

Abstract
The Distalless Dlx3 homeodomain gene is expressed in terminally differentiated murine epidermal cells, and there is evidence to support an essential role as a transcriptional regulator of the terminal differentiation process in these cells. In an attempt to determine the factors that induce Dlx3 gene expression, we have cloned the 1.2-kilobase pair proximal region of murine gene and analyzed its cis-regulatory elements and potential trans-acting factors. The proximal region of the Dlx3 gene has a canonical TATA box and CCAAT box, and the transcription start site was located 205 base pairs upstream from the initiation of translation site. Serial deletion analysis showed that the region between −84 and −34 confers the maximal promoter activity both in undifferentiated and differentiated primary mouse keratinocytes. Gel retardation assays and mutational analysis demonstrated that the transcriptional regulator NF-Y (also referred to as CBF) binds to a CCAAT box motif within this region and is responsible for the majority of the Dlx3 promoter activity. In addition, an Sp1-binding site was located immediately upstream of transcription start site that acts as a positive regulatory element of the Dlx3 promoter, independent of the CCAAT box motif. Importantly, elements residing between +30 to +60 of the Dlx3 gene are responsible for the Ca2+-dependent induction of Dlx3 during keratinocyte differentiation.
During epidermal differentiation, mitotically active basal keratinocytes cease to proliferate, detach from the basement membrane, and migrate through the spinous and granular layers to the outermost terminally differentiated cornified layer of the skin. This cornification process is tightly associated with a stepwise program of transcriptional regulation and is concurrent with the sequential induction and repression of structural and enzymatic differentiation-specific markers (1). This process can be achieved in mouse keratinocytes cultivated in vitro by increasing the Ca2+ concentration from 0.05 to 0.12 mm in the culture medium (2), which produces a situation that mimics the endogenous Ca2+ gradient present in the skin (3). The Ca2+ signaling differentiation pathway is associated with increased phospholipase C activity (4) and activation of protein kinase C (PKC)1 (5). Previous work has demonstrated an essential role of PKC signaling in the late stages of epidermal differentiation. Activation of PKC has been shown to be necessary for expression of late differentiation markers loricrin and profilaggrin and for the suppression of the spinous-specific markers K1 and K10 (6).
Dlx33, a murine ortholog of the Drosophila Distalless homeodomain protein (7), is a member of the Dlx vertebrate family. This family comprises to date six genes identified both in mouse and human and found to be organized as three convergently transcribed pairs, each closely linked to one of the four mammalian Hox clusters (8–13). Disruption of the DLX3 coding sequence has been associated with a human disorder, Tricho-Dento-Osseous syndrome. This inherited autosomal dominant disorder is characterized by defects in ectodermal derivatives such as hair and teeth and craniofacial bone abnormalities (14). Dlx3 is expressed in the granular layer of the epidermis and in the hair matrix cells of the hair follicle (15, 16), and there is evidence strongly supporting the critical role of the Dlx3 homeoprotein in the regulation of expression of late epidermal differentiation genes (15). In vitro studies have shown that Dlx3 binds to an AT-rich region and acts as a positive transcriptional regulator (17), which is activated during the Ca2+ shift in keratinocytes induced to differentiate in culture (18). In transgenic mice, ectopic expression of Dlx3 in basal cells is accompanied by the cessation of cell proliferation and the up-regulation of expression of late epidermal differentiation structural genes including profilaggrin (15). A potential binding site for Dlx3 has been identified in the profilaggrin gene, suggesting that the observed up-regulation of this gene in the transgenic mice may result from a direct effect of Dlx3 (15). Altogether, these data strongly support a role for Dlx3 as a determinant factor in the activation of expression of granular markers during the terminal differentiation of keratinocytes.
During the process of terminal epidermal differentiation, many genes expressed in the keratinocyte are regulated at the transcriptional level (1). The transcription factors AP1 and AP2 have been characterized as primary regulatory factors of keratinocyte gene expression (19–24). PKC is an upstream component of the pathway that regulates AP1 in many systems and may play a role in the epidermal differentiation expression of K5, K1, loricrin, profilaggrin, and involucrin (19, 21–24). Members of the POU family of transcription factors such as Oct1, Oct2, Oct6, Skn1a, and Skn1i have also been implicated as regulators of epidermal genes (25–28).
In order to elucidate the role of Dlx3 in the cascade of transcriptional events that ultimately leads to terminal differentiation, we have cloned and characterized the mouse Dlx3 promoter. Deletional promoter analysis was utilized to delineate the sequences that regulate the transcription of Dlx3 in differentiated and undifferentiated keratinocytes. In turn, these cis-acting elements were used to identify the transcription factors that regulate the Dlx3 promoter. Importantly, we have identified a region residing between +10 and +60 that responds to the Ca2+ shift used to differentiate the keratinocytes in culture.
MATERIALS AND METHODS
Cloning of Upstream Regulatory Region of the Mouse Dlx3 Gene
To clone the upstream sequence of the Dlx3 gene we used the Genome-Walker Kit (CLONTECH). It is designed to amplify a specific genomic DNA using an adapter sequence attached to the genomic DNA and specific oligomers based on known sequence of the gene of interest. We made gene-specific oligomers located 45 bp upstream of the translation start site and used an oligomer located 30 bp further upstream as a nested primer. After a second round of PCR, a 754-bp DNA fragment was obtained and sequenced. To obtain a larger 5′-regulatory sequence, genome walking was performed using the oligomers based on the newly cloned DNA upstream sequence for the Dlx3 gene. After PCR, we obtained a 617-bp DNA fragment. The two overlapping DNA fragments were cloned into the XbaI and SalI sites of the pCAT-basic vector (Promega).
Plasmids Construction
The −1213 to +160 upstream DNA fragment of the Dlx3 gene was inserted into a promoterless CAT plasmid (pCAT-basic; Promega) and designated as p1213CAT. All 5′-deleted clones were constructed by PCR using the oligonucleotides DEL1 (5′-cgcgtcgacCACAGGTCGGTCATTCAGGAC-3′; −1029 to −1009), DEL2 (5′-cgcgtcgacGACTTCCTGAAGAACACAGAA-3′; −827 to −807), DEL3 (5′-cgcgtcgacTCCAGTAGGGACTTGCAGGCC-3′; −692 to −672), DEL4 (5′-cgcgtcgacCGGAGAAACCCTGTCTCAAAAA-3′; −572 to −552), DEL5 (5′-cgcgtcgacGCCACTTTCT GTCTGTCATTT-3′; −437 to −417), DEL6 (5′-cgcgtcgacCTCCGCACAGCCAACCCCTCC-3′; −284 to −264), DEL7 (5′-cgcgtcgacTTAGGGGTAACAACAAAGAGG-3′; −162 to −142), DEL8 (5′-cgcgtcgacAGACTTGCAGCCAATCAGCGC-3′; −84 to −64), DEL9 (5′-cgcgtcgacTGAGTCTATAACCGGCTGGCC-3′; −34 to −14), and 160R (5′-cgctctagaAACGGGCGGAGGAGCCCAGGT-3′; +160 to +140) with the p1213CAT vector as the template. The lower-case letters correspond to extensions to the Dlx3-specific sequence with the underlined letters corresponding to restriction enzyme sites for SalI and XbaI. Each PCR product was digested with SalI and XbaI and inserted into the pCAT-basic vector. The 3′-deleted clones, p60(+)CAT and p30(+)CAT, were constructed using the oligonucleotides 60R (5′-cgctctagaGCCGGCTGTGCCTCAGTCGCT-3′; +60 to +40) and 30R (5′-cgctctagaTCTCCGTGTCCCAAGCCACAG-3′; +30 to +10), respectively, and DEL9. The shortened 3′-deleted clone, p10(+)CAT, was constructed using two oligonucleotides 10F (5′-cgcgtcgacTGAGTC-TATAACCGGCTGGCCGGGCGGAGCTGGCAGCATTTGACT-3′) and 10R (5′-cgctctagaAGTCAAATGCTGCCAGCTCCGCCCGGCCAGCCG-GTTATAGACTCA-3′), which were annealed, digested with SalI and XbaI, and inserted into the pCAT-basic vector. The other 3′-deleted clones, p1213(+)60CAT, p1213(+)30CAT, and p1213(+)10CAT, were constructed using the oligonucleotides +60R, +30R, +10R, and (5′-cg-cgtcgacATTACCCGGGAACTGGACTTTGAGA-3′, −1213 to −1189), respectively. The general PCR conditions to generate the deletional fragments were 30 cycles of denaturation at 94 °C for 45 s, annealing at 55 °C for 45 s, and extension at 72 °C for 1 min. To make the p84mSPCAT and p34mSPCAT constructs containing mutant Sp1-binding sites, site-directed mutagenesis was performed using the QuickChange site-directed mutagenesis kit and the following conditions suggested in the protocol from the manufacturer (Stratagene). The two oligonucleotides DlxSpm5 (5′-CCGGCTGGCCGatatGAGCTGGCAG-3′), and DlxSpm3 (5′-CTGCCAGCTcatatCGGCCAGCCGG-3′) were designed and used for mutagenesis, with lowercase letters being the modified nucleotides. The CCAAT box mutant clone, p84mCAT, was constructed using the oligonucleotide M6 (5′-AGACTTGCAtaaggaCA-GCGCGCAGG-3′) and 160R by PCR. The 3′-deletion mutant constructs were made by PCR using oligonucleotides containing each mutant site (5′-cgctctagaGCCGGCTGTCGGTCAGTCGCTatacGCCTC-3′) for M1C-AT, (5′-cgctctagaGCCGGCTGTCGGTCAGTCtagtCGTGCCTC-3′) for M2CAT, (5′-cgctctagaGCCGGCTGTCGGTCAactaCTGCGTGCCTC-3′) for M3CAT, (5′-cgctctagaGCCGGCTGTCGagacGTCGCTGCGTGCCT-C-3′) for M4CAT, (5′-cgctctagaGCCGGCTtgatGTCAGTCGCTGCGTG-CCTC-3′) for M5CAT, (5′-cgctctagaGCCGaagtTCGGTCAGTCGCTGC-GTGCCTC-3′) for M6CAT, (5′-cgctctagaaattGCTGTCGGTCAGTCGC-TGCGTGCCTC-3′) for M7CAT, (5′-cgctctagaGCCGGCTGTCGagacact-aCTGCGTGCCTC-3′) for M34CAT, (5′-cgctctagaGCCGaagtTCGGTCA-actaCTGCGTGCCTC-3′) for M36CAT, (5′-cgctctagaGCCGaagtTCGag-acGTCGCTGCGTGCCTC-3′) for M46CAT, and (5′-cgcgtcgacATTACC-CGGGAACTGGACTTTGAGA-3′, for −1213 to −1189). The lowercase and underlined letters correspond to restriction enzyme sites for SalI and XbaI, and lowercase letters are the modified nucleotides. Each PCR product was digested with SalI and XbaI and inserted into the pCAT-basic vector.
Cell Culture
Primary mouse keratinocytes were isolated from BALB/c newborn mouse skins and grown in Eagle’s minimal essential medium lacking Ca2+, with 8% Chelex-treated fetal bovine serum (2, 18). Ca2+ concentrations were determined by analysis in an atomic absorption spectrophotometer. Unless otherwise indicated, the Ca2+ concentration of the medium was adjusted to 0.05 mm to maintain a basal cell-like population of undifferentiated cells.
Transfection and CAT Assays
Primary mouse keratinocytes were transfected using the Lipofectin reagent (Life Technologies, Inc.). Typically, 1 μg of each CAT construct was used to transfect cells plated and cultured in 6-well plates coated with rat tail collagen (0.1 mg/ml). After 4 h of incubation, cells were treated with 15% glycerol in KSF medium (Life Technologies, Inc.) for 3.5 min and then maintained either in medium with Ca2+ concentration of 0.05 or 1.4 mm. CAT activities were determined 48 h after transfection and normalized to protein value at A595. CAT activities were measured by the fluor diffusion CAT assay using Econofluor premixed scintillation fluid and [3H]acetyl coenzyme A (NEN Life Science Products) in the linear range of the assay. Results are expressed as the counts/min transferred to the organic solvent phase. Each transfection was done in duplicate, and the experiment was repeated at least three times.
Mapping of the Transcriptional Start Site
To determine the +1 site, we used the rapid amplification of cDNA ends method, as described by Frohman et al. (29). The steps of reverse transcription, tailing, and PCR were performed using the 5′-rapid amplification of cDNA ends system (Life Technologies, Inc.). The gene-specific oligomer was located at +124 bp from the translation initiation site of Dlx3. The final PCR products were cloned and sequenced to determine the extreme 5′ mRNA sequence.
Preparation of Nuclear Extracts
Nuclear extracts were prepared from undifferentiated and differentiated primary mouse keratinocyte cultures as described by Andrews and Faller (30). All steps were carried out at 4 °C. The cells (1 × 107) were harvested, washed three times with ice-cold phosphate-buffered saline, and pelleted. The keratinocytes were resuspended in 0.4 ml of buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mM KCl, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride), incubated on ice for 15 min, and then homogenized with a Dounce homogenizer B pestle for about 20–25 strokes. The homogenate was centrifuged at 14,000 rpm, and the nuclear pellet was resuspended in 0.2 ml of buffer B (20 mm HEPES, pH 7.9, 25% (v/v) glycerol, 0.42 m NaCl, 1.5 mm MgCl2, 0.2 mM EDTA, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride) and incubated on ice for 30 min. After centrifugation at 14,000 rpm for 20 min at 4 °C, the supernatant fraction was aliquoted and stored at −70 °C. The protein concentration was determined using the Bradford assay following the methods described for the Bio-Rad Protein Assay.
Gel Retardation Analysis
Gel retardation analysis was carried out according to Ausubel et al. (31). 32P-End-labeled DNA fragments (about 1 ng of DNA) were incubated for 15 min at 4 °C with nuclear extracts in a total volume of 15 μl of binding buffer (10 mm HEPES, pH 7.9, 60 mm KCl, 1 mm EDTA, 10% glycerol, 10 mm MgCl2) containing 2μg of poly(dI-dC). The products of the DNA-protein binding reaction were separated by electrophoresis on a nondenaturing 6% polyacrylamide gel in low salt TBE buffer containing 44 mm Tris, 44 mm boric acid, and 1 mm EDTA. DNA-protein complexes and unbound DNA probe were visualized in the gel by autoradiography on x-ray film. For the competition experiments, various amounts of unlabeled DNA were added at the beginning of the binding reaction. For gel mobility supershift analysis, the binding reactions were performed as described above, except that the nuclear extracts were incubated for 30 min at 4 °C with variable amounts of specific antisera prior to the addition of the radio-labeled probe. The antisera raised against NF-1, C/EBP, Sp1, Sp2, Sp3, Sp4, c-Jun, c-Fos, JunB, and JunD were purchased from Santa Cruz Biotechnology, Inc.; the NF-YA antibody was purchased from Rockland Inc.
RNA Blotting
Total RNA was isolated from keratinocytes cultured in medium containing 0.05, 0.12, or 1.4 mm Ca2+ using Trizol following the instructions provided by the manufacturer (Life Technologies, Inc.). The RNA samples (2 μg) were separated in a 1.2% agarose/methylmercury hydroxide gel (32), transferred to a nylon membrane, and hybridized according to Church and Gilbert (33). The blot was hybridized with a 1.1-kilobase pair mouse Dlx3 cDNA probe (16), and a rat glyceraldehyde-3-phosphate dehydrogenase probe (34) was used as control, labeled with 32P by random priming (Stratagene), and washed at high stringency (0.1× SSC, 0.1% SDS at 65 °C). The hybridizing bands were visualized by autoradiography on x-ray film and quantitated with a TD932 Macbeth densitometer using linear exposure.
UV Cross-linking Assay
The 5′-end-labeled probe was incubated with primary mouse keratinocyte nuclear extract under standard mobility shift assay conditions in the presence of 500 ng of poly(dI-dC) in a 30-μl binding reaction at room temperature. The reaction mixture was irradiated with an UV transilluminator (254 nm, 2400 microwatts/ cm2) for 20 min at 4 °C, and the complex was loaded on a 10% SDS-polyacrylamide gel. The gel was dried, and the proteins directly involved in the DNA interaction were detected by autoradiography.
RESULTS
Induction of Dlx3 Expression by Ca2+ in Mouse Keratinocytes Cultured in Vitro
Northern blot analysis of total RNA from mouse keratinocytes cultured in 0.05 mm Ca2+ medium revealed very low levels of Dlx3 transcript (Fig. 1). However, after 12 h, Dlx3 expression increased 3.5-fold in keratinocytes induced to differentiate by Ca2+ increase (to 0.12 mm). As shown in Fig. 1, the expression increased in a time-dependent manner in 0.12 mm Ca2+ medium, and by 12 h an 8-fold increased expression of Dlx3 was observed in keratinocytes cultured at 1.4 mm.
Fig. 1. Dlx3 expression is up-regulated during differentiation induced by Ca2+.
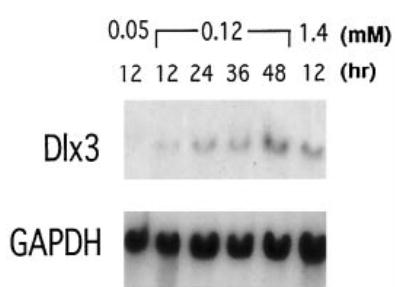
Northern blot analysis of 2 μg of total RNA from primary mouse keratinocytes cultured in 0.05, 0.12, and 1.4 mm Ca2+ containing medium for the indicated times (in hours after Ca2+ addition). A 1.1-kilobase pair Dlx3 cDNA fragment was used as a hybridization probe. The same RNA samples were hybridized with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probe for normalization.
Cloning and Analysis of the Upstream Sequence of the Murine Dlx3 Gene
We cloned two overlapping genomic DNA sequences of the Dlx3 upstream regulatory region (754 bp and 617 bp, see “Materials and Methods”), which combined were cloned as 1416 bp of upstream sequence from the translation start site (Fig. 2). The transcription start site was determined using the rapid amplification of cDNA ends procedure (29) and was located 205 bp upstream from the translation start site. Genomic DNA sequence analysis revealed that the upstream region of Dlx3 gene contains canonical CCAAT and TATA boxes. Comparison of the proximal promoters of the Xenopus, human, and mouse Dlx3 genes showed a striking degree of conservation. Particularly the mouse and human upstream regions exhibit about 91% homology in the region located between −104 and +61 (data not shown), suggesting the possibility of conserved mechanisms of regulation for Dlx3 orthologs during evolution.
Fig. 2. Upstream sequence of the mouse Dlx3 gene.

The major transcription start site is indicated with a bold arrow. The canonical CCAAT box and TATA box are boxed. The putative transcription factor binding sites are underlined. The translation start site is in italics. The numbers on the left indicate the nucleotide number with respect to the transcription start site.
Deletion Analysis of the Dlx3 Promoter
Comparing the sequence of the −1213 to +160 fragment of the Dlx3 gene with the data base for consensus transcription factor binding sequences, putative sites for AP1, GRE, OCT1, NFκB, and Sp1 were identified (Fig. 2). This information was taken into consideration when serial deletion plasmids were constructed by PCR in order to identify the regulatory elements with a functional role in Dlx3 gene expression. These constructs were transfected into primary mouse keratinocytes, and promoter activity was determined by measuring CAT activity in cells maintained in an undifferentiated state (cultured in 0.05 mm Ca2+) or in cells differentiated in vitro by addition of Ca2+ (1.4 mm Ca2+) (Fig. 3). An increase in activity of all constructs was observed upon differentiation. This increase is coincident with the results obtained for the endogenous gene shown in Fig. 1 by Northern blot analysis. In 0.05 mm Ca2+, the p84CAT construct showed the highest activity, almost 3-fold increased activity when compared with the p162CAT. The p34CAT construct containing the TATA box and putative Sp1-binding site showed a 3-fold decrease in CAT activity when compared with the p84CAT, even though the activity is still higher than that of the full-length promoter (p1213CAT). These results indicated that there are, at least, one negative regulatory and one positive regulatory region in the upstream sequence of Dlx3 gene, which are located between −162/−84 and −84/−34, respectively. The p1213CAT construct showed about 3.5-fold increased transcriptional activity by Ca2+ addition. Similar induction by Ca2+ addition was measured for each deletion construct, and as shown in Fig. 3, the activity of the p34CAT construct is still at least 2-fold higher in 1.4 mm Ca2+ than in 0.05 mm Ca2+
Fig. 3. Effect of 5′-deletions on the transcriptional activity of the mouse Dlx3 promoter.
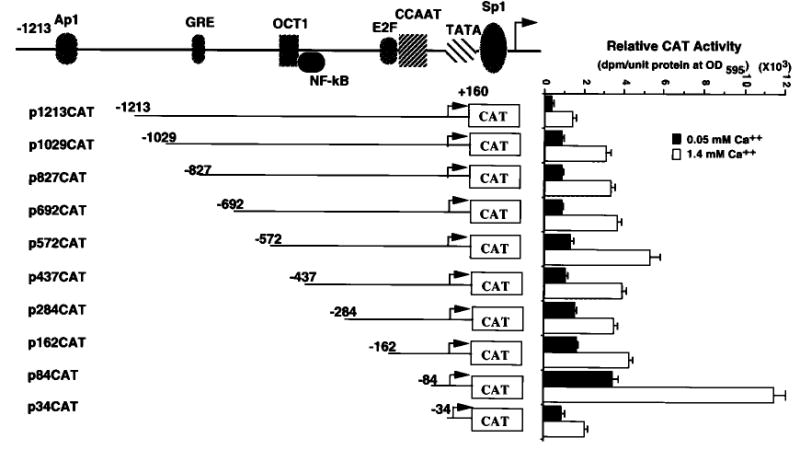
Primary mouse keratinocytes were transfected with each indicated deletion construct. The transfected cells were incubated with growth medium containing 0.05 or 1.4 mm Ca2+ concentration, and the CAT activity of each plate of cells was measured. CAT activity was normalized to the protein concentration measured by the Bradford assay. The bars represent the average normalized CAT activity of duplicate plates from three experiments for each construct.
The Transcription Factor NF-Y Binds to the CCAAT Box and Plays an Important Role in the Dlx3 Promoter
By deletional analysis, we showed that the region between −84 and −34 played a major regulatory role in the activity of the Dlx3 promoter. To examine if a protein bound to this region and to define the nucleotides required for the formation of the complex, the 50-bp oligonucleotide (−84 to −34; d oligonucleotide) was used as a probe in gel shift analysis, and three overlapping oligomers (d1–d3) were used as competitors (Fig. 4A). One major complex was found, and by competition analysis, binding was located to the proximal part of the region (between −84 and −59) (Fig. 4B). Better resolution of the specific nucleotides required for formation of the complex was obtained using oligonucleotides with specific 3-bp mutations (Fig. 4C). Competition gel shift analysis with wild-type (d1) and mutant oligonucleotides (M1–M6) showed that competitors M1, M2, and M3 bound to the specific complex (Fig. 4D). In contrast, oligonucleotides M4, M5, and M6 (which has the combined mutation of M4 and M5) failed to compete. These results indicated that the complex was due to binding in the CCAAT region (Fig. 5B). To determine the nature of the binding, oligonucleotides that have been reported to bind selectively the CCAAT box binding proteins were used as competitors (Fig. 5A). The CCAAT box binding factors include NF-Y (also known as CBF), the C/EBP family, and NF-1 (35). As shown in Fig. 5B, the albumin C element, which is known to bind NF-Y (36), fully competed the formation of the complex (Fig. 5B, left panel, 3rd lane). In contrast, the consensus NF-1 and C/EBP oligonucleotides did not compete for formation of the complex. The same pattern of competition was found using nuclear extracts either from undifferentiated or differentiated keratinocytes (Fig. 5B, right panel). These results suggested that the nuclear protein that bound to the CCAAT box motif in the Dlx3 promoter was NF-Y. To confirm this directly, supershift experiments were performed with specific antisera (Fig. 5C). By using the labeled wild-type d1 oligonucleotide, either anti-NF-Y, anti-C/EBP, or anti-NF-1 antisera were added in increasing amounts to undifferentiated mouse primary keratinocyte nuclear extracts. Anti-NF-Y antibody supershifted the complex with each amount, whereas the anti-C/EBP or anti-NF-1 antibody did not affect the mobility. These observations indicated that the specific complex contained the NF-Y transcription factor.
Fig. 4. Delineation of a binding site within the region between −84 and −34 that binds nuclear factors from keratinocytes.
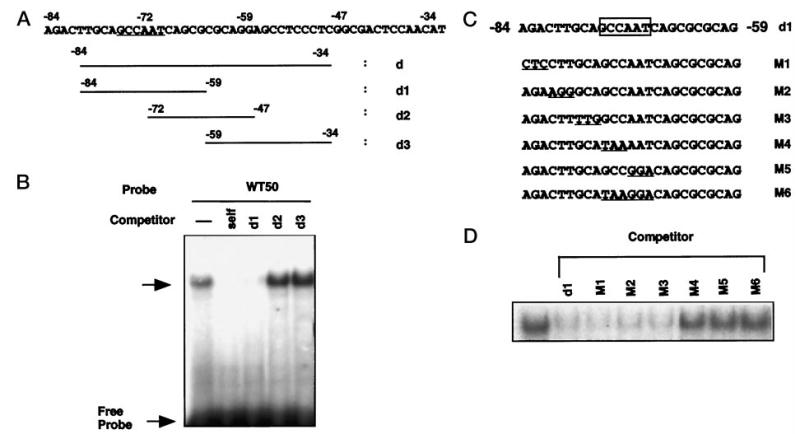
A, the DNA sequence between −84 and −34 (oligomer d) and oligomers used in the competitive gel shift assay (d1–d3); the CCAAT box is underlined. B, the d oligomer (−84/−34) was labeled and used as a probe in gel shift assay with nuclear extracts of keratinocytes (3 μg). In competition assays, the nuclear extracts were mixed with each competing oligonucleotide (100-fold molar excess) prior to addition of the 32P-labeled probe. The data presented in this figure are representative of electromobility shift assays that were performed at least three separate times with the same results. C, the DNA sequence for oligonucleotide d1 is shown, along with the location of the 3-bp mutations. The putative CCAAT motif is boxed. The serial 3-bp mutated sequences are underlined in each mutated oligomer (M1–M6). D, competition binding assay with 32P-labeled d1 oligomer used as a probe and competitor oligomers at 100-fold molar excess (d1 and M1–M6). Identical results were observed with multiple nuclear extract preparations.
Fig. 5. Identification of the nuclear protein from undifferentiated and differentiated mouse keratinocytes that binds to the CCAAT box of Dlx3 promoter.
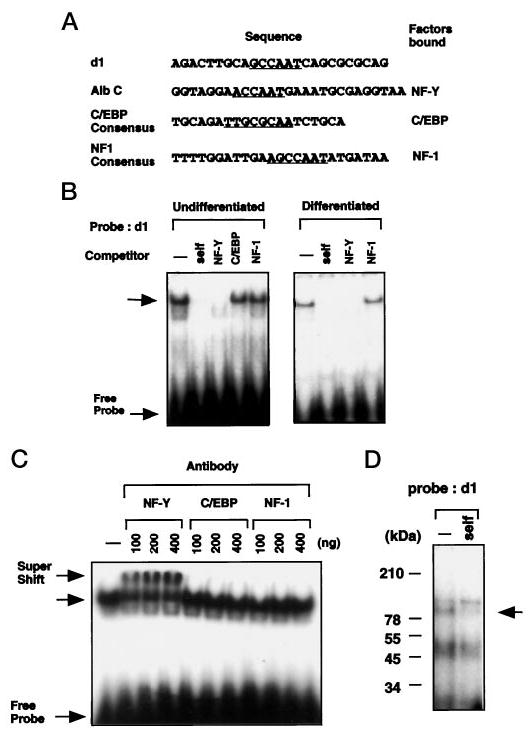
A, DNA sequences used in gel shift analyses. The d1 sequence along with the albumin C element (NF-Y), C/EBP, and NF1 consensus sequences are shown. B, nuclear extract from undifferentiated (left panel) and differentiated (right panel) primary mouse keratinocytes (3 μg) were used to shift a 32P-labeled d1 probe. The competitors, albumin C element (Alb C), consensus C/EBP, and NF-1 (Santa Cruz Biotechnology), were added at 100-fold molar excess. Top arrow indicates the protein-DNA complex formed between nuclear extract and d1 probe. C, nuclear extracts from primary mouse keratinocytes (3 μg) were incubated with 0.1, 0.2, or 0.4 μg of antibodies against the NF-Y A-subunit, C/EBP, or NF-1 for 30 min on ice. 32P-Labeled d1 oligomer was added, and the preparation was separated by electrophoresis. The top arrow indicates supershifted complex when using antibody against NF-Y. D, UV cross-linking assay was performed to estimate the molecular weight of the protein(s) binding to the CCAAT box of the Dlx3 promoter (see “Materials and Methods”). For the competition analysis, the nonradioactive competitor oligonucleotides were added at 100-fold molar excess. Arrow indicates the specific complex formed. The numbers on the left are the molecular masses of protein standards.
NF-Y is a heterotrimeric transcription factor composed of three subunits, A, B, and C (37–43). The subunits A and C together form a heterodimer, which then interacts with the subunit B to form the heterotrimeric NF-Y protein. This heterotrimeric NF-Y then binds to DNA to form an NF-Y-DNA complex. To examine whether the complex consisted of all three subunits, we checked the molecular weight of the complex using the UV cross-linking assay (Fig. 5D). The complex (indicated by arrow) which competed with self-oligonucleotide (d1) had an estimated molecular mass of ~100 kDa. This molecular mass coincides with the approximate expected combined molecular mass of subunits NF-YA, -B, and -C (35, 25, and ~40 kDa, respectively) (44). This result strongly supported that NF-Y was binding to the CCAAT box motif of Dlx3 promoter in its heterotrimeric form.
The function of the Dlx3 promoter CCAAT box was assessed by mutational analysis. Primary mouse keratinocytes were transfected with the wild-type p84CAT construct, the mutant p84mCAT construct, and the p34CAT construct (Fig. 6A). The CAT activity conferred by each construct was assayed in transiently transfected cells grown in 0.05 and 1.4 mm Ca2+ (Fig. 6B). The CAT activity of the mutant construct was reduced to 40 and 25% as compared with the wild-type p84CAT construct in the undifferentiated and differentiated cells, respectively. These results underscore the functional importance of the CCAAT box for the activity of the Dlx3 promoter.
Fig. 6. Functional analysis of the Dlx3 promoter CCAAT box motif.
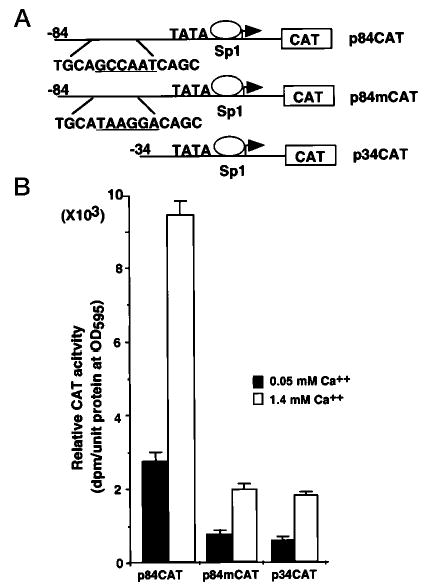
A, the diagram illustrates the p84CAT and p34CAT deletion constructs and the sequence of the mutation substituting the CCAAT box motif. B, primary mouse keratinocytes were transfected with p84CAT, p84mCAT, and p34CAT and incubated with medium containing 0.05 or 1.4 mm Ca2+. After 48 h, cells were harvested. The CAT activity was normalized to the protein concentration measured by the Bradford assay. The bars represent the average normalized CAT activity of three experiments done on duplicate plates for each construct.
Functional Sp1-binding Site in the Dlx3 Promoter
In addition to CCAAT box sequence, analysis of the proximal promoter region revealed a putative recognition site for Sp1 transcription factor, which was located between the TATA box and transcription start site (−13 to −3). To examine this putative Sp1-binding site, gel shift analysis was performed using an oligonucleotide corresponding to −21 to −2 on the Dlx3 promoter (dsp), and a mutant form of this oligonucleotide (dspM). Nuclear extracts from undifferentiated and differentiated primary mouse keratinocytes were used in the gel shift analysis (Fig. 7A). As shown in Fig. 7B, the complexes formed were competed with wild-type dsp and Sp1 consensus oligonucleotides but not with the mutant form of dsp (dspM). In gel shift assays using antibodies corresponding to each of the Sp1-like family members, the Sp1 and Sp3 antisera supershifted the two complexes formed between the Sp1-binding site of Dlx3 promoter and nuclear proteins (Fig. 7C). These results indicate that both of these Sp1-like factors are able to bind the Sp1 site on the Dlx3 promoter.
Fig. 7. Identification of nuclear proteins binding to the GC-rich site of Dlx3 promoter.
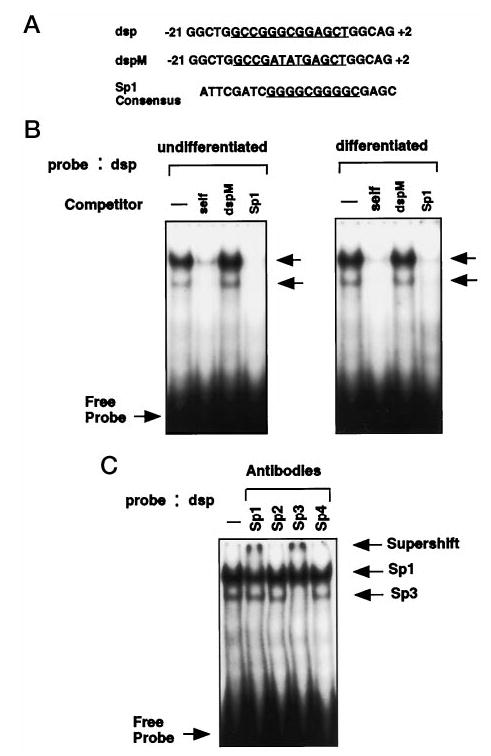
A, the sequences of oligonucleotides containing the GC-rich putative Sp1-binding site (underlined), mutant Sp1-binding site (underlined), and consensus Sp1-binding site (underlined) are shown. B, 3 μg of nuclear extracts from undifferentiated (left panel) or differentiated (right panel) keratinocytes were used to shift 32P-labeled dsp oligonucleotide as described. Nonradioactive competitor Sp1 consensus (Santa Cruz Biotechnology) and dspM oligonucleotides were added at a 100-fold molar excess. Arrows indicate the protein-DNA complex formed between nuclear extract (3 μg) and the dsp probe. C, antibodies to Sp1, Sp2, Sp3, and Sp4 (Santa Cruz Biotechnology) were used to test which members of Sp1 family are associated with the Sp1-binding site of Dlx3 promoter. The arrows indicate the specific complex formed and the supershifted complexes after addition of antibodies raised against Sp1 and Sp3 proteins.
DNA constructs containing the mutant Sp1-binding site (Spm) in the p84CAT and p34CAT clones were transfected into mouse keratinocytes to examine the function of this region (Fig. 8). The CAT activity of p84spmCAT and p34spmCAT was reduced to 55 and 66% of the wild types, respectively, in undifferentiated keratinocytes and to 62 and 72% of each respective wild-type in differentiated keratinocytes (Fig. 8). These results showed that the Sp1-binding site of Dlx3 promoter plays a positive regulatory role, and its function is not dependent on the CCAAT box-binding protein, NF-Y.
Fig. 8. Mutational analysis of the Sp1-binding site in the Dlx3 promoter.
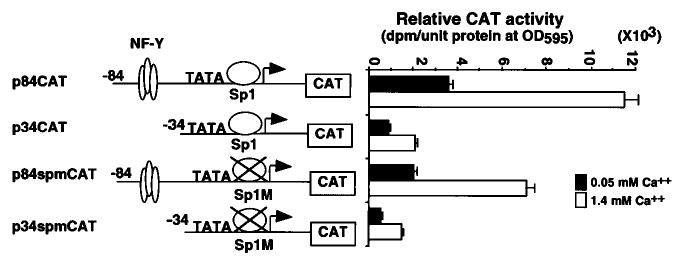
The diagram (left) illustrates the p84CAT, p34CAT, p84spmCAT, and p34spmCAT deletion and mutant constructs. The sequence of the mutation of the Sp1-binding site is the same shown in Fig. 7A. Primary mouse keratinocytes were transfected with p84CAT, p34CAT, p84spmCAT, or p34spmCAT, incubated with growth medium containing 0.05 or 1.4 mm Ca2+, and harvested after 48 h. The CAT activity was normalized to the protein concentration measured by the Bradford assay. The bars represent the average normalized CAT activity of duplicate plates for each construct (right panel). All experiments in this figure were repeated three times with similar results.
Defining the Region of the Dlx3 Promoter Responsible for Ca2+ Inducibility
Despite the finding that the binding sites for NF-Y and Sp1 play an important role in the transcriptional regulation of the Dlx3 gene, mutation of these sequences did not affect the activation of Dlx3 by Ca2+ (Fig. 6 and 8). Serial 3′-deletion constructs were made on the p34CAT construct, which contains the 160 bp of untranslated region (Fig. 9A). The constructs were made in an attempt to delineate the region responsible for the Ca2+ inducibility. Each construct was transfected into primary mouse keratinocytes, and CAT activity was measured in 0.05 and 1.4 mm Ca2+ conditions (Fig. 9A). The p60(+)CAT deletion retained the capability of induction by Ca2+, but the p10(+)CAT deletion showed no induction of CAT activity with Ca2+ increase (Fig. 9A). Similar 3′-deletions were made on the p1213CAT construct. These constructs were transfected into primary mouse keratinocytes (Fig. 9B), and the CAT activity indicated that the p1213(+)160CAT as well as the p1213(+)60CAT were induced 2.5-fold, whereas the p1213(+)30CAT was induced only 1.5-fold. In contrast, the p1213(+)10CAT deletion was not induced with Ca2+. Our findings indicated that the region located between +10 and +60 was indispensable for induction by Ca2+ of the Dlx3 promoter.
Fig. 9. Deletion analysis and function of sequence residing downstream of the transcription initiation site.
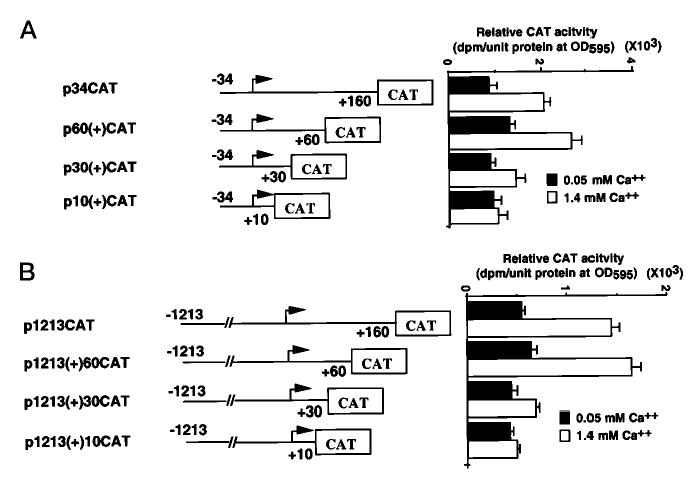
A, p34CAT constructs with decreasing downstream promoter deletions were transfected into primary mouse keratinocytes and incubated with growth medium containing either 0.05 or 1.4 mm Ca2+. CAT activity was assayed 48 h after transfection. B, p1213CAT constructs with decreasing downstream promoter deletions were transfected into keratinocytes and assayed as described above. The bars represent the average normalized CAT activity of duplicate plates for each construct. All experiments in the figure were repeated in triplicate with similar results.
Characterization of the Ca2+-responsible Region of Dlx3 Promoter
Based on the results of the serial deletion analysis of the Dlx3 promoter (Fig. 9), the region between +30 and +60 has the element(s) that mediate the Ca2+ induction. A mutational analysis was performed to determine the specific nucleotides that conform the responsive element. Serial and double 4-bp mutations were made in the region between +30 and +60 in the context of the p1213(+)60CAT construct. These constructs were transfected into primary mouse keratinocytes, cultured for 48 h in 0.05 or 1.4 mm Ca2+ concentrations, and the CAT activity was measured. As shown in the Fig. 10A, the M1CAT, M2CAT, M5CAT, and M7CAT showed reduced promoter activity at 0.05 and 1.4 mm Ca2+, but the fold activation by Ca2+ did not change significantly when compared with the p1213(+)60CAT. In contrast, the M4CAT and M6CAT showed increased promoter activity in 0.05 and 1.4 mm Ca2+ and slightly increased fold activation by Ca2+. These results suggest that these sites mainly affect the basal promoter activity of Dlx3. Interestingly, the M3CAT construct showed dramatic reduction in the fold activation by Ca2+, similar to that of p1213(+)30CAT. These results indicated that the nucleotides (+42-CGAC-+45) are crucial for the activation by Ca2+. Double mutant constructs (M34CAT, M36CAT, and M46CAT, Fig. 10A) were made and assayed for CAT activity. The double mutant constructs containing the M3 site (M34CAT and M36CAT) showed reduced fold activation by Ca2+, but the M46CAT mutation had no effect. These results again delineated the sequence (+42-CGAC-+45) as primarily responsible for the Ca2+ induction of Dlx3 promoter.
Fig. 10. Characterization of Ca2+-responsible region of Dlx3 promoter.
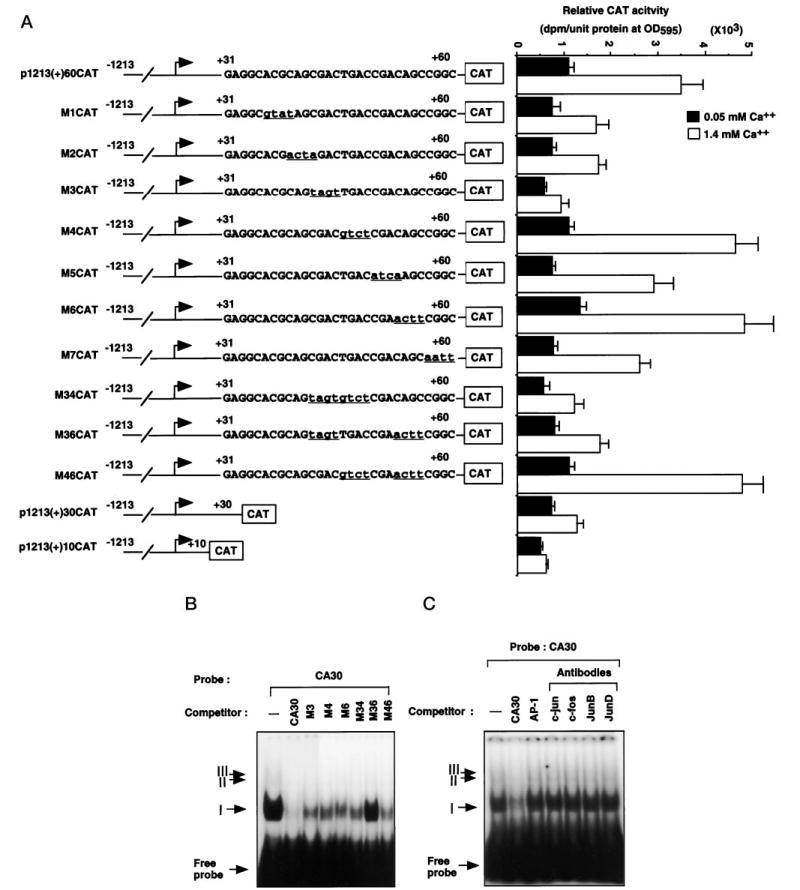
A, the region between +30 and +60 of Dlx3 promoter was mutated with serial or double 4-bp modified bases by the PCR method in the p1213(+)60CAT. Each mutated construct, p1213(+)60CAT, p1213(+)30CAT, and p1213(+)10CAT, was transfected into primary mouse keratinocytes. Underlined lowercase letters indicate the modified nucleotides. CAT assay was performed the same as in Fig. 3. The bars represent the average normalized CAT activity of duplicate plates for each construct. All experiments in the figure were repeated in triplicate with similar results. B, gel shift assay was performed using the oligonucleotide corresponding to the region between +30 and +60 (CA30) as a probe and 3 μg of nuclear extract of primary mouse keratinocytes. Incubations were carried out in the presence or absence of a 100-fold excess of unlabeled wild-type and mutant oligonucleotides. Arrows indicate the protein-DNA complexes, and roman numbers indicate each individual complex (I–III). C, supershift analysis was carried out using the antibodies against the AP1 family. Nuclear extracts were preincubated with 2 μg of each antibody as indicated, followed by incubation with labeled CA30 probe. The 3rd lane corresponds to the gel shift analysis performed in the presence of 100-fold excess unlabeled AP1 consensus oligonucleotide. All experiments in this figure were repeated at least three times with similar results.
We were interested in determining by gel shift analysis if there were nuclear proteins that bound the +30/+60 region (CA30). The CA30 oligonucleotide was used as a probe in gel shift mobility assays with nuclear extracts from mouse primary keratinocytes. One major complex I and two weak bands II and III were identified (Fig. 10B). These complexes were competed with 100-fold excess of unlabeled cold CA30 oligomer. To examine the relationship between the sequences affecting the Dlx3 promoter activity and these complexes, the oligomers containing each mutant site were used as competitors. The M3 oligomer competed the complexes II and III, but did not completely compete the major complex I. The M4, M6, and M34 oligomers showed similar results for the complex I but did not compete the complexes II and III. In contrast, none of the complexes were competed by the M36 oligomer that contains mutations in the M3 and M6 sites. These results indicated that the M3 and M6 sites are important in the binding of the major complex I and that the M4 site is partially involved in the formation of this complex.
The region between +30 and +60 contains an element (+46-TGACCGA-+52) with sequence homology to a consensus AP1-binding site. It has been suggested that the AP1 transcription factor is involved in the Ca2+ inducibility of epidermal differentiation-specific markers (45). To determine the possible participation of AP1 in the protein-DNA complex formation with CA30, the AP1 consensus oligonucleotide and antibodies against AP1 family members were used in competition and supershift assays. As shown in Fig. 10C, the complexes formed by CA30 were not competed by AP1 consensus oligonucleotide, and the supershift assays corroborated that AP1 family members (c-Jun, c-Fos, JunB, and JunD) do not bind to this region. Therefore, the sequence (+42-CGAC-+45) present in the CA30 region is responsible for the transcriptional up-regulation of Dlx3 promoter by Ca2+, through the binding of nuclear factors that are not members of the AP1 family of transcription factors.
DISCUSSION
The cascade of events that leads to terminal differentiation can be triggered in primary mouse keratinocytes by the elevation of extracellular Ca2+. The expression of the Dlx3 homeobox gene is restricted to the suprabasal layer of the epidermis (18), and this expression is dramatically increased in primary mouse keratinocytes induced to differentiate by Ca2+ in vitro. Evidence obtained in transgenic mice suggests the role of Dlx3 as a positive transcriptional activator of differentiation-specific epidermal structural genes. In an attempt to decipher the different stages of the pathway that culminate in terminal differentiation, we have cloned and characterized the upstream promoter sequence of the murine Dlx3 gene, and we determined the regulatory elements necessary for expression of the gene in keratinocytes. By sequence comparison we have found that the region close to the transcription start site of the mouse Dlx3 gene (−110 to +61) has striking homology with that of the Xenopus and human dlx3 genes. In transgenic mice, 470 bp of the Xenopus Dlx3 promoter conferred an expression pattern to the β-galactosidase reporter that was indistinguishable from that of the endogenous gene, including the Ca2+ response (46). Thus, it could be hypothesized that the cis- and trans-regulatory elements important in the regulation of the Dlx3 orthologs have been conserved through evolution and are contained within the proximal promoter region.
To understand the mechanism(s) that control the regulation and expression of the Dlx3 promoter during epidermal differentiation, our studies examined the expression of Dlx3 promoter/CAT constructs in primary mouse keratinocytes to identify putative regulatory elements that are required for Dlx3 expression. The data presented here demonstrate the existence of one positive regulatory region in the Dlx3 promoter located between −84 and −34. We determined that the CCAAT box in this region exerts a significant positive influence on the Dlx3 promoter activity in undifferentiated and differentiated keratinocytes. Competition gel shift analysis and supershift analysis further demonstrated that the transcription factor NF-Y is the protein that binds specifically to the CCAAT box motif. Mutation of this motif dramatically reduced transcription of the Dlx3 promoter. Several genes also contain canonical sites for CCAAT-binding proteins that have been described to be important in early functions of preinitiation complex formation (36). CCAAT boxes are typically located within 100 bp of the transcription start sites, and CCAAT box binding factors include NF-Y (also known as CBF), C/EBP family members, and NF-1 (35). NF-Y was originally identified as a ubiquitously expressed protein that binds to the Y box motif, defined as an inverted CCAAT box motif in all major histocompatibility complex class II genes (47). NF-Y is a heterotrimeric transcription factor composed of three subunits (37–43). Formation of a complex between the A and C subunits is required to bind the B subunit, and together, the heterotrimeric complex binds DNA (48). The activation domains of both B and C subunits, which are rich in glutamine and hydrophobic residues, share protein sequence homology with each other and with the glutamine-rich activation domain of the transcription factor Sp1 (48).
NF-Y has been demonstrated to act as both a positive (49–51) and a negative (52) regulator of transcription. Several studies suggest that NF-Y acts by stabilizing the binding of additional factors to adjacent regulatory elements, such as RFX in the major histocompatibility complex class II promoter (53, 54). NF-Y also interacts with transcription factors binding upstream elements and basal transcription machinery, such as HNF4 and TAF100 (42, 55). Recently it was shown that the interaction of NF-Y with cAMP-response element-binding protein may be responsible for the gene-specific transcriptional activity of the insulin cAMP-response element (56), and similar results have been reported for the albumin promoter (36). Since the expression of Dlx3 gene is epithelial specific and the activity of the Dlx3 promoter is low in the fibroblast cell line NIH 3T3 (data not shown), NF-Y and some interacting regulatory factors may be involved in conferring tissue- and differentiation-specific expression to the Dlx3 gene.
In this study, we also demonstrate that the Sp1-binding site located between −21 and +2 positively regulates, and contributes to, the basal activity of the Dlx3 promoter in undifferentiated and differentiated keratinocytes. The Sp1 consensus motif is highly GC-rich; therefore, certain Sp1-binding sites can overlap with other GC-rich consensus transcription factor binding motifs such as that for Ap-2. The ratio of Sp1/Ap2 might be determinant in the differentiation-specific gene expression (57). Many epidermal specific gene promoters contain consensus sites for ubiquitously expressed regulators such as Sp1 family members. The Sp1 family consists of four members (Sp1–Sp4; see Refs. 58–60). Sp1 is involved with tissue/cell type-specific expression of certain genes in epithelial cells. In most of these cases, Sp1 alone does not affect tissue specificity but requires cooperation with other transcription factors, such as Ets in the promoter of human transglutaminase 3 (61), Ap-2 in the K3 keratin gene (57), Egr-1 in the tissue factor gene (62), and NF-1 in the mouse vas deferens protein promoter (63).
From the family of Sp1-related transcription factors, studies have shown that Sp3 acts as an antagonist to Sp1. The human transcobalamin II gene is controlled by the relative ratios of Sp1 and Sp3 (64), and activation of the human papilloma virus type 16 promoter correlates with the ratios of Sp1/Sp3 during epithelial differentiation (65). We have shown that the Sp1 consensus motif on the Dlx3 promoter can bind specifically Sp1 and Sp3, providing the possibility of antagonistic effects on transcription depending on the specific levels of each of these factors throughout the terminal differentiation process.
An essential aspect of the transcriptional regulation of the Dlx3 promoter is the elucidation of the mechanism for inducing its expression by increases in external Ca2+ during differentiation. Until now, the prevailing evidence points to the involvement of PKC isozymes in the induction of keratinocyte differentiation markers by Ca2+, although the exact mechanism is unclear. In this study we show that the region located between +10 and +60 is important for Ca2+ inducibility of the Dlx3 gene. By mutational and gel shift analysis of the +30/+60 sequence, we found that the crucial element responsible for Ca2+ inducibility is located between +42 and +45 (CGAC) and that nuclear factor(s) are involved in the up-regulation of Dlx3 expression by Ca2+. It has been reported that the AP1 and Ets transcription factors are involved in the regulation of the human SPRR1A keratinocyte terminal differentiation marker (45). However, in the case of the Dlx3 gene, the AP1 transcription factor does not bind to the +42/+45 or +46/+52 sequences, which strongly supports that it is not the determinant for the Ca2+ inducibility of Dlx3. Future work will determine the specific nature of the nuclear factor(s) involved in the binding to this regulatory element. The identification of a Ca2+ response element in the Dlx3 gene is the first link between the extracellular signal and the transcriptional control of a regulatory gene involved in keratinocyte differentiation, and represents an important step in the elucidation of the molecular mechanisms underlying this developmental program.
Acknowledgments
We thank Dr. Ulrike Lichti and members of the Laboratory of Skin Carcinogenesis for the Ca2+ determination and for providing reagents. We are grateful to Drs. Peter Steinert, Janine Bryan, Andres Buonanno, and Thomas Sargent for reading and providing comments on the manuscript. We also thank Dr. Shyh-Ing Jang for helpful suggestions and advice and Dr. Peter Steinert for encouragement and support throughout this work.
Footnotes
The abbreviations used are: PKC, protein kinase C; CAT, chloramphenicol acetyltransferase; PCR, polymerase chain reaction; bp, base pair.
References
- 1.Fuchs E, Byrne C. Curr Opin Genet & Dev. 1994;4:725–736. doi: 10.1016/0959-437x(94)90140-x. [DOI] [PubMed] [Google Scholar]
- 2.Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. J Cell Biol. 1989;109:1207–1217. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menon GK, Grayson S, Elias PM. J Invest Dermatol. 1985;84:508–512. doi: 10.1111/1523-1747.ep12273485. [DOI] [PubMed] [Google Scholar]
- 4.Punnonen K, Denning M, Lee E, Li L, Rhee SG, Yuspa SH. J Invest Dermatol. 1993;101:719–726. doi: 10.1111/1523-1747.ep12371682. [DOI] [PubMed] [Google Scholar]
- 5.Nishizuka Y. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 6.Dlugosz AA, Yuspa SH. J Cell Biol. 1993;120:217–225. doi: 10.1083/jcb.120.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen SM, Bro¨nner G, Ku¨tter F, Ju¨rgens G, Ja¨ckle H. Nature. 1989;338:432–434. [Google Scholar]
- 8.Robinson GW, Wray S, Mahon KA. New Biol. 1991;3:1183–1194. [PubMed] [Google Scholar]
- 9.Porteus MH, Bulfone A, Ciaranello RD, Rubenstein JLR. Neuron. 1991;7:221–229. doi: 10.1016/0896-6273(91)90260-7. [DOI] [PubMed] [Google Scholar]
- 10.Simeone A, Acampora D, Pannese M, D’Esposito M, Stornaiuolo A, Gulisano M, Mallamaci A, Kastury K, Druck T, Huebner K, Boncinelli E. Proc Natl Acad Sci U S A. 1994;91:2250–2254. doi: 10.1073/pnas.91.6.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura S, Stock DW, Wynder KL, Bolekens JA, Takeshita K, Nagai BM, Chiba S, Kitamura T, Freeland TM, Zhao Z, Minowada J, Lawrence JB, Weiss KM, Ruddle FH. Genomics. 1996;38:314–324. doi: 10.1006/geno.1996.0634. [DOI] [PubMed] [Google Scholar]
- 12.Morasso MI, Yonescu R, Griffin CA, Sargent TD. Mamm Genome. 1997;8:302–303. doi: 10.1007/s003359900427. [DOI] [PubMed] [Google Scholar]
- 13.Quinn LM, Johnson BV, Nicholl J, Sutherland GR, Kalionis B. Gene (Amst) 1997;187:55–61. doi: 10.1016/s0378-1119(96)00706-8. [DOI] [PubMed] [Google Scholar]
- 14.Price JA, Bowden DW, Wright JT, Pettenati MJ, Hart TC. Hum Mol Genet. 1998;7:563–569. doi: 10.1093/hmg/7.3.563. [DOI] [PubMed] [Google Scholar]
- 15.Morasso MI, Markova NG, Sargent TD. J Cell Biol. 1996;135:1879–1887. doi: 10.1083/jcb.135.6.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson GW, Mahon KA. Mech Dev. 1994;48:199–215. doi: 10.1016/0925-4773(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 17.Feledy J, Morasso MI, Jang SI, Sargent TD. Nucleic Acids Res. 1999;27:764–770. doi: 10.1093/nar/27.3.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morasso M, Jamrich M, Sargent TD. Dev Biol. 1994;162:267–276. doi: 10.1006/dbio.1994.1084. [DOI] [PubMed] [Google Scholar]
- 19.Casatorres J, Navarro JM, Blessing M, Jorcano JL. J Biol Chem. 1994;269:20489–20496. [PubMed] [Google Scholar]
- 20.Leask A, Byrne C, Fuchs E. Proc Natl Acad Sci U S A. 1991;88:7948–7952. doi: 10.1073/pnas.88.18.7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu B, Rothnagel JA, Longley MA, Tsai SY, Roop DR. J Biol Chem. 1994;269:7443–7449. [PubMed] [Google Scholar]
- 22.DiSepio D, Jones A, Longley MA, Bundman D, Rothnagel JA, Roop DR. J Biol Chem. 1995;270:10792–10799. doi: 10.1074/jbc.270.18.10792. [DOI] [PubMed] [Google Scholar]
- 23.Welter JF, Crish JF, Agarwal C, Eckert RL. J Biol Chem. 1995;270:12614–12622. doi: 10.1074/jbc.270.21.12614. [DOI] [PubMed] [Google Scholar]
- 24.Jang SI, Steinert PM, Markova NG. J Biol Chem. 1996;271:24105–24114. doi: 10.1074/jbc.271.39.24105. [DOI] [PubMed] [Google Scholar]
- 25.Blumemberg, M (1993) in Molecular Biology of the Skin: The Keratinocyte (Darmon, M., and Blumemberg, M., eds) pp. 1–32, Academic Press, New York
- 26.Faus I, Hsu HJ, Fuchs E. Mol Cell Biol. 1994;14:3263–3275. doi: 10.1128/mcb.14.5.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen B, Schonemann MD, Flynn SE, Pearse RV, Singh H, Rosenfeld M. Science. 1993;260:78–82. doi: 10.1126/science.7682011. [DOI] [PubMed] [Google Scholar]
- 28.Welter JF, Gali H, Crish JF, Eckert RL. J Biol Chem. 1996;271:14727–14733. doi: 10.1074/jbc.271.25.14727. [DOI] [PubMed] [Google Scholar]
- 29.Frohman MA, Dush MK, Martin GR. Proc Natl Acad Sci U S A. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrews NC, Faller DV. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ausubel, F. M., Brent, R., Kingston, R., Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K. (1987) Current Protocols in Molecular Biology, pp. 12.2.1–12.2.10, Greene Publishing Associates and John Wiley & Sons, New York
- 32.Sargent TD, Jamrich M, Dawid IB. Dev Biol. 1986;114:238–246. doi: 10.1016/0012-1606(86)90399-4. [DOI] [PubMed] [Google Scholar]
- 33.Church GM, Gilbert W. Proc Natl Acad Sci U S A. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fort P, Marty L, Piechaczyk M, El Sabrouty S, Dani C, Jeanteur P, Blanchard JM. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gronostajski RM. Nucleic Acids Res. 1986;14:9117–9129. doi: 10.1093/nar/14.22.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milos PM, Zaret KS. Genes Dev. 1992;6:991–1004. doi: 10.1101/gad.6.6.991. [DOI] [PubMed] [Google Scholar]
- 37.Hatamochi A, Golumbek PT, Van Schaftingen E, de Crombrugghe B. J Biol Chem. 1989;263:5940–5947. [PubMed] [Google Scholar]
- 38.Osawa H, Robey RB, Printz RL, Granner DK. J Biol Chem. 1996;271:17296–17303. doi: 10.1074/jbc.271.29.17296. [DOI] [PubMed] [Google Scholar]
- 39.Dorn A, Bollenkens J, Staub A, Benoist C, Mathis D. Cell. 1987;50:863–872. doi: 10.1016/0092-8674(87)90513-7. [DOI] [PubMed] [Google Scholar]
- 40.Maity SN, Vuorio T, de Crombrugghe B. Proc Natl Acad Sci U S A. 1990;87:5378–5382. doi: 10.1073/pnas.87.14.5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Huijsduijnen RH, Li XY, Black D, Matthes H, Benoist C, Mathis D. EMBO J. 1990;9:3119–2127. doi: 10.1002/j.1460-2075.1990.tb07509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coustry F, Maity SN, de Crombrugghe B. J Biol Chem. 1995;270:468–475. doi: 10.1074/jbc.270.1.468. [DOI] [PubMed] [Google Scholar]
- 43.Sinha S, Maity SH, Lu J, de Crombrugghe B. Proc Natl Acad Sci U S A. 1995;92:1624–1628. doi: 10.1073/pnas.92.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maity SN, Golumbek PT, Karsenty G, de Crombrugghe B. Science. 1988;241:582–585. doi: 10.1126/science.3399893. [DOI] [PubMed] [Google Scholar]
- 45.Sark MWJ, Fisher DF, de Meijer E, van de Putte P, Backendorf C. J Biol Chem. 1998;273:24683–24692. doi: 10.1074/jbc.273.38.24683. [DOI] [PubMed] [Google Scholar]
- 46.Morasso MI, Mahon KA, Sargent TD. Proc Natl Acad Sci U S A. 1995;92:3968–3972. doi: 10.1073/pnas.92.9.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glimcher LH, Kara CJ. Annu Rev Immunol. 1992;10:13–49. doi: 10.1146/annurev.iy.10.040192.000305. [DOI] [PubMed] [Google Scholar]
- 48.Coustry F, Maity SN, Sinha S, de Crombrugghe B. J Biol Chem. 1996;271:14485–14491. doi: 10.1074/jbc.271.24.14485. [DOI] [PubMed] [Google Scholar]
- 49.Lamb KA, Johnson LR, Rizzino A. Mol Reprod Dev. 1997;48:301–309. doi: 10.1002/(SICI)1098-2795(199711)48:3<301::AID-MRD1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 50.Osawa H, Robey RB, Printz RL, Granner DK. J Biol Chem. 1996;271:17296–17303. doi: 10.1074/jbc.271.29.17296. [DOI] [PubMed] [Google Scholar]
- 51.Katula KS, Wright KL, Paul H, Surman DR, Nuckolls FJ, Smith JW, Ting JPY, Yates J, Cogswell JP. Cell Growth Differ. 1997;8:811–820. [PubMed] [Google Scholar]
- 52.Moriuchi H, Moriuchi M, Cohen JI. Virology. 1995;214:256–258. doi: 10.1006/viro.1995.9932. [DOI] [PubMed] [Google Scholar]
- 53.Hinkley C, Perry M. Mol Cell Biol. 1992;12:4400–4411. doi: 10.1128/mcb.12.10.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reith W, Kobr M, Emery P, Durand B, Siegrist CA, Mach B. J Biol Chem. 1994;269:20020–20025. [PubMed] [Google Scholar]
- 55.Ueda A, Takeshita F, Yamashiro S, Yoshimura T. J Biol Chem. 1998;273:19339–19347. doi: 10.1074/jbc.273.30.19339. [DOI] [PubMed] [Google Scholar]
- 56.Eggers A, Siemann G, Blume R, Knepel W. J Biol Chem. 1998;273:18499–18508. doi: 10.1074/jbc.273.29.18499. [DOI] [PubMed] [Google Scholar]
- 57.Chen TT, Wu RL, Castro-Munozledo F, Sun TT. Mol Cell Biol. 1997;17:3056–3064. doi: 10.1128/mcb.17.6.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kingsley C, Winoto A. Mol Cell Biol. 1992;12:4251–4561. doi: 10.1128/mcb.12.10.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hagen G, Muller S, Beato M, Suske G. Nucleic Acids Res. 1992;20:5519–5525. doi: 10.1093/nar/20.21.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sogawa K, Imataka H, Yamasaki Y, Kusume H, Abe H, Kuriyama YF. Nucleic Acids Res. 1993;21:1527–1532. doi: 10.1093/nar/21.7.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JH, Jang SI, Yang JM, Markova NG, Steinert PM. J Biol Chem. 1996;271:4561–4568. [PubMed] [Google Scholar]
- 62.Cui MZ, Parry GCN, Oeth P, Larson H, Smith M, Huang RP, Adamson ED, Mackman N. J Biol Chem. 1996;271:2731–2739. doi: 10.1074/jbc.271.5.2731. [DOI] [PubMed] [Google Scholar]
- 63.Darne CH, Morel L, Claessens F, Manin M, Fabre S, Veyssiere G, Rombauts W, Jean CL. Mol Cell Endocrinol. 1997;132:13–23. doi: 10.1016/s0303-7207(97)00116-0. [DOI] [PubMed] [Google Scholar]
- 64.Li N, Seetharam S, Seetharam B. J Biol Chem. 1998;273:16104–16111. doi: 10.1074/jbc.273.26.16104. [DOI] [PubMed] [Google Scholar]
- 65.Apt D, Watts RM, Suske G, Bernard HU. Virology. 1996;224:281–291. doi: 10.1006/viro.1996.0530. [DOI] [PubMed] [Google Scholar]