

Abstract
A new method for detecting chimeras and other anomalies within 16S rRNA sequence records is presented. Using this method, we screened 1,399 sequences from 19 phyla, as defined by the Ribosomal Database Project, release 9, update 22, and found 5.0% to harbor substantial errors. Of these, 64.3% were obvious chimeras, 14.3% were unidentified sequencing errors, and 21.4% were highly degenerate. In all, 11 phyla contained obvious chimeras, accounting for 0.8 to 11% of the records for these phyla. Many chimeras (43.1%) were formed from parental sequences belonging to different phyla. While most comprised two fragments, 13.7% were composed of at least three fragments, often from three different sources. A separate analysis of the Bacteroidetes phylum (2,739 sequences) also revealed 5.8% records to be anomalous, of which 65.4% were apparently chimeric. Overall, we conclude that, as a conservative estimate, 1 in every 20 public database records is likely to be corrupt. Our results support concerns recently expressed over the quality of the public repositories. With 16S rRNA sequence data increasingly playing a dominant role in bacterial systematics and environmental biodiversity studies, it is vital that steps be taken to improve screening of sequences prior to submission. To this end, we have implemented our method as a program with a simple-to-use graphic user interface that is capable of running on a range of computer platforms. The program is called Pintail, is released under the terms of the GNU General Public License open source license, and is freely available from our website at http://www.cardiff.ac.uk/biosi/research/biosoft/.
Analysis of the 16S rRNA gene is currently fundamental to an understanding of bacterial taxonomy, phylogeny, and diversity (3, 5). Sequence anomalies, if undetected, can generate misleading impressions of environmental diversity and complicate attempts to reconstruct bacterial evolutionary trees. It is vital, therefore, that public repositories such as those managed by EMBL (9), GenBank (2), and the Ribosomal Database Project (RDP) (3) contain reliable sequences if correct conclusions are to be made within studies that rely on 16S rRNA sequence analysis.
Unfortunately, corrupt sequences, such as chimeras formed during PCR amplification (12, 14, 15, 20, 21) or anomalies produced by other steps in the sequencing process, have long been present in the public databases. Poor sequencing methodology often produces highly degenerate sequences; these are easy to spot. More insidious are other sequencing errors that cannot be detected by a visual inspection of the sequence alone. Chimeras, sometimes referred to as jumping PCR products, shuffle genes, or in vitro recombination products have been a recognized PCR amplification problem for some time (17), with damage or degradation to the DNA template and contamination with other templates being likely causes of their formation (14). Chimeras have been shown to occur in PCR-amplified gene libraries with frequencies of up to 30% or more (12, 20, 21) and therefore pose a potentially significant problem.
Chimeric anomalies have long been recognized, and several computational methods have been developed over the years to detect and analyze suspect sequences (6, 7, 10, 11, 13, 16). Historically, the RDP's Chimera_Check program (13) has been used most widely, although the more recent Bellerophon program (7) appears to be gaining in popularity. However, existing tools for chimera detection, although often effective, have limitations (8, 11, 16, 21). Also, most of these tools have not been developed into sufficiently accessible computer programs that can be used easily by researchers regardless of computing background. One reason for the widespread use of RDP's Chimera_Check program is that it has a user-friendly interface and is available to anyone with a web browser.
Most importantly, the problem of chimeras and other sequence anomalies is still underestimated by the research community. Despite recent papers highlighting the problem, some very obvious anomalies continue to be submitted to sequence repositories. Until the extent of this problem is known, the impetus to improve screening procedures prior to submission and to better curate those that have been submitted is unlikely to come.
The aim of the current study was twofold: (i) to develop a 16S rRNA sequence anomaly-detecting method currently used in our laboratory into a new software tool that is sufficiently user friendly and reliable to be used easily by as many researchers as possible, and (ii) to use this tool to estimate the true level of sequence corruption within public repositories. To this end, we present our software to the wider community and detail the results from a survey of selected bacterial taxa, as defined by the RDP database.
MATERIALS AND METHODS
Developing detection method.
All software was written in the Java computer language, using Sun's Java software development kit, J2SE SDK 1.4.2 (Java Technology [http://java.sun.com/]). The final program, called Pintail, was tested on RedHat 9.0 Linux, Microsoft Windows XP, and Apple Mac OS X, version 10.2. Pintail, along with its source code and help files, is freely available from http://www.cardiff.ac.uk/biosi/research/biosoft/ and is released under the terms of the GNU General Public License (http://www.gnu.org/copyleft/gpl.html). The program uses ClustalW (19) to generate sequence alignments.
Our method works by aligning a query sequence (Sq) with a trusted subject sequence (Ss) and then analyzing differences between query and subject over the entire length of the 16S rRNA gene, by employing a sliding window of specified size w progressing a fixed number of bases l at a time along the resulting alignment Sqs of length n. The total number of windows will be m = ⌈n − w + 1/l⌉, where ⌈⌉ signifies the ceiling of the enclosed expression, i.e., the smallest whole number greater than or equal to the value of the expression. At the ith window wi (1 ≤ i ≤ m), the percentage of mismatched bases is calculated, giving rise to an observed percentage difference oi that can be thought of as an uncorrected measure of evolutionary distance between query and subject within wi. The resulting set of observed percentage differences Oqs = {oi: o1, o2,…, om} when plotted provide a visual representation of the variation in evolutionary distance between Sq and Ss over the length of the 16S rRNA gene. The core algorithm for generating Oqs can be summarized as follows.
Algorithm 1.
(i) Input query sequence Sq, the sequence to be checked for anomalies. (ii) Input subject sequence Ss, a reliable sequence closely related to the query. (iii) Globally align Sq with Ss using ClustalW to generate alignment Sqs of length n. (iv) By sliding a window of size w with step l along Sqs, determine the percentage of mismatched bases oi within window wi as described above and compute the resulting data set Oqs = {oi: o1, o2,…, om} of the observed percentage differences detected between Sq and Ss. (v) Plot Oqs against base position i to display graphically the changes in evolutionary distance between Sq and Ss over their mutual length n.
Note that the mean of the observed percentage differences, expressed as (Σioi)/m, is essentially a measure of the overall uncorrected evolutionary distance between the two sequences. Although this value will not be exactly the same as that derived by a simple global alignment, for simplicity we will use the term “overall evolutionary distance” to refer to this mean, as the distinction between the two concepts is irrelevant as far as the rest of the paper is concerned.
Expected percentage differences.
To assess whether the observed percentage difference plot indicates an anomalous query, a method was developed for predicting expected percentage differences that one might expect if both query and subject were reliable. To generate expected percentage differences Eqs = {ei: e1, e2, …, em} for any pair of sequences Sq and Ss, it was necessary to map accurately the hypervariable regions within the 16S rRNA gene sequence. This was done as follows.
All type strain sequences of ≥1,200 nucleotides were downloaded from the RDP web site (3) as a single aligned file, with Escherichia coli U00096 included as a reference sequence. At the time of this study, RDP release 9, update 22 (September 2004), was current, with 4,383 full-length type strain sequences available for downloading.
We totalled the number of each nucleotide residue r{r: A, C, G, T/U} at each base position j (1 ≤ j ≤ 1,542) within the RDP aligned type strain sequences, using E. coli U00096 as a reference (hence, 1,542 base positions). From these raw counts, we identified the frequency fjr of the most common residue r at each base position j within the alignment (ignoring gap characters). Note that when position j is most variable, each of the four possible residues is equally likely to occur. By a simple correction, pj = (fjr − 0.25)/0.75 relative frequencies were converted into probabilities, and so the entire type strain data set was described by the probability profile P = {pj: p1, p2, …, p1,542}, which reflects the probability of a 16S rRNA sequence being conserved at any particular residue position.
If pj describes residue conservation at position j, then qj = 1 − pj describes residue variability at that position. In other words, Q = {qj: q1, q2, …, q1564} is a probability profile that reflects the variability of a 16S rRNA sequence at any particular residue position. Thus, profile Q can be used to map accurately the hypervariable regions within the 16S rRNA gene. The expected percentage differences Eqs can be generated from Q by applying the following algorithm.
Algorithm 2.
(i) By sliding a window of size w with step l along the probability profile Q, determine the average probability ai for each window wi such that the resulting data set Qav = {ai: a1, a2,…, am} is a set of average probabilities that can be related directly to the observed percentage differences data set Oqs generated by Algorithm 1. (ii) Define a fitting coefficient α as the overall evolutionary distance between query and subject, as defined by (Σioi)/m, divided by the mean of data set Qav. Thus, 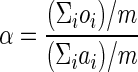 . (iii) Multiply each element of Qav by α to generate the expected percentage differences Eqs (i.e., ei = ai · α). (iv) Plot Eqs alongside Oqs.
. (iii) Multiply each element of Qav by α to generate the expected percentage differences Eqs (i.e., ei = ai · α). (iv) Plot Eqs alongside Oqs.
Algorithm 2 generates expected percentage differences for any query and subject pair. By plotting the expected values Eqs against their observed values Oqs generated by algorithm 1, a visual assessment of the quality of sequence Sq with respect to sequence Ss can be made. In addition, subtracting ei from oi for each position i generates a series of deviations, the standard deviation of which quantifies the overall deviation of Oqs from Eqs. We refer to this standard deviation as the deviation from expectation (DE) statistic. Thus, 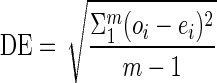 .
.
Calibrating the method.
Of the 4,383 type strain sequences from the RDP, 2,361 contained at least one degenerate base. As a means of discarding potentially unreliable records, these degenerate sequences were removed, leaving an RDP aligned data set of 2,022 sequences, plus the E. coli reference. The type strains were then analyzed by applying the following two procedures.
Procedure 1.
(i) Applying algorithms 1 and 2, each sequence in the data set was compared to each other, resulting in a DE value for each comparison. (ii) All DE values were plotted against their corresponding overall evolutionary distances. (iii) Obvious outlier DE values were identified from the plot. (iv) Sequences responsible for the outlier DE values were then identified. Since each DE value was generated by a pair of sequences, the sequence responsible for the high DE value was identified by using a ranking system that scored sequences according to the number of times they were involved the generation of a DE outlier.
Identified sequences were then investigated by applying procedure 2.
Procedure 2.
(i) A National Center for Biotechnology Information (NCBI) BlastN search (1) was undertaken with each query sequence to identify its nearest neighbors within the public database. (ii) A suitable nearest neighbor was chosen for comparison (labeled the first subject). Sequences originating from different research groups, and hence a different 16S rRNA gene library from that which had generated the query, were preferred. (iii) The first subject was compared to the query using the Pintail program, and the output was assessed for evidence of any sequence anomaly. (iv) To confirm the reliability of the first subject, and hence the conclusions drawn, a second nearest neighbor was selected again from a separate study. This second subject was compared to the first subject by using Pintail, and output was checked. (v) Finally, as a final check, the query was compared to the second subject.
It can be seen that, ideally, only three comparisons are necessary per query sequence to unambiguously identify an anomaly. In practice, this was not always possible, either because a lack of suitable database entries meant that the only nearest neighbors available were those generated by the same author(s) and thus were probably from the same gene library or because the best available nearest neighbor was only distantly related to the query. Under such circumstances, up to nine nearest neighbors were compared to the query sequence and each other, and the final conclusion was made after assessing the overall trend in the resulting matrix of pairwise comparisons. Where necessary, the NCBI's BLAST 2 SEQUENCES program (bl2seq) (18) was used to resolve uncertainties.
Procedures 1 and 2 were applied to the type strain data, and outlier DE values found to be generated by anomalous sequences were excluded from subsequent analysis. The median, upper quartile, and 95, 99, 99.9, and 100% quantiles of the corrected DE plot were then determined for each 1% interval along the x axis of the plot. In this way, the corrected DE plot could be described in terms of a series of quantile plots and could be included within the final Pintail program. Thus, a DE value subsequently generated by Pintail could be compared to DE values previously generated from the type strain comparisons, and conclusions could be drawn as to the likelihood of the new DE value being generated by a pair of nonanomalous sequences.
Testing Pintail with known chimeras.
The Pintail program was tested with 50 known bacterial chimeric sequences originally identified by Hugenholtz and Huber (8) and listed in the RDP database, release 9, update 22. A further five archaeal sequences listed by Hugenholtz and Huber (8) but not included on the RDP website were also tested. Each chimera was analyzed by following procedure 2.
Screening selected bacterial phyla.
Using the RDP's online hierarchy browser, all bacterial phyla containing up to 200 sequence records were downloaded as separate aligned files. For each aligned data set, procedure 1 was applied to identify putatively anomalous sequences. In this screening, outlier DE values were defined as those falling above the 99.9% quantile line calculated from the type strain data. Anomalous sequences identified in this way were checked by procedure 2.
Procedure 1 was also applied to the 2,739 almost-complete (≥1,200-nucleotide) sequence records making up the Bacteroidetes phylum as defined by RDP, release 9, update 22. In this much larger single analysis, potentially anomalous sequences were confirmed by application of a simplified version of procedure 2 (i.e., steps i to iii only).
RESULTS
Implementation of methodology.
The development of the methodology described in this paper culminated in the computer program Pintail, the operation of which is now described. Figure 1 shows a screenshot of Pintail, showing the outcome of a typical analysis. The query sequence Sq (in this instance, a chimera) was entered into the top-left text box, and the subject sequence Ss (a reliable sequence, identified by BlastN as closely related to the query) was entered into the bottom-left text box. The results of the analysis are displayed in the panel on the right and show graphically that the query is indeed a chimera with its 3′ end phylogenetically more distant from the subject sequence than its 5′ end. Figure 2 illustrates in more detail typical graphs generated by the program, with panels A to C showing the output from a reliable query sequence being compared with equally reliable subject sequences of various evolutionary distances. Conversely, panels D to F show typical plots obtained when the query sequence is chimeric. The trends shown in panels D to F are very characteristic of chimeras. Other anomalies, such as missing sequence data or blocks of degenerate bases, are easily recognized from much sharper plot variations, which are particularly noticeable when smaller sampling window sizes are employed.
FIG. 1.

Program screenshot illustrating a typical analysis. In this example, query AY693838 (top left) is compared with subject AJ551147 (bottom left), generating a plot of evolutionary distances that demonstrate high similarity between these two sequences at the 5′ end only. AY693838, introduced into the NCBI on 30 August 2004, is classified by the RDP as belonging to the proposed new OP11 phylum. AJ551147, in contrast, belongs to the β-Proteobacteria genus Janthinobacterium.
FIG. 2.

Typical 16S rRNA gene sequence comparison plots generated by Pintail (all graphs generated with window size 300 and step size 25). (A to C) Plots between pairs of trusted sequences of increasing evolutionary distance, while D to F show examples where the query sequence is a chimera. Observed percentage differences between sequences are plotted as black lines. Gray lines show the expected percentage differences for the sequence pairs. Light gray shading indicates expected percentage differences ±5%. Escherichia coli ATCC 11775T (X80725) is compared to Escherichia vulneris ATCC 33821T (X80734) (A), Pseudomonas aeruginosa LMG 1242T (Z76651) (B), and Aquifex pyrophilus (T) Kol5a (M83548) (C). (D to F) Three typical chimeric patterns. (D) The three-fragment Nitrospira chimeric sequence AY373422 (estimated breakpoints, 340 and 740) is compared to its BLAST identified nearest neighbor, X82559. (E) The three fragment chimeric record U10877 generated from Riemerella anatipestifer (T) ATCC 11845 is shown to diverge from the sequence of its nearest neighbor, R. anatipestifer strain 115/02 (AY856450) around E. coli positions 790 to 1130. (F) The two-fragment Fusobacteria chimeric sequence AY548989 (estimated breakpoint, 800) is compared to the sequence from its nearest neighbor, AY548984.
Each graph generated by the program consists of four plots. The plot of observed percentage differences (Oqs, shown as a black line in Fig. 2) shows the change in percentage difference between query and subject as the sampling window moves along the alignment. In all examples shown in Fig. 2, a window size w of 300 nucleotides was used, moving along the alignment l for 25 bases at a time. This combination was found to be most suitable for displaying overall trends. Reducing window size to ≤100 bases supplies more detail and is useful for estimating chimeric breakpoints.
The mean of the observed percentage differences displayed by the program is roughly equivalent to the uncorrected evolutionary distance between query and subject. From this mean, the expected percentage differences (Eqs) which might be expected for sequences of this evolutionary distance were calculated. These expected percentage differences are displayed as a second plot line within the program's output graph (Fig. 1) and as gray lines in Fig. 2. Similarly, two further expected lines were plotted based on the mean observed percentage differences ±5% and represent graphically this level of variation around the expected line as an area shaded light gray (Fig. 2).
The expected line (Eqs plot) helps to indicate if and where the observed line deviates from what might be expected from reliable sequences with the same overall evolutionary distance as the query and subject. The DE statistic calculated by the program quantifies this deviation. The higher the DE value, the greater will be the departure of the observed data from that expected of trusted sequences. To aid interpretation, the DE statistic is best viewed in the context of reliable query-versus-subject comparisons sharing similar evolutionary distances. So the program summarizes the DE values obtained between type strains of the same evolutionary distance as exhibited between query and subject; from this information, the probability that the observed DE value is likely to have been generated by two reliable sequences is inferred (Fig. 1).
Development of methodology and testing the underlying assumption.
The assumption underlying the method implemented in Pintail is that two reliable (i.e., nonanomalous) 16S rRNA sequences of known overall evolutionary distance will vary by roughly the same amount over the length of the gene, allowing for the effects of the hypervariable regions when homologous bases are compared. Given the empirical nature of the methodology, it was necessary to test this assumption.
One test was to select pairs of reliable sequences at random, apply the method, and assess the output for any contradiction of our assumption. Figure 2A to C illustrates typical results obtained this way. However, this approach was inevitably limited in scope. To test the assumption more thoroughly and at the same time calibrate our method, we needed to consider a much larger data set of reliable sequences. To do this necessitated finding a way of quantifying our observations so that a more automated checking procedure could be employed. This led to the concept of expected percentage differences and the deviation from expectation statistic, described in Materials and Methods and now considered in more detail below.
(i) Expected percentage differences.
To generate expected percentage differences for any two sequences, it was necessary to take account of the regions of conservation and variability inherent in the 16S rRNA gene and the evolutionary distance represented by sequence dissimilarity between the two sequences. As Fig. 2A to C illustrates, the character of the observed percentage difference plot was informed by both of these concepts. Therefore, we needed to model 16S rRNA intragene variability and then use this model to predict expected percentage differences from overall evolutionary distance (as represented by the mean of the observed percentage differences).
Type strain sequences, a priori, can be considered reliable in that they will normally have been generated from pure cultures and therefore will have been less prone to the errors common to environmental samples, due to quality and purity of the template. RDP release 9, update 22, contains 4,383 type strain sequences with a length of ≥1,200 nucleotides. We downloaded all 4,383 records from the RDP website retaining the RDP's alignment, along with a reliable Escherichia coli record (U00096) as a reference sequence. From this, we were able to allocate to each base position in the E. coli reference sequence a frequency for the most common nucleotide residue (A, C, G, or T/U) (Fig. 3A). For example, a position that is occupied by an adenine in all type strain sequences would have a frequency of 1. Conversely, a position where all four bases are equiprobable would have a frequency of 0.25.
FIG. 3.

Illustrating variable regions within the 16S rRNA gene and location of chimeric breakpoints. (A) The frequency of occurrence of the most common nucleotide residue at each base position within the 16S rRNA gene, as determined from RDP-listed 4,383 type strains, with E. coli U00096 as a reference. These frequencies are measures of variability within the gene. (B) Smoothing the data, by taking the mean frequency within a window of 50 bases, moving one base at a time along the gene, creates the plot shown in panel B. The locations of the hypervariable regions are labeled, with gray bars on the x axis defining these regions as V1 to V9 (the Comparative RNA Web Site [http://www.rna.icmb.utexas.edu/]). (C) Histogram of all chimera breakpoints identified in this study and that of Hugenholtz and Huber (8).
Smoothing these data revealed peaks and troughs which corresponded to the known hypervariable and conserved regions for the 16S rRNA gene (Fig. 3B), matching peaks and troughs in observed percentage difference plots. Converting these frequencies to a probability profile—allocating a probability to each 16S rRNA base position—created a profile of 16S rRNA intragene variability for use in the final program. Expected percentage differences for any two sequences were generated from this profile by multiplying each probability by the fitting coefficient α to ensure the resulting data set had the same mean as the observed data.
(ii) DE statistic.
Subtracting a set of expected values from corresponding observed data points generated a set of error values, the standard deviation of which summarized the extent to which observation deviated from expectation. This is how the DE statistic was derived and used in this study as a way of summarizing any analysis of sequence pairs as a single value.
We were now in a position to automate our method and consider a much larger data set of reliable sequences. The 4,383 type strain sequences initially served as the data set; however, since our method detects any sequence anomaly, it quickly became apparent that high levels of type strain degeneracy were hampering our survey and needed to be discounted. Only 2,022 of 4,383 type strain sequences were completely without degenerate base characters. Of the remaining 2,361 sequences, levels of degeneracy as high as 483 bases were detected, although 2,173 had ≤50 degenerate characters. Further analysis concentrated on the 2,022 degeneracy-free sequences, since these were considered to be least likely to have anomalies.
(iii) Calibration.
Pairwise comparisons of the 2,022 sequences without degeneracies generated 2,043,231 DE values. Plotting all these against the mean of the observed percentage differences for each comparison (Fig. 4) revealed that most DE values, and hence most comparisons, clustered together. However, a number of outlier clusters quite distinct from the main cluster were also observed (Fig. 4A), and investigation showed the same 15 sequences were responsible for these outliers (Table 1).
FIG. 4.

DE values generated from type strain data set containing 2,022 16S rRNA gene sequences without any degenerate base positions (see text). DE value was generated for each of the 2,043,231 pairwise sequence comparisons and plotted against evolutionary distance between sequences. (A) The data set prior to the removal of the 15 anomalous sequences (see text); (B) the plot after removal; (C) the quantile values used to describe these data and incorporated into the Pintail program as a means of calibration.
TABLE 1.
Anomalous Bacteria 16S rRNA gene sequence records from type strains
| Accession no. | Name | Location of anomaly relative to E. coli (bases) | Description |
|---|---|---|---|
| D17751 | Leucobacter komagatae IFO15245T | 60-220 | Anomaly near 5′ end; likely sequencing error |
| D21342 | Microbacterium imperiale IFO 12610T | 230 | Anomaly at 5′ end; likely sequencing error |
| D21344 | Microbacterium laevaniformans IFO 14471T | 90-220 | Anomaly near 5′ end; likely sequencing error |
| AJ242532 | Arthrobacter flavus CMS-19Y | 1,130-1,420 | Anomaly near 3′ end; likely sequencing error |
| AJ233946 | Nannocystis exedens Na e1 | 730-840 | Anomaly near middle; likely sequencing error |
| D21245 | Luteococcus japonicus IFO12422 | 240, 680-790 | Anomaly at 5′ end and in middle; likely sequencing errors |
| AF195797 | Thermoanaerobacter subterraneus SEBR 7858; LA61 | 800-960 | Anomaly near middle; likely sequencing error |
| D21343 | Microbacterium lacticum IFO 14135T | 70-240 | Anomaly near 5′ end; likely sequencing error |
| Z49116 | Halanaerobium saccharolyticum subsp. senegalense DSM 7379 | 1,320-1,450 | Anomaly near 3′ end; likely sequencing error |
| D21339 | Microbacterium arborescens IFO 3750T | 230 | Practically identical to D21342 |
| D21341 | Microbacterium dextranolyticum IFO 14592T | 60-240 | Anomaly near 5′ end; likely sequencing error (only visible with RDP alignment) |
| AB013297 | Vibrio rumoiensis S-1 | 500? | Anomaly near 5′ end; likely sequencing error (only visible with RDP alignment) |
| D17527 | Kineococcus aurantiacus IFO 15268 | 70-240 | Anomaly near 5′ end; likely sequencing error |
| AJ272391 | Lactobacillus psittaci | 790 | Two-fragment chimera with 5′ end practically identical to Lactobacillus jensenii (AF243159) and 3′ end practically identical to Lactobacillus vaginalis (AF243154) |
| U10877 | Riemerella anatipestifer ATCC 11845 | 790, 1,130 | Three-fragment chimera with middle fragment of γ-Proteobacteria origin; fragments one and three derive from the same Bacteroidetes origin |
Application of procedure 2 (Fig. 5) showed 2 of these 15 sequences to be chimeric. Record AJ272391 (classified as Lactobacillus psittaci) is a two-fragment chimera with a 5′ end practically identical to that of Lactobacillus jensenii (AF243159) and a 3′ end similarly close to that of Lactobacillus vaginalis (AF243154). Record U10877 (classified as Riemerella anatipestifer ATCC 11845T) is a three-fragment chimera with fragments 1 and 3 deriving from a member of the Bacteroidetes and fragment 2 of γ-Proteobacteria origin (Fig. 2E). It is worth noting here that ATCC 11845T has subsequently been resequenced as record U60101 and that analysis of this record shows no anomaly. The remaining 13 sequences contained anomalies most likely to be sequencing errors. Eight originated from the same research group, and all contained some sort of sequencing error in the first 220 to 240 bases at the 5′ end. Intriguingly, two of these anomalies were observed when the original 2,022-type strain RDP alignment was used but not when checked with ClustalW. Further investigation by eye confirmed these anomalies to be real, confirming the RDP alignment to be the more accurate than the ClustalW alignment.
FIG. 5.

Illustrating procedure 2 for unambiguously confirming a chimeric sequence (all graphs were generated with window size 300 and step size 25). (A) In this example, the query, an Acidobacteria sp. (AF523990), is compared to its nearest neighbor (AF523976) identified by BlastN search, and an anomaly at the 5′ end is identified. (B) AF523976 is next compared to its nearest neighbor, AY234512, to confirm that it is reliable. No anomaly is detected. (C) As a final check, AF523990 is compared to AY234512; as expected, the 5′ end anomalous feature is seen. (D) To determine whether this anomaly is chimeric, the identified 5′ region is excised, a BLAST search is undertaken, and the identified nearest neighbor (in this case Actinobacteria X68459) is compared to AF523990. Again, an anomaly is detected, but this time the reverse of that seen in panel A, clearly indicating our query to be a chimera. (E) Comparing X68459 with its neighbor, AF498683, confirms its reliability, and as expected, (F) comparing the original query with AF498683 generates the same profile as that seen in panel D. The chimeric breakpoint can be estimated by superimposing A on D.
When the 15 anomalous sequences were removed from the data set, the plotted DE values clustered together as one group (Fig. 4B). Figure 4C shows the same data reduced to a series of quantile plots, which were used to estimate the probability of the query sequence being anomalous, as indicated in Fig. 1.
Testing program with known chimeras.
We tested our approach with 39 chimeric 16S rRNA sequences identified by Hugenholtz and Huber (8) and applied procedure 2 as summarized in Fig. 5. All were confirmed as chimeric by our method. In addition, we found that Hugenholtz and Huber had incorrectly characterized record AF254401 as a two-fragment chimera, whereas our method reveals it to be a three-fragment chimera (Fig. 6). AF254401 sequence up to E. coli position 340 is of Firmicutes origin (closely matching AF323775). Bases from 341 to 1,080 come from an unknown source, the closest match being AF323760, previously identified as from the OP9 phylum (8) but remaining unclassified by the RDP. The remainder of AF254401 derives from the Spirochaetes phylum and closely matches M88719.
FIG. 6.

Analysis of the three-fragment chimera AF254401 (all graphs were generated with window size 100 and step size 25). The query is shown compared to AF323775 (A), AF323760 (B), and M88719 (C).
We also tested an additional 15 chimeras identified by Hugenholtz and Huber and listed within the RDP hierarchy browser but not included in their paper (8). We confirmed 12 to be chimeric. However, we could not find evidence that X84498, AF333535, or AY082475 were chimeric (although with AY082475 there was evidence of a possible sequencing anomaly at the extreme 5′ end), and a series of comparisons using bl2seq (18) under a range of parameter settings failed to contradict this analysis.
Database analysis.
The RDP website hierarchy browser (3) classifies 16S rRNA sequence records according to the current Bergey's 16S rRNA-based classification system (5). We used this facility to obtain aligned sequence files for 19 phyla, amounting to 1,399 records in all. Phyla were selected purely by size, with any phylum containing ≤200 sequences chosen. Thus, all were selected without prior knowledge of any sequence anomalies.
Initial screening by DE value, as detailed in procedure 1, identified 73 putatively anomalous sequences. Application of procedure 2 showed 70 of these 73 to be unambiguously anomalous and distributed within 16 of the 19 phyla (Fig. 7; Table 2). The three false positives all occurred within the Aquificae and were caused by the absence of sufficiently closely related subject sequences for comparison with the query sequences concerned.
FIG. 7.

Distribution of sequence anomalies with the nineteen Bacteria phyla, as defined by the Ribosomal Database Project (3). Numbers in brackets after the phylum (or candidate division) name are the total number of sequences within that phylum present in RDP release 9, update 22, of September 2004.
TABLE 2.
Anomalous sequences identified by this study
| Accession no. | Phylum | Approx. break position relative to E. coli (bases) | Details |
|---|---|---|---|
| AY268103 | Aquificae | 560 | PCR or sequencing error with first ∼465 bases the reverse complement of what they should be |
| AB183857 | Aquificae | 425 | Anomaly at 5′ end, though origin unknown; either chimera or sequencing error |
| AF018191 | Aquificae | 1,080 | Two-fragment chimera, with both fragments Aquificae in origin |
| AJ237665 | Thermotogae | 930 | Two-fragment chimera, with 3′ end Firmicutes in origin |
| L10662 | Thermodesulfobacteria | Degenerate sequence; several large blocks of N bases | |
| Z15060 | Deinococcus-Thermus | Degenerate sequence; one large block of N bases | |
| X58340 | Deinococcus-Thermus | Degenerate sequence; several large blocks of N bases | |
| AF317775 | Nitrospira | Degenerate sequence; one large block of N bases | |
| AF317779 | Nitrospira | Degenerate sequence; one large block of N bases | |
| L14619 | Nitrospira | Degenerate sequence; several large blocks of N bases | |
| AY661410 | Nitrospira | 320, 540 | Two- or possibly three-fragment chimera, with 3′ end of unknown origin |
| AF543500 | Nitrospira | 250 | Sequencing anomaly only visible when RDP alignment used |
| AY373422 | Nitrospira | 340, 740 | Three-fragment chimera, with 5′ end γ-Proteobacteria, middle α-Proteobacteria, and 3′ end unknown in origin |
| AY661421 | Nitrospira | 370 | Two-fragment chimera, with 3′ end unclassified (candidate division OP5 according to NCBI) |
| AF485343 | Nitrospira | 1,080 | Two-fragment chimera, with 3′ end unclassified; record now replaced in database |
| AY297986 | Nitrospira | 700 | Two-fragment chimera, with 5′ end Firmicutes in origin; record already marked as chimeric in database |
| AY796049 | Nitrospiraa | 790 | Two-fragment chimera with 5′ end β-Proteobacteria in origin |
| AY762631 | Nitrospiraa | 660, 940 | Three-fragment chimera derived from two parents, with middle fragment of unclassified origin |
| X86774 | Nitrospira | 790, 1,220 | Three-fragment chimera derived from two parents, with middle fragment γ-Proteobacteria in origin; already identified as chimera |
| AF543509 | Nitrospira | 500, 790 | Three-fragment chimera, with middle fragment also Nitrospira in origin |
| AB176700 | Nitrospira | 500 | Two-fragment chimera, with 5′ end of unknown origin |
| AF543503 | Nitrospira | 540 | Two-fragment chimera, with both fragments Nitrospira in origin |
| AF543511 | Nitrospira | 760 | Two-fragment chimera, with both fragments Nitrospira in origin |
| L22045 | Nitrospira | Degenerate sequence; one large block of N bases | |
| M79383 | Nitrospira | Degenerate sequence; one large block of N bases | |
| Y10652 | Chlorobi | Degenerate with Ns clustered at 5′ and 3′ ends, giving superficial appearance of a chimera | |
| Y10643 | Chlorobi | Degenerate with Ns clustered at 5′ and 3′ ends, giving superficial appearance of a chimera | |
| Y10651 | Chlorobi | Degenerate with Ns clustered at 5′ and 3′ ends, giving superficial appearance of a chimera | |
| Y10647 | Chlorobi | Degenerate sequence; one large block of N bases and numerous other Ns | |
| Y10640 | Chlorobi | Degenerate with Ns clustered at 5′ and 3′ ends, giving superficial appearance of a chimera | |
| AY661796 | Chlamydiae | Sequencing anomaly only visible when RDP alignment used | |
| AY661795 | Chlamydiae | Sequencing anomaly only visible when RDP alignment used | |
| AB179510 | Acidobacteria | 940-1,100 | Two-fragment chimera, with 3′ end of unclassified origin; lack of clear break due to number of degenerate bases |
| AY326570 | Acidobacteria | 600-1,000 | Two-fragment chimera, with 3′ end of unclassified origin; no obvious reason for lack of clear break |
| AF523990 | Acidobacteria | 370 | Two-fragment chimera, with 5′ end of Actinobacteria origin |
| AJ536862 | Acidobacteria | 280 | Two-fragment chimera, with 5′ end of unclassified origin |
| Y07575 | Acidobacteria | 560 | Two-fragment chimera, with 3′ end of unknown origin |
| AY548989 | Fusobacteria | 800 | Two-fragment chimera, with 3′ end δ-Proteobacteria in origin. |
| AJ289180 | Fusobacteria | 930, 1,210 | Three-fragment chimera, with 5′ end Fusobacteria, middle Spirochaetes, and 3′ end Bacteroidetes in origin |
| AJ441248 | Fusobacteria | ∼580 | Two-fragment chimera, with 3′ end of unclassified origin; exact position of break unclear due to lack of full-length subjects |
| AY548992 | Fusobacteria | 280, 790 | Three-fragment chimera, with 5′ end ɛ-Proteobacteria, middle Fusobacteria, and 3′ end ɛ-Proteobacteria in origin |
| AJ441228 | Fusobacteria | 150, 930 | Two-, possibly three-fragment chimera with 5′ end unknown, middle unclassified, and 3′ end ɛ-Proteobacteria in origin |
| AY548985 | Fusobacteria | 1,140 | Two-fragment chimera, with 3′ end of Spirochaetes origin |
| AF287807 | Fusobacteria | 350 | Two-fragment chimera, with both fragments of Fusobacteria origin |
| AF287808 | Fusobacteria | 920 | Two-fragment chimera, with both fragments of Fusobacteria origin |
| AF366272 | Fusobacteria | 1,160 | Two-fragment chimera, with both fragments of Fusobacteria origin |
| AF385542 | Fusobacteria | 350 | Very similar to AF287807 |
| Z94005 | Verrucomicrobia | 1,025, 1,150 | Region 1,025-1,150 is alien to sequence but no close match found within database; unusual nature of plot suggests sequencing error |
| AJ401133 | Verrucomicrobia | 550 | Two-fragment chimera, with both fragments of Verrucomicrobia origin |
| AJ401131 | Verrucomicrobia | 920 | Two-fragment chimera, with 5′ end of unknown origin |
| AF316731 | Verrucomicrobia | 300 | Two-fragment chimera, with 5′ end of unclassified origin |
| AJ401123 | Verrucomicrobia | 590 | Two-fragment chimera, with both fragments of Verrucomicrobia origin |
| AB179538 | Verrucomicrobia | 570 | Two-fragment chimera, with 3′ end of unknown origin |
| AF351215 | Verrucomicrobia | 1,080 | Two-fragment chimera, with 3′ end of δ-Proteobacteria origin |
| AJ617868 | Verrucomicrobiaa | 1,080 | Two-fragment chimera, with 3′ end of δ-Proteobacteria origin |
| AF234140 | Gemmatimonadetes | Degenerate sequence; one large block of N bases | |
| AF009987 | Gemmatimonadetes | Degenerate sequence; two large blocks of N bases | |
| AY218634 | Gemmatimonadetes | 700 | Two-fragment chimera, with both fragments of Gemmatimonadetes origin |
| AY221051 | Gemmatimonadetes | ∼900 | Two-fragment chimera, with both fragments of Gemmatimonadetes origin; break point uncertain due to quality of available subject sequences |
| AJ582052 | Gemmatimonadetes | 600, 950 | Likely sequencing error |
| AY218706 | Gemmatimonadetes | 275 | Likely sequencing error at 5′ end |
| AF368188 | OP10 | 1,100 | Two-fragment chimera, with 3′ end of probable Bacteroidetes origin |
| AF368185 | OP10 | ∼260, 970 | Likely three fragment chimera with 5′ and 3′ ends originating from some unknown source |
| AF368184 | OP10 | ∼260, 970 | Same as AF368185 |
| AY693838 | OP11 | 520 | Two-fragment chimera, with 5′ end of β-Proteobacteria origin |
| AY218572 | OP11 | 660 | Two-fragment chimera, with 5′ end of ɛ-Proteobacteria origin |
| AJ582211 | OP11 | ∼560 | Likely two-fragment chimera, with 3′ end of unknown origin |
| AF513093 | TM7 | 900 | Two-fragment chimera, with 3′ end of unclassified origin |
| AJ318135 | TM7 | 1,090 | Two-fragment chimera, with 3′ end of Actinobacteria origin |
| AY592328 | WS3 | 380 | Two-fragment chimera, with 5′ end of Actinobacteria origin |
| AY217439 | Dehalococcoides | 500, 675 | Likely sequencing error |
| AY133080 | Dehalococcoides | 1,400 | Likely sequencing error |
| AY548991 | ɛ-Proteobacteriab | 320 | Two-fragment chimera, with 5′ of γ-Proteobacteria origin; 3′ of ɛ-Proteobacteria origin |
| AJ441247 | Unclassified Bacteriab | 380 | Two-fragment chimera, with 5′ end of δ-Proteobacteria origin, 3′ end of Chloroflexi origin |
| AY762628 | β-Proteobacteriab | 780 | Two-fragment chimera, with both fragments of β-Proteobacteria origin |
| AY762632 | β-Proteobacteriab | 780 | Practically identical to AY762628 |
| AJ582208 | Unclassified Bacteriab | 540 | Two-fragment chimera, with 5′ of Firmicutes origin and end 3′ of Gemmatimonadetes origin |
| AY218710 | Unclassified Bacteriab | 630 | Sequencing error in which first ∼152 bases are the reverse complement |
| AY280419 | Unclassified Bacteriab | 520 | Two-fragment chimera, with 5′ of Bacteroidetes origin, and 3′ end of WS3 origin |
| AB007420 | Actinobacteriab | 40-250 | Likely sequencing error |
Added after September release (not included in calculations).
Uncovered during analysis (not included in calculations).
Of the 70 confirmed anomalies, 45 were clearly chimeric. A further 15 anomalies were highly degenerate. The remaining 10 contained other sequence anomalies, such as that found within the Aquificae record AY268103, the 5′ end of which, up to E. coli position 560, was the reverse complement of 16S rRNA.
The Pintail program identified 22 of the 45 chimeras as derived from parents belonging to different phyla. For example, sequence AF523990 is part Acidobacteria and part Actinobacteria. A further 16 chimeras contained one parent of either unknown (no close record in current database) or unclassified (RDP was unable to classify according to Bergey's classification) origin. Thirteen out of 45 were formed from parents belonging to the same phylum.
While most chimeras were composed of two fragments from unrelated source sequences, nine three-fragment chimeras were also detected. A striking example of this is the Fusobacteria sequence AJ289180 with its 5′ end originating from a Fusobacterium, the middle region being of Spirochaetes origin, and the 3′ end belonging to a member of the Bacteroidetes.
Table 2 lists a further 10 anomalous sequences discovered during our investigations but not included in our original 19-phylum data set. All but two are obvious chimeras. One is another example of the 5′ end being a reverse complement of the correct sequence. Three of these records were submitted to the public repositories during our study.
The Bacteroidetes phylum, as identified by RDP release 9, update 22, was also screened by applying procedure 1 and steps i to iii of procedure 2. Of the 2,739 near-complete sequences checked, 159 (5.8%) were identified as likely anomalies. Of these, 12 were highly degenerate, 104 appeared to be chimeric, 21 contained missing sequence blocks due to assembly errors, and the remainder were miscellaneous anomalies.
Chimera breakpoints.
Approximate breakpoints for chimeras in this study were determined by analyzing the plots produced by Pintail. Reducing window size to 50 to 100 was most effective in providing sufficient visual detail to make this assessment. Breakpoints were most easily assessed when both parent sequences were identified (e.g., Fig. 5), since their corresponding observed percentage difference plots could easily be superimposed on one another and breakpoints could be identified where the lines crossed.
Identified breakpoint positions were combined with values identified by Hugenholtz and Huber (8) and plotted alongside the known hypervariable regions within the 16S rRNA gene (Fig. 3C). Most were found to fall between hypervariable regions. Given that variability of each 16S rRNA base position can be described in terms of the frequency of the most common residue at that position (Fig. 3A), the overall median and 95% confidence interval notches of these frequencies were 0.931 ± 0.013. In contrast, the median of those frequencies corresponding to breakpoint positions was significantly higher at 0.975 ± 0.015.
DISCUSSION
It has long been recognized that corrupt sequences are present within the public repositories. What has not been known is how many there may be. Of the 19 phyla studied, 5% of records were found to be corrupt; most of these (78.6%) were chimeras or similarly insidious sequencing errors. Eleven of the 19 phyla investigated contained obvious chimeras with chimeric content, ranging from 0.8 to 11.8% of the total. Six phyla contained sequence anomalies presumably generated during sequencing. Five phyla contained records with highly degenerate sequences. In total, 16 of the 19 phyla considered contained some sort of substantial sequence anomaly.
Since the 19 selected phyla might not be representative of the full database, a separate analysis of the entire Bacteroidetes phylum was carried out. With 2,739 near-complete 16S rDNA sequences, this well-characterized taxon is the fourth-largest phylum currently within the RDP, with half of these records (50.1%) derived from uncultured sources. In all, 5.8% of the Bacteroidetes sequences were anomalous. Excluding degeneracy (7.5%), these anomalies were likely either chimeras (65.4%), assembly errors (13.2%), or other miscellaneous anomalies (13.8%).
Extrapolating these results to the public database as a whole this would suggest, at a conservative estimate, 1 in 20 sequences have substantial errors. We believe these figures underestimate the true number of anomalous records, given that we concentrated our efforts on uncovering the more obvious sequence anomalies.
This study confirms that anomalous sequences continue to be added to the public databases; of the chimeras identified in this study, 27.7% were submitted to the NCBI during 2004 alone (Fig. 8), and 91.5% of these were submitted in the last 5 years. These figures reflect recent interest in many of the phyla considered in this study and the steady yearly increase in sequence submissions generally. They also highlight the ongoing nature of the problem. Indeed, we noted five chimeric additions to the RDP database while our study progressed (two were added to Nitrospira, one was added to Verrucomicrobia, and two were added to the β-Proteobacteria, a taxon not otherwise investigated in this study).
FIG. 8.

First appearance in the NCBI database of the anomalous records identified by this study.
It is fair to say that many researchers have been insufficiently cognizant of the problem of sequence anomalies within the public databases. This situation is changing, however, as evidenced by the renewed burst of activity in generating software tools for recognizing chimeras. Within the last year or so, three new tools have been introduced (6, 7, 10), presumably driven by these authors' desire, like ours, to screen sequences generated through their own researches. Certainly, our experiences with chimeric sequences within 16S rRNA clone libraries led us to develop Pintail.
It is important that the extent of sequence anomalies within public repositories is fully realized. The research community's phylogenetic view of the bacterial world is increasingly informed by 16S rRNA information (3, 5, 15). At least half of the 53 phyla named in 2003 are currently known only from 16S rRNA gene sequences amplified from the environment by PCR (15), and this number is growing (4). It is notable that, of the six proposed new taxa analyzed in this study, four harbored chimeras, some of which were extreme. For example, a third of the OP11 sequence AY693838 derives from a β-proteobacterium. Another OP11 sequence, AY218572, is almost half an ɛ-proteobacterial. The 5′ end of WS3 bacterium AY592328 is from the Actinobacteria.
In all, 48.9% of identified chimeras were derived from bacteria belonging to different phyla (a particularly striking example being AJ289180, a jumble of Fusobacteria, Spirochaetes, and Bacteroidetes). This figure is undoubtedly an underestimate as, for a further 35.6%, either we could not identify the source (no suitable subject record in the database) or the source was as yet unclassified. Some of these chimeras were so extreme that it is surprising that they have not been detected before. We find this worrying, as our concern is that there are far more subtle chimeras in the database, constructed from close phylogenetic neighbors, that have less chance of being spotted and that could give rise to all sorts of spurious intrataxon clustering errors.
Our study also shows that a significant proportion of chimeras were generated from three fragments, often from three separate sources (consider AJ289180, above). Chimeras with more than three fragments may also be possible, since the positions of chimeric breakpoints in conserved regions suggested that there are several areas within the 16S rRNA gene where splicing may occur (Fig. 3C).
The methodology presented here depends on the type strain 16S rRNA database used. Clearly, current type strain sequences are not representative of all members of the Bacteria; our RDP-derived type strain database reflects past cultivation successes and there is a definite slant towards members of the Bacteria of medical interest. Furthermore, as this study shows, the quality of some type strain sequences is not good. Nevertheless, our method was effective over a wide phylogenetic range and could even be applied to Archaea sequences, as analysis of those archaeal chimeras listed in Hugenholtz and Huber's paper (8) proved. Since we used sequence alignments from the RDP database that currently only lists members of the Bacteria, our model and calibration data were constructed from members of this domain only. However, there is no theoretical reason why a more comprehensive model incorporating Archaea sequences could not be created or indeed generate models for specific domains, phyla, or other taxa to improve sensitivity. Note also that although this study concentrated on near-complete 16S rDNA sequence records, partial sequences can also be analyzed by Pintail in the same manner (although for very short partial sequences, a smaller sampling window will be necessary to give meaningful results).
DE values generated from type strain data, once anomalous sequences were removed, proved useful in calibrating our method; that is, placing observed DE values in the context of sequences identified as reliable. This raises the possibility of screening database records on a much larger scale than that tackled in this study. How should the research community tackle the problem of monitoring anomalous sequences in databases? Curators have a role to play. For example, we found three chimeras within the NCBI, labeled as such, yet not similarly flagged within the RDP database (although this is an understandable omission, given the RDP's automated nature). But the practicalities of current database management are such that the curators' contribution must be limited. Primary responsibility must and indeed should lie with researchers submitting sequences. To this end, software tools must be available and used by researchers to assist in screening PCR-generated sequences for anomalies before database deposition. Chimera_Check (13) and Bellerophon (7) are currently the programs most commonly used for detecting chimeric anomalies. Both require a database of sequences to be used, in addition to the query sequence, a requirement that can be both time consuming to prepare and prone to error. It is hoped that Pintail's simpler requirements, along with its user-friendly interface and its ability to run on all major computer platforms, will encourage greater screening of sequence data before and after submission to the public repositories. Unless chimeras and other anomalous sequences can be eliminated from public databases, microbial ecologists will have an erroneous picture of natural prokaryotic biodiversity.
Acknowledgments
This study was supported by grant BBS/B/11494 from the Biotechnology and Biological Sciences Research Council (BBSRC).
REFERENCES
- 1.Altschul, S., T. Madden, A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson, D. A., I. Karsch-Mizrachi, D. J. Lipman, J. Ostell, B. A. Rapp, and D. L. Wheeler. 2000. GenBank. Nucleic Acids Res. 28:15-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole, J., B. Chai, T. Marsh, R. Farris, Q. Wang, S. Kulum, S. Chandra, D. McGarrell, T. Schmidt, G. Garrity, and J. Tiedje. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox, J. L. 2005. Ribosomal gene milestone met, already left in dust. ASM News 71:6-7. [Google Scholar]
- 5.Garrity, G. M., M. Winters, A. W. Kuo, and D. Searles. 2002. Taxonomic outline of the prokaryotes, p. 49-66. Bergey's manual of systematic bacteriology, 2nd ed. Springer-Verlag, New York, N.Y.
- 6.Gonzalez, J. M., J. Zimmerman, and C. Saiz-Jimenez. 2005. Evaluating putative chimeric sequences from PCR-amplified products. Bioinformatics 21:333-337. [DOI] [PubMed] [Google Scholar]
- 7.Huber, T., G. Faulkner, and P. Hugenholtz. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317-2319. [DOI] [PubMed] [Google Scholar]
- 8.Hugenholtz, P., and T. Huber. 2003. Chimeric 16S rDNA sequences of diverse origin are accumulating in the public databases. Int. J. Syst. Evol. Microbiol. 53:289-293. [DOI] [PubMed] [Google Scholar]
- 9.Kanz, C., P. Aldebert, N. Althorpe, W. Baker, A. Baldwin, K. Bates, P. Browne, A. van den Broek, M. Castro, G. Cochrane, K. Duggan, R. Eberhardt, N. Faruque, J. Gamble, F. G. Diez, N. Harte, T. Kulikova, Q. Lin, V. Lombard, R. Lopez, R. Mancuso, M. McHale, F. Nardone, V. Silventoinen, S. Sobhany, P. Stoehr, M. A. Tuli, K. Tzouvara, R. Vaughan, D. Wu, W. Zhu, and R. Apweiler. 2005. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 33:D29-D33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klepac-Ceraj, V., M. Bahr, B. C. Crump, A. P. Teske, J. E. Hobbie, and M. F. Polz. 2004. High overall diversity and dominance of microdiverse relationships in salt marsh sulphate-reducing bacteria. Environ. Microbiol. 6:686-698. [DOI] [PubMed] [Google Scholar]
- 11.Komatsoulis, G. A., and M. S. Waterman. 1997. A new computational method for detection of chimeric 16S rRNA artifacts generated by PCR amplification from mixed bacterial populations. Appl. Environ. Microbiol. 63:2338-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopczynski, E. D., M. M. Bateson, and D. M. Ward. 1994. Recognition of chimeric small-subunit ribosomal DNAs composed of genes from uncultured microorganisms. Appl. Environ. Microbiol. 60:746-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maidak, B. L., J. R. Cole, T. G. Lilburn, C. T. Parker, Jr., P. R. Saxman, R. J. Farris, G. M. Garrity, G. J. Olsen, T. M. Schmidt, and J. M. Tiedje. 2001. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 29:173-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paabo, S., D. M. Irwin, and A. C. Wilson. 1990. DNA damage promotes jumping between templates during enzymatic amplification. J. Biol. Chem. 265:4718-4721. [PubMed] [Google Scholar]
- 15.Rappe, M. S., and S. J. Giovannoni. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369-394. [DOI] [PubMed] [Google Scholar]
- 16.Robison-Cox, J. F., M. M. Bateson, and D. M. Ward. 1995. Evaluation of nearest-neighbor methods for detection of chimeric small-subunit rRNA sequences. Appl. Environ. Microbiol. 61:1240-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shuldiner, A., A. Nirula, and J. Roth. 1989. Hybrid DNA artifact from PCR of closely related target sequences. Nucleic Acids Res. 17:4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatusova, T. A., and T. L. Madden. 1999. Blast 2 sequences—a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174:247-250. [DOI] [PubMed] [Google Scholar]
- 19.Thompson, J., D. Higgins, and T. Gibson. 1994. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, G. C.-Y., and Y. Wang. 1996. The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 142:1107-1114. [DOI] [PubMed] [Google Scholar]
- 21.Wang, G. C.-Y., and Y. Wang. 1997. Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl. Environ. Microbiol. 63:4645-4650. [DOI] [PMC free article] [PubMed] [Google Scholar]