MutY BER glycosylases in this strain explains the observed accumulation of 8-oxodG within the genomic DNA.
Conclusion
The deletion of the Nei BER enzyme in E. coli results in a differential sensitivity of this cell line toward chromate. The high level of Sp measured in the genomic DNA of this strain almost certainly contributes to the chromate sensitivity of these cells. On the basis of human mutation data and BER homologies between bacteria and humans, these data suggest that the Sp lesion may play a major role in chromate-induced lung tumors. The high degree of homology between bacterial and mammalian BER enzymes suggests that polymorphisms in the mammalian NEIL BER enzymes may be molecular epidemiological markers to assess chromate sensitive individuals.
Acknowledgments
Funding for this study was provided by the National Institute of Environmental Health Sciences Grant (ES10437) to K.D.S. B.D.M. is partially supported by an NCRR, NIH Grant P20RR017670. We thank Dr. Fernando Cardozo-Pelaez, Dave Cox, and Celeste Bolin for assistance with 8-oxodG measurements. We also thank Dr. Bryn Bridges and Dr. Andy Timms for their kind gift of the MutM−, MutY−, and the MutM−/ MutY− double mutant E. coli strains.
Footnotes
Abbreviations: 8-oxodG, 7,8-dihydro-8-oxo-2′-deoxyguanosine; Sp, spiroiminodihydantoin; Gh, guanidinohydantoin; HPLC-ESI-MS, high-performance liquid chromatography–electrospray ionization–mass spectrometry; Nei, DNA glycosylase endonuclease VIII; NEIL, Nei-like DNA glycosylase; SIM, selected ion monitoring; BER, base excision repair.
References
- 1.Luo W, Muller JG, Burrows CJ. The pH-dependent role of superoxide in riboflavin-catalyzed photooxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 2.Tretyakova NY, Niles JC, Burney S, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced reactions of synthetic oligonucleotides containing 8-oxoguanine. Chem Res Toxicol. 1999;12:459–466. doi: 10.1021/tx980235c. [DOI] [PubMed] [Google Scholar]
- 3.Joffe A, Geacintov NE, Shafirovich V. DNA lesions derived from site selective oxidation of guanine by carbonate radicals. Chem Res Toxicol. 2003;16:1528–1538. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 4.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 5.Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high valent chromium complex: A possible mechanism of chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 6.Muller JG, Duarte V, Hickerson RP, Burrows CJ. Gel electrophoretic detection of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine via oxidation by Ir(IV) Nucleic Acids Res. 1998;26:2247–2249. doi: 10.1093/nar/26.9.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 8.Duarte V, Muller JG, Burrows CJ. Insertion of dGMP and dAMP during in vitro DNA synthesis opposite an oxidized form of 7,8-dihydro-8-oxoguanine. Nucleic Acids Res. 1999;27:496–502. doi: 10.1093/nar/27.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slade PG, Hailer MK, Martin BD, Sugden KD. Guanine-specific oxidation of double stranded DNA by Cr(VI) and ascorbic acid forms spiroiminodihydantoin and 8-oxo-2′-deoxygua-nosine. Chem Res Toxicol. 2005;18:1140–1149. doi: 10.1021/tx050033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugden KD, Martin BD. Guanine and 7,8-dihydro-8-oxoguanine specific oxidation in DNA by chromium(V) Environ Health Perspect. 2002;110:725–728. doi: 10.1289/ehp.02110s5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 12.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 13.Henderson PT, Delaney JC, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;42:914–921. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 14.Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation products of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hailer MK, Slade PG, Rosenquist TA, Martin BD, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair. 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Timms AR, Bridges BA. Reversion of the tyrosine ochre strain Escherichia coli WU3610 under starvation conditions depends on a new gene tas”. Genetics. 1998;148:1627–1635. doi: 10.1093/genetics/148.4.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dr. Bryn Bridges, personal communication.
- 18.Bridges BA, Sekiguchi M, Tajiri T. Effect of MutY and MutM/Fpg-1 mutations on starvation-associated mutation in Escherichia coli: Implications for the role of 7,8-dihydro-8-oxoguanine. Mol Gen Genet. 1996;251:352–357. doi: 10.1007/BF02172526. [DOI] [PubMed] [Google Scholar]
- 19.Gifford CM, Wallace SS. The genes encoding endonuclease VIII and endonuclease III in Escherichia coli are transcribed as the terminal genes in operons. Nucleic Acids Res. 2000;28:762–769. doi: 10.1093/nar/28.3.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolin C, Stedeford T, Cardozo-Pelaez F. Single extraction protocol for the analysis of 8-hydroxy-2′-deoxyguanosine and the associated activity of 8-oxoguanine DNA glycosylase. J Neurosci Methods. 2004;136:69–76. doi: 10.1016/j.jneumeth.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 21.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 22.David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 23.Collins AR, Cadet J, Moller L, Poulsen HE, Vina J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys. 2004;423:57–65. doi: 10.1016/j.abb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 24.Cantoni O, Costa M. Analysis of the induction of alkali sensitive sites in the DNA by chromate and other agents that induce single strand breaks. Carcinogenesis. 1984;5:1207–1209. doi: 10.1093/carcin/5.9.1207. [DOI] [PubMed] [Google Scholar]
- 25.Shi X, Mao Y, Knapton AD, Ding M, Rojanasakul Y, Gannett PM, Dalal NS, Liu K. Reaction of Cr(VI) with ascorbate and hydrogen peroxide generates hydroxyl radicals and causes DNA damage: Role of Cr(IV)-mediated Fenton-like reaction. Carcinogenesis. 1994;15:2475–2478. doi: 10.1093/carcin/15.11.2475. [DOI] [PubMed] [Google Scholar]
- 26.Misra M, Alcedo J, Wetterhahn KE. Two pathways for chromium(VI)-induced DNA damage in 14 day chick embryos: Cr-DNA binding in liver and 8-oxo-2′-deoxyguanosine in red blood cells. Carcinogenesis. 1994;15:2911–2917. doi: 10.1093/carcin/15.12.2911. [DOI] [PubMed] [Google Scholar]
- 27.Miller CA, Costa M. Characterization of DNA-protein complexes induced in intact cells by the carcinogen chromate. Mol Carcinog. 1988;1:125–133. doi: 10.1002/mc.2940010208. [DOI] [PubMed] [Google Scholar]
- 28.Cupo DY, Wetterhahn KE. Binding of chromium to chromatin and DNA from liver and kidney of rats treated with sodium dichromate and chromium(III) chloride in vivo. Cancer Res. 1985;145:1146–1151. [PubMed] [Google Scholar]
- 29.Zhitkhovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- 30.Feng Z, Hu W, Rom WN, Costa M, Tang MS. Chromium(VI) exposure enhances polycyclic aromatic hydrocarbon-DNA binding at the p53 gene in human lung cells. Carcinogenesis. 2003;24:771–778. doi: 10.1093/carcin/bgg012. [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Medvedovic M, Dixon K. Mutational specificity in a shuttle vector replicating in chromium(VI)-treated mammalian cells. Environ Mol Mutagen. 1999;33:313–319. doi: 10.1002/(sici)1098-2280(1999)33:4<313::aid-em8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 32.Takao M, Kanno S, Kobayashi K, Zhang QM, Yonei S, Horst GTJ, Yasui A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homolog. J Biol Chem. 2002;44:42205–42213. doi: 10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 33.Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]