

Abstract
Deletion of perR in Listeria monocytogenes results in a small-colony phenotype (ΔperRsm) that is slow growing and exhibits increased sensitivity to H2O2. At a relatively high frequency, large-colony variants (ΔperRlg) arise, which are more resistant to H2O2 than the wild-type and ultimately dominate the culture. Transcriptional analysis revealed that the kat gene (catalase) is up-regulated in both types of mutants and that the highest level is apparent in ΔperRsm mutants, demonstrating PerR regulation of this gene. Overexpression of the catalase gene in the wild-type background resulted in a slower-growing strain with a smaller colony size similar to that of ΔperRsm. By combining a bioinformatic approach with experimental evidence, other PerR-regulated genes were identified, including fur, lmo0641, fri, lmo1604, hemA, and trxB. The transcriptional profile of these genes in both mutant backgrounds was similar to that of catalase in that a higher level of expression was observed in ΔperRsm than in the wild type or ΔperRlg. Murine studies revealed that the virulence potential of the ΔperRsm mutant is substantially reduced compared to that of the wild-type and ΔperRlg strains. Collectively, the data demonstrate that the ΔperRsm mutant represents the true phenotype associated with the absence of PerR, which is linked to overexpression of regulated genes that negatively affect bacterial homeostasis both in vitro and in vivo. A subsequent secondary mutation occurred at a high frequency, which resulted in phenotypic reversion to a large-colony phenotype with increased fitness that may have obstructed the analysis of the role of PerR in the physiology of the bacterial cell.
Reactive oxygen species (ROS), such as hydroxyl radicals, superoxide anions (O2−), and hydrogen peroxide (H2O2), are an inevitable consequence of aerobic growth (35). If excessive amounts are present, these highly reactive compounds can damage DNA, proteins, and lipid membranes (26). Aerobic and facultative bacteria have evolved highly sophisticated inducible responses in order to survive these deleterious effects. The important ROS-inducible systems include superoxide dismutase, catalase (Kat), alkyl hydroperoxide reductase (AhpCF), and proteins required for protection and repair (22).
In aerobically growing cells, specific transcriptional regulators are required to activate defenses against the significant amounts of O2− and H2O2 which are generated (34). In Escherichia coli one system is mediated by OxyR, a peroxide-sensing regulator with a regulon which includes genes involved in peroxide metabolism and protection (including ahpCF, katG, and dps) (34). This regulator is widely distributed in most gram-negative and some gram-positive bacteria. However, in Bacillus subtilis and Staphylococcus aureus protection against ROS is mediated principally by PerR (17, 22, 24). The PerR regulon of B. subtilis has been well characterized and includes Kat, AhpCF, MrgA (DNA protection), the HemA operon (heme biosynthesis), and PerR itself (7, 8, 17, 35). These systems mount a response that protects cells when they are challenged with peroxide stress. Unlike OxyR, PerR is a metalloregulatory protein consisting of two metal-binding domains. The zinc ion has a structural role, with either Fe or Mn complexed in a regulatory role (23).
Interestingly, Fur (iron uptake and homeostasis) (2) and ZosA (zinc uptake during peroxide stress) (20) also form part of the PerR regulon in B. subtilis. Metal ion homeostasis can also have a significant impact on the effect of ROS. For example, unregulated uptake of iron, like that in a Fur mutant, can lead to the formation of toxic hydroxyl radicals (1, 40). As a result, metal ion uptake must be tightly regulated and is controlled by four main metalloregulatory proteins in B. subtilis, namely, Fur, PerR, Zur (zinc uptake and homeostasis), and MntR (manganese uptake and homeostasis) (25, 39).
Listeria monocytogenes is a gram-positive pathogen which can cause serious infections in susceptible individuals (47). The ability to respond to stress plays an important role in the virulence of this pathogen (5, 11, 33, 46). L. monocytogenes is extremely aerotolerant and hence must be able to resist the action of ROS. A homologue of each of the four main metalloregulators in B. subtilis has been identified in Listeria. Establishment of an infection requires tightly controlled gene regulation, and any disruption of the series of events may decrease the fitness and result in a less virulent strain (21, 42, 43). In a previous study we found that eliminating PerR resulted in a mutant which was more resistant to the action of H2O2 but was marginally less virulent than the wild type (42). We also observed that creation of the PerR mutant resulted in two phenotypically distinct colony types, one much smaller than the wild type and one whose size was equivalent to that of the wild type. In that study, we did not discriminate between colony types in further analyses, and, given the higher growth rates of the large-colony phenotype, it is likely that in this initial work we analyzed the phenotype of the large-colony mutant.
In the current study, we established that the small-colony phenotype represents the “true” phenotype associated with loss of PerR and that the large-colony revertant represents a subsequent, unidentified mutation. We also examined the consequences of PerR deletion for bacterial fitness and identified a number of PerR-regulated genes in Listeria. We analyzed the expression of these genes in the two mutant (small- and large-colony) backgrounds. The H2O2 sensitivity of both mutants was also examined because of the well-defined role of PerR in defense against peroxide stress. The ability of these mutants to establish an infection compared to the ability of the wild type was analyzed using a mouse model of infection. Overall, this work demonstrated that in contrast to the findings of previous reports, L. monocytogenes PerR mutants are actually less resistant to peroxide stress and that the ability to cause infection in the mouse model is dramatically affected.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used are listed in Table 1. E. coli strains were grown in Luria-Bertani medium, and L. monocytogenes strains were grown in brain heart infusion broth (BHI) (Oxoid) at 37°C. When appropriate, antibiotics (Sigma Chemical Company, St.Louis, Mo.) were used in L. monocytogenes cultures at the following concentrations: tetracycline, 10 μg/ml; and chloramphenicol, 10 μg/ml, unless otherwide stated. For solid media agar was added to a concentration of 1.5%. Nisin (Sigma Chemical Company, St.Louis, Mo.) was solubilized in dimethyl sulfoxide and acetic acid and was added to sterilized media.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Characteristics | Source or reference |
|---|---|---|
| Listeria monocytogenes strains | ||
| EGDe | Serotype 1/2a | W. Goebel |
| EGDe ΔperRsm | Wild type with a deletion in perR, small-colony phenotype | 42 |
| EGDe ΔperRlg | Wild type with a deletion in perR, phenotypically reverted to large-colony phenotype | 42 |
| EGDe-NICE | EGDe derivative with the nisRK genes integrated on the chromosome; expression host for nisin-induced expression system | Sheila Ryan |
| EGDe-NICE(pNZ8048) | EGDe-NICE containing the cloning vector pNZ8048 | This study |
| EGDe-NICE(pNZ8048-kat) | EGDe-NICE containing pNZ8048 expressing kat under the control of the nisin-induced promoter | This study |
| Plasmidsa | ||
| pNZ8048 | Cmr; nisin-inducible expression vector | 14 |
| pNZ8048-kat | pNZ8048 with kat under the control of the nisA promoter | This study |
| pPTPL | Tetr; promoter probe vector | 36 |
| pPTPL-kat | pPTPL with the kat promoter driving the expression of β-galactosidase | This study |
| pPL2 | Cmr; integrates at PSA phage attachment site within a tRNAArg gene on the chromosome | 29 |
| perR+lg | ΔperRlg with a full copy of the perR gene integrated at tRNAArg-attB | This study |
| perR+sm | ΔperRsm with a full copy of the perR gene integrated at tRNAArg-attB | This study |
| Primersb | ||
| KatF | TTACCGCGAATTCCGGGGCT | |
| KatR | ACAAACGGTCTAGAGAAGGA | |
| pKatF | CTAAGAGATCTCTTTTTATAAGC | |
| pKatR | CGTTAAATTTTTTCTAGATGT | |
| NZKatF | ATACCCATGGCAGATAGAAA | |
| NZKatR | ACTCCAACCTTCTAGACCAA | |
| CompF | CATTCGGATCCATATTTAGAGC | |
| CompR | CATATCACTGCAGCAACTATCT |
Tetr, tetracycline resistance; Cmr, chloramphenicol resistance.
Restriction sites incorporated into primer sequences are underlined.
DNA manipulation.
Gel extraction was performed using a QIAGEN gel extraction kit (QIAGEN). Plasmid DNA was isolated using a QIAGEN QIAprep Spin Miniprep kit (QIAGEN). T4 DNA ligase, PCR reagents, and restriction enzymes were purchased from Roche Diagnostics GmbH (Mannheim, Germany)and were used according to the manufacturer's instructions. PCRs were performed using a Hybaid (Middlesex, United Kingdom) PCR express system, and products were cloned into pPTPL and pNZ8048 and sequenced using plasmid-specific primers (Lark Technologies Inc., Essex, United Kingdom). Oligonucleotide primers (Table 1) for PCR were synthesized by Sigma-Genosys Biotechnologies.
Growth curves.
Overnight cultures were centrifuged (12,000 rpm for 5 min), washed, and resuspended in an equal volume of one-quarter-strength Ringer's solution (Merck). A 2% inoculum was added to 10 ml BHI. Two hundred microliters was added to a 96-well plate, and growth was determined automatically at 600 nm using a Spectra Max 340 spectrophotometer (Molecular Devices, Sunnyvale, Calif.) for 24 h at 37°C. Viability was assessed by plate counting. Overnight cultures prepared as described above were inoculated (2%) into 10 ml BHI. Bacterial counts were determined at this stage (time zero) and then every 3 h for 12 h. Briefly, 100 μl was removed, serially diluted, and plated onto BHI agar plates, which were incubated at 37°C overnight. The wild-type, ΔperRsm, and ΔperRlg strains were passaged for nine consecutive days. Initially, overnight cultures (2%) were inoculated into BHI. These cultures were passaged into fresh BHI (passage 1) on the following day and for the next 8 days. A sample was removed from each passage, serially diluted, and plated onto BHI agar plates for enumeration. Overnight cultures (2%) were also inoculated into BHI with nisin (45 μg nisin powder/ml), and growth was monitored automatically as described above.
Complementation of deletion mutants.
pPL2 was used for complementation of ΔperRlg and ΔperRsm. This plasmid is a site-specific phage integration vector which integrates within a tRNAArg gene on the chromosome. The entire perR gene and flanking regions were amplified from the L. monocytogenes EGDe genome using the proofreading enzyme Pwo DNA polymerase (Roche). Primers CompF and CompR were designed to incorporate BamHI and PstI restriction sites, respectively. The resulting PCR product was gel extracted, digested with the enzymes described above, and cloned into similarly digested vector pPL2. This construct was electroporated into DH5α, and transformants were selected using Luria-Bertani plates with 15 μg of chloramphenicol/ml. Plasmids were extracted using a QIAGEN QIAprep Spin Miniprep kit. The presence of the correct insert was confirmed by sequencing (Lark) using the T3 and T7 primers. Plasmid preparations were ethanol precipitated and resuspended in 5 μl (final volume) of elution buffer (10 mM Tris-Cl, pH 8.5). This preparation was electroporated into competent ΔperRlg and ΔperRsm cells, and transformants were selected on BHI agar plates containing 7.5 μg of chloramphenicol/ml. pPL2 with no insert was transformed into mutant cells as a control. Integration at the correct location on the chromosome was confirmed by PCR using a primer that annealed 5′ of the integration site and a primer that annealed to the cloned insert.
Hydrogen peroxide sensitivity assays.
Overnight cultures were inoculated into fresh BHI and grown to the early log phase (optical density at 600 nm [OD600], ∼0.20). Cells were harvested and washed with one-quarter-strength Ringer's solution. Hydrogen peroxide was added to BHI to a final concentration of 50 mM (bactericidal), and 1 ml was used to resuspend the pellets. Samples were taken every 30 min for 2 h, and survivors were determined by dilution in Ringer's solution and plating onto BHI agar. Overnight cultures (2%) were also inoculated into BHI with 22 mM H2O2 (bacteriostatic), and growth was monitored automatically as described above. For the disk assay overnight cultures were inoculated into fresh BHI and grown to the early log phase (OD600, ∼0.2). Four hundred microliters was added to 4 ml of sloppy agar (0.75%), poured onto 20 ml of BHI agar (1.5%), and left to solidify. A sterile blank disk was placed in the center of the agar, to which 20 μl of 33% hydrogen peroxide was added. The plates were incubated at 37°C overnight.
Catalase activity assay.
Strains grown statically overnight at 37°C were resuspended in 1/20 the original culture volume and lysed using a mini bead beater. Debris was pelleted by centrifugation at 12,000 × g at 4°C for 10 min. The total protein concentration was determined by the method of Lowry et al. (32) following protein precipitation with 10% trichloroacetic acid. Catalase activity was determined using the method described by Beers and Sizer (4). Briefly, 0.5 ml of H2O2 (59 mM H2O2 freshly diluted in 50 mM potassium phosphate buffer, pH 7.0) was added to 1 ml of the crude extract, and the absorbance at 240 nm was measured every 15 s for 1 min. The specific activity of catalase (expressed in micromoles of H2O2 decomposed per minute per milligram of total protein) was calculated as follows:
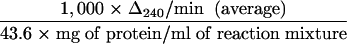 |
RNA extraction.
RNA was extracted from log- and stationary-phase cells using the Macaloid method (41). Briefly, cells were spun and lysed using a combination of glass beads, 10% sodium dodecyl sulfate, and a mini bead beater. RNA was isolated from the cellular debris using Macaloid clay and phenol-chloroform (Sigma Chemical Company, St. Louis, Mo.). The RNA was purified further by extraction with phenol-chloroform and chloroform-isoamyl alcohol (Sigma Chemical Company, St. Louis, Mo.). When necessary, the RNA was treated with DNase using a DNA free kit (Ambion). The absence of contaminating DNA was confirmed using control primers. For overexpression studies RNA was isolated from EGDe-NICE(pNZ8048-kat) and EGDe-NICE(pNZ8048) (control) following nisin induction. Cultures were grown to an OD600 of approximately 0.2 and preinduced with 4.5 μg of nisin powder/ml for 1 h. This was followed by induction with 45 μg of nisin powder/ml for 30 min. RNA was isolated at this stage.
Transcriptional analysis.
The RNA was converted to cDNA using Expand reverse transcriptase and random primer p(dN)6 (Roche). Kat-specific primers were used to analyze the cDNA, and samples were taken at regular intervals and electrophoresed on agarose gels. Primers for the rrnA gene (15) were used as controls to ensure that equal-intensity cDNA was added to all PCR mixtures. The promoter of the Kat gene was amplified using pKatF (incorporating a BglII site) and pKatR (incorporating an XbaI site). The resulting PCR product was digested with the BglII and XbaI restriction enzymes and cloned into similarly digested pPTPL. Electrotransformation of L. monocytogenes with wild-type, ΔperRsm, and ΔperRlg backgrounds with this construct was performed by using protocols outlined by Park and Stewart (38). β-Galactosidase assays were performed as described by Miller, with modifications described by Israelsen et al. (27). Briefly, cells were spun and resuspended in LacZ buffer. Cells were lysed by adding chloroform and 0.1% sodium dodecyl sulfate and were incubated at 37°C. The reaction was started by addition of o-nitrophenyl-β-d-galactopyranoside (Sigma Chemical Company, St. Louis, Mo.). Upon development of a yellow color the reaction was stopped using NaCO3. β-Galactosidase activity was calculated using spectrophotometric data.
Overexpression of catalase.
The catalase gene was amplified from L. monocytogenes EGDe using NZKatF (incorporating the Kat start codon and an NcoI site) and NZKatR (incorporating the Kat stop codon and an XbaI site). The resultant PCR product was digested with the NcoI and XbaI restriction enzymes and cloned into similarly digested pNZ8048. This resulted in generation of a fusion between the nisin-inducible nisA promoter on pNZ8048 and the kat gene. This event was confirmed by sequence analysis. The construct was electroporated into a wild-type strain carrying a copy of the nisRK genes (EGDe-NICE) on thechromosome, which generated EGDe-NICE(pNZ8048-kat). As a control, pNZ8048 was also introduced into this wild-type background to generate EGDe-NICE(pNZ8048).
Bioinformatics.
A number of potential PerR-regulated genes were identified using the search pattern function on the listilist homepage (http://genolist.pasteur.fr/listilist/). The PerR box (target DNA sequence to which PerR has been shown to bind in vitro) from B. subtilis (16) was used to search the Listeria genome. Searches were restricted to within 350 bp upstream of the start codon. Members of the PerR regulon in B. subtilis are likely candidates and were considered regardless of the number of mismatches with the consensus sequence. Subsequent reverse transcription (RT)-PCR analysis could confirm if identified genes were PerR regulated.
Virulence assay.
Overnight cultures were centrifuged, washed once with phosphate-buffered saline (PBS), resuspended, and subsequently diluted in PBS. In vivo survival was determined by inoculating 8- to 12-week old BALB/c mice intraperitoneally with 3 × 105 CFU in 200 μl PBS. The mice were euthanized on the appropriate day postinfection, and bacteria were harvested from the spleen or liver by homogenization of organs in PBS, serial dilution of the organ homogenates on BHI agar, and incubation overnight at 37°C.
RESULTS
Physiological analysis of mutants.
In a previous study we observed that disruption (using pORI19 plasmid integration) or deletion (splicing by overlap extension procedure [SOEing]) of perR resulted in a strain that produced much smaller colonies on BHI agar than the wild type (42). However, interspersed among the small colonies were larger colonies whose sizes were similar to those of the wild type (Fig. 1A). Subsequent PCR analysis confirmed that there was a perR deletion in both colony types, suggesting that the larger colonies did not represent genotypic revertants or contamination with wild-type cells (Fig. 1B).
FIG. 1.

(A) Deletion of the perR gene by the SOEing procedure in Listeria resulted in two colony sizes. (B) Confirmation of the deletion event in both colony types by PCR using primers located outside the deleted region. A wild-type (wt) colony was used as a control. (C) Growth of the wild type, the ΔperRsm mutant, and the ΔperRlg mutant in BHI at 37°C as determined by measuring the OD600. (D) Growth of the wild type, the ΔperRsm mutant, and the ΔperRlg mutant in BHI at 37°C as determined by plate counting. The error bars indicate the standard deviations from the means of triplicate experiments.
The growth of the ΔperRsm and ΔperRlg mutants in BHI (pH 7.0) at 37°C was compared to that of the wild type (Fig. 1C). Under these conditions no difference in either the exponential or stationary phase was detected for the ΔperRlg mutant. However, a significant growth defect was observed for the ΔperRsm mutant; this mutant had a lower growth rate, and the final numbers of cells were lower. In order to determine which mutant represents the “primary” PerR mutant, we selected a single colony of each type and grew it in BHI for 9 days (Fig. 2). The colony sizes of the wild type and the ΔperRlg mutant remained constant throughout the experiment. However, the OD valves and plate counts for the ΔperRsm mutant were lower than those for either the wild type or the ΔperRlg mutant after 24 h, and only small colonies were recovered. However, on subsequent days the appearance of wild-type-sized colonies was observed with increasing frequency. By day 6 more than 80% of the colonies were large (ΔperRlg), and the large-colony variants remained the dominant population in subsequent transfers. The original PerR deletion event was confirmed in these colonies, suggesting that the ΔperRlg phenotype is a result of a secondary mutation which alleviates the growth defect associated with the initial PerR mutation event.
FIG. 2.

Wild type, ΔperRsm mutant, and ΔperRlg mutant were passaged for nine consecutive days in BHI at 37°C. Data for the wild type and the ΔperRlg mutant are not shown for purposes of clarity. While the graph represents one experiment, the experiment was repeated two more times, and similar results were obtained.
Hydrogen peroxide sensitivity assay.
Increased resistance to peroxide stress has been reported to be associated with the loss of PerR in a number of bacteria (6, 8, 24, 28, 43). Indeed, in a previous study we found that plasmid disruption of perR in L.monocytogenes resulted in a strain that was able to respond to peroxide stress better than the wild type. This was later confirmed with a deletion mutant (42). However, in hindsight, we did not discriminate between the original small-colony mutant and the large-colony revertant in that study. To resolve this problem, we compared the abilities of the wild type, a ΔperRlg mutant, and a ΔperRsm mutant to respond to hydrogen peroxide. At 50 mM H2O2, the levels of both the wild type and the ΔperRsm mutant declined significantly over a 90-min period (Fig. 3A). The decline associated with the ΔperRsm mutant was much more rapid than that associated with the wild type, indicating that there was increased sensitivity to H2O2. However, not only was the ΔperRlg mutant able to tolerate this level of H2O2, but some growth was observed (Fig. 3A). Furthermore, at 22 mM H2O2, which is inhibitory for the wild type, the ΔperRlg mutant was capable of growth, albeit with an extended lag phase (Fig. 3B). The ΔperRsm mutant was not capable of growth under these conditions, confirming that it is more sensitive to peroxide stress than the ΔperRlg mutant. The H2O2 responses of the two mutants compared to the wild type were also assessed using a disk assay. The zones of inhibition observed for the ΔperRsm mutant were larger than those observed for both the wild type and the ΔperRlg mutant, indicating increased sensitivity to this stress. For the ΔperRlg mutant the zones of inhibition were smaller than the zones of inhibition for the wild type, indicating that there was increased H2O2 resistance. Interestingly, reintroduction of a complete copy of the perR gene using the pPL2 plasmid restored the H2O2 response of both mutants to wild-type levels, which confirmed the phenotypes of these mutants (Fig. 3C). Thus, the increased H2O2 tolerance reported previously to be representative of a Δper mutant (42) did not reflect the true phenotype; rather, it reflected the phenotype of the ΔperRlg revertant, which was associated with a secondary mutation. We concluded that the true ΔperR phenotype is actually that of theΔperRsm mutant (i.e., increased H2O2 sensitivity).
FIG. 3.

(A) Effect of 50 mM hydrogen peroxide on EGDe (▪) and the ΔperRlg (•) and ΔperRsm (▵) mutants. The cells were in the exponential growth phase. The error bars indicate the standard deviations from the means of triplicate experiments. (B) Growth of L.monocytogenes EGDe (wild type) (▪) and the ΔperRlg (•) and ΔperRsm (▵) mutants in BHI containing 22 mM hydrogen peroxide. (C) Resistance of the wild type (Wt), the ΔperRlg mutant, the ΔperRsm mutant, and complemented strains to H2O2, as assessed using a disk assay. (D) Levels of catalase activity in the wild-type, ΔperRlg, and ΔperRsm strains. The catalase specific activities are expressed in micromoles of H2O2 decomposed per minute per milligram of total protein. Standard deviations were calculated from the means (n = 2). Similar results were obtained in two independent experiments.
Transcriptional analysis.
One of the principal differences between the ΔperRsm and ΔperRlg mutants is in their abilities to respond to H2O2 stress. Since catalase is the principal detoxifier of H2O2, we examined the expression of the kat gene in both mutants using RT-PCR and promoter analysis. RNA was extracted from aerobically grown wild-type, ΔperRsm, and ΔperRlg cells in both the log and stationary phases. Interestingly, expression of the gene was significantly up-regulated in both mutants compared to the wild type, and the highest level was apparent in the ΔperRsm mutant. The results were confirmed using real-time PCR analysis (data not shown). This finding was obtained with both log-phase cells (Fig. 4A) and stationary-phase cells (Fig. 4B).
FIG. 4.

RT-PCR analysis of gene expression. Control primers were used to ensure that the concentrations of template cDNAs were equal. Samples were removed at various PCR cycles and visualized on agarose gels. Transcription data were confirmed by promoter studies. Levels of β-galactosidase (βGAL) activity were calculated under the conditions used for RNA extraction. (A) Log-phase cells. (B) Stationary-phase cells. Wt, wild type.
These results were subsequently confirmed using promoter studies. The kat promoter regions from the wild type and both mutants were cloned into the promoter probe vector pPTPL and reintroduced into the appropriate background for further analysis. β-Galactosidase activity was measured for all three strains under the conditions used for RNA extraction. The results confirmed that kat expression was elevated in both mutant backgrounds in log-phase cells (Fig. 4A) and stationary-phase cells (Fig. 4B). As with the RT-PCR results, a greater degree of up-regulation was observed in the ΔperRsm strain. The kat promoter region was sequenced in order to identify any changes which might explain this altered expression, but no differences were identified. Subsequent sequencing of the entire kat gene in all three strains did not reveal any alteration, suggesting that the cause of this change in expression lies at a different locus.
Overexpression of catalase.
To determine whether the increased expression of kat observed in the ΔperRsm mutant could be responsible for the small-colony phenotype and the increased peroxide sensitivity, kat was cloned into the expression plasmid pNZ8048, which placed the gene under control of the nisin-inducible Pnis promoter using the NICE (nisin-controlled expression) system (12, 14). RT-PCR analysis was used to confirm that in the presence of the nisin inducer EGDe-NICE containing pNZ8048-kat had substantially higher levels of catalase than the control EGDe-NICE strain containing pNZ8048 with no insert (Fig. 5A). It was observed that EGDe-NICE(pNZ8048-kat) had a slightly slower growth rate than the control in the presence of nisin (Fig. 5B). This growth defect was similar to that seen with the ΔperRsm mutant. Induction with nisin also led to smaller colonies on BHI agar plates for EGDe-NICE(pNZ8048-kat) after 24 h (Fig. 5C). However, unlike those of the ΔperRsm mutant, these colonies eventually reached the wild-type size. The control strain always produced wild-type-sized colonies. We also found that overexpression of catalase led to slightly decreased resistance to H2O2 compared to the control, as determined by a disk diffusion assay (data not shown). Therefore, many, although not all, of the phenotypes associated with the ΔperRsm mutation can be attributed to the increased expression of catalase.
FIG. 5.

(A) RT-PCR analysis of gene expression. Control primers were used to ensure that the concentrations of template cDNAs were equal. Samples were removed at various PCR cycles and visualized on agarose gels. (B) Growth of EGDe-NICE(pNZ8048) and EGDe-NICE(pNZ8048-kat) in BHI with 45 μg nisin powder/ml at 37°C. The error bars indicate the standard deviations from the means of triplicate experiments. (C) Growth of EGDe-NICE(pNZ8048) (left panel) and EGDe-NICE(pNZ8048-kat) (right panel) on BHI agar with 45 μg nisin powder/ml.
Bioinformatic analysis of the PerR regulon.
We were interested in determining whether the up-regulation of kat was unique to this gene or whether there was also up-regulation of other members of the PerR regulon. We used the consensus PerR box (TTATAATnATTATAA) defined in B. subtilis (16) to search the L. monocytogenes EGDe genome for potential members of the PerR regulon. This led to identification of a large number of candidates. Only one gene, fur, was identified as having a PerR box identical to that in the B. subtilis consensus sequence. Fourteen sequences with two mismatches were identified, and many genes were likely candidates for regulation by PerR. However, allowing two and three mismatches to the consensus sequences resulted in 206 and 1,862 hits, respectively, in the Listeria genome. Many sequences could be eliminated based on the position of the PerR box, but the remainder required further bioinformatic analysis. For the purposes of this study we considered only the genes already defined as members of the PerR regulon in Bacillus.
(i) Catalase.
The previously characterized kat (30) gene in Listeria exhibits 49% identity with katA of Bacillus. A PerR box with one mismatch to the consensus sequence was identified 53 bp upstream of the annotated start codon (Fig. 6).
FIG. 6.

(A) Alignment of PerR boxes (boldface type) from putative PerR-regulated genes. Boxes were identified by searching the L. monocytogenes EGDe genome sequence using the PerR consensus sequence from B. subtilis. The numbers in parentheses indicate the numbers of mismatches with the consensus sequence (underlined). (B) RT-PCR analysis was used to confirm PerR regulation of all identified genes from log-phase cultures. Analysis of kat expression was included as a control. Primers for a housekeeping gene were used to ensure that the concentrations of template cDNAs were equal. Samples were removed at various PCR cycles and visualized on agarose gels. Wt, wild type.
(ii) Alkyl hydroperoxide reductase.
ahpC and ahpF encode the two subunits of alkyl hydroperoxide reductase and are cotranscribed in Bacillus. In Listeria the closest homolog of ahpC (32% identity) is lmo1604 (similar to 2-Cys peroiredoxin). A PerR box with one mismatch was identified 99 bp upstream of the annotated start codon (Fig. 6). However, there is no adjacent ahpF homolog in Listeria, and the closest homolog, trxB (thioredoxin reductase; 27% identity), is located some distance away at a separate locus. In addition to ahpF Bacillus also has a trxB gene which exhibits 73% homology with trxB in Listeria. This gene was not found to be PerR regulated in Bacillus. However, we included this gene due to the absence of ahpC and ahpF in Listeria and the presence of a PerR box, albeit with four mismatches 320 bp upstream of the annotated start codon (Fig. 6).
(iii) Ferric uptake regulator.
fur in Listeria exhibits 76% identity with the corresponding gene in Bacillus. A PerR box with a perfect match to the consensus sequence was identified 91 bp upstream of the annotated start codon (Fig. 6).
(iv) ZosA.
The closest homolog of the P-type ATPase in Listeria is lmo0641 (54% identity). A PerR box with two mismatches to the consensus sequence was identified 102 bp upstream of the annotated start codon (Fig. 6).
(v) Metalloregulation DNA-binding stress protein (MrgA). The closest homolog of the metalloregulation DNA-binding stress protein (MrgA) gene in Listeria is the nonheme iron-binding ferritin gene (fri). This gene has been shown to be a stress- and starvation-induced gene controlled by sigma-B (37). mrgA exhibits 35% identity with fri. A PerR box with two mismatches to the consensus sequence was identified 50 bp upstream of the annotated start codon (Fig. 6).
(vi) HemAXCDBL.
The six-gene HemAXCDBL operon is involved in heme biosynthesis in Bacillus. There is a similar operon in Listeria, but it contains only five genes. The closest homologue of hemX in Listeria is located at a separate locus. hemA exhibits 47% identity with the corresponding gene in Listeria. However, no PerR box wasidentified upstream of the annotated start codon in L.monocytogenes.
The expression of all of the genes described above was analyzed in the two mutant backgrounds. As stated above, the kat gene was found to be up-regulated in both the ΔperRlg and ΔperRsm mutants compared to the wild type, but a greater degree of up-regulation was observed in the ΔperRsm mutant. The same phenomenon was also noted for many of the other genes identified as members of the PerR regulon in L. monocytogenes. fur, fri, lmo0641, and lmo1604 all displayed expression patterns similar to that of catalase. trxB and hemA were found to be up-regulated in the ΔperRsm mutant compared to the wild type. However, the levels of these genes were comparable in the wild-type and ΔperRlg backgrounds.
Virulence studies.
In a previous study we identified a role for PerR in the pathogenesis of Listeria (42). However, we did not discriminate between the two colony types, and the perR mutant exhibited a 10-fold reduction in splenic numbers compared to the wild type on day 3 of infection. In the current study we compared the virulence of ΔperRlg and ΔperRsm mutants and the virulence of the wild type using the intraperitoneal mouse model of infection (Fig. 7). The numbers of bacteria in the spleens of infected mice were determined after 3 days. The levels of both perR mutants were lower than those of the wild type in the spleens of infected animals (P < 0.05); however, the ΔperRsm mutant was most significantly affected. The numbers of cells of this mutant recovered from the spleens were 100-fold less than the numbers of wild-type cells. Repeat experiments verified that ΔperRsm mutants were isolated at significantly lower levels than the wild type and ΔperRlg mutants (data not shown). Therefore, the secondary mutation resulting in the large colonies is beneficial for Listeria in vivo. However, this mutation does not fully restore virulence to wild-type levels, and the rate of appearance of large colonies from the ΔperRsm mutant in vivo did not differ from the rate encountered in complex broth. These results demonstrate that a loss of PerR activity severely reduces virulence and confirm that the perR gene plays a significant role in the virulence of Listeria.
FIG. 7.

Effect of deletion of the perR gene on survival of L. monocytogenes EGDe in vivo. Mice were inoculated intraperitoneally with either the wild type or the mutants, and the numbers of bacteria recovered from the spleen were determined 3 days postinoculation. The error bars indicate the standard deviations from the means (n = 4).An asterisk indicates that the mean is significantly different from the wild-type (Wt) mean (P < 0.05).
DISCUSSION
The ferric uptake repressor (Fur) superfamily is composed of three DNA-binding metalloregulatory proteins that act as repressors of gene expression (Fur, Zur, and PerR). Zur is responsible for zinc uptake and homeostasis (13, 18, 19), whereas Fur is responsible for balancing the intracellular concentration of iron to maintain appropriate levels for normal growth (2). PerR regulates a number of genes which play a critical role in defense against peroxide stress and ROS, and it responds to either Fe or Mn ions (20, 23). In this study we examined the physiological and genetic consequences of deleting PerR and determined the effects on the ability of Listeria to cause disease.
We determined that deletion of perR results in two distinct colony types, small colonies (ΔperRsm) and colonies that are similar in size to the wild type yet maintain the perR deletion (ΔperRlg) (42). Initial observations only showed that these colonies differ in size, as the texture, hemolytic activity, color, and motility were the same for both colony types. Physiological analyses further separated these two colony types since the ΔperRsm mutant grew at a slower rate than either the wild type or the ΔperRlg mutant. Slower growth rates of a B. subtilis perR mutant have been reported previously (8, 10, 22). Repeated passaging of the ΔperRsm mutant gave rise to larger colonies that were similar in size to the wild type (ΔperRlg), which ultimately dominated the culture. Therefore, deletion of the perR gene in Listeria initially resulted in a strain which produced smaller colonies, had a decreased growth rate, and exhibited increased sensitivity to peroxide stress. Subsequently, larger colonies with increased resistance to peroxide stress appeared, whose growth rate was similar to that of the wild type. Although the genetic change responsible for these larger colonies has not been elucidated (see below), it restored some of the lost fitness to the bacterium. Significantly, previous work with B. subtilis also identified two colony sizes following perR deletion (10). This study did not identify the precise genetic change that resulted in this phenomenon. However, the authors did determine that the larger colonies lacked catalase activity. Clearly, it is important to comprehend the true nature of the phenotype associated with perR deletion in order to understand the significance of this regulon for in vitro and in vivo growth and to inform future transcriptomic and proteomic analyses.
One of the principal physiological differences between the ΔperRsm and ΔperRlg mutants is the response to H2O2 stress. Transcriptional analysis (RT-PCR and promoter studies) revealed that there was up-regulation of catalase in both backgrounds compared to the wild type. Surprisingly, there were significantly higher levels of the transcript in the ΔperRsm mutant, even though it was less resistant to H2O2 stress than the ΔperRlg mutant. Therefore, we postulated that creation of a perR mutant results in an increase in catalase to levels that are somehow toxic to the cell, resulting in smaller colonies and an inability to resist peroxide stress. Indeed, in support of this hypothesis, we determined that significant overexpression of catalase in wild-type L. monocytogenes resulted in a small-colony phenotype and decreased resistance to H2O2. It is interesting that many previous reports have suggested that deletion of PerR in gram-positive bacteria results in increased resistance to H2O2 (6, 8, 24, 28, 43). We concluded here that this is not the true phenotype associated with a PerR mutation in L. monocytogenes and that care must be taken when ΔperR colonies are selected for further study. Interestingly, deletion of PerR in Synechocystis sp. resulted in a strain which responded to H2O2 stress in a manner similar to the wild type (31). The increased H2O2 sensitivity of a ΔperRsm mutant may be due to the higher levels of Fur present in this mutant. Fur is required to derepress iron uptake genes under iron limitation conditions. Iron is cofactor for many important enzymes in the cell, including catalase. The increased level of Fur may prevent derepression of iron uptake genes, resulting in iron starvation. Any remaining iron within the cell is incorporated only into essential proteins. This may hinder the ability of the catalase enzyme to work efficiently.
It is clear from our work that a subsequent genetic alteration gives rise to large colonies (ΔperRlg) from the small-colony background. The large-colony mutants exhibit a relative reduction in levels of kat expression, alleviation of the growth defect, and increased resistance to H2O2. We speculated that this phenomenon might result from secondary mutation of the kat locus. Sequence analysis revealed that no base changes had occurred in the kat promoter or structural gene in either ΔperRsm or ΔperRlg mutants, suggesting that the genetic events that give rise to increased fitness in a ΔperRlg mutant reside outside the kat locus, perhaps in another, as-yet-unidentified regulator of catalase expression.
Indeed, increased transcription in the ΔperRsm mutant background appears to extend beyond the kat locus to affect other PerR-regulated genes. We used a bioinformatic approach (3) to identify a number of genes as putative members of the PerR regulon and examined their expression in both mutant backgrounds. The identification of PerR-regulated genes in L.monocytogenes adds to the recent report of a PerR-regulated iron-binding protein (Fri) in this pathogen (37). The PerR-regulated genes identified in this study are also part of the PerR regulon in B. subtilis and include kat and ahpCF (defense against peroxide stress), mrgA (protection of DNA), hemAXCDBL(heme biosynthesis), zosA (zinc uptake), and fur (regulation of iron homeostasis) (19, 23). It was significant that the expression of these genes in the two ΔperR mutants followed a pattern similar to that displayed by kat. Most of the genes were up-regulated in both backgrounds, and a greater increase in expression was observed in the ΔperRsm mutant. It is interesting that the bioinformatic search also identified a number of putative regulators as likely candidates for regulation by PerR. Strong matches with the consensus sequence were identified upstream of the start codons of lmo0416, lmo0106, lmo2146, and lmo2165. It is possible that in the absence of PerR one of these regulators results in the altered gene expression observed in a ΔperRlg mutant. However, this requires further investigation.
Studies with S. aureus have shown that PerR is an essential regulator that is required for virulence in a murine subcutaneous skin abscess model of infection (24). We previously demonstrated a role for PerR in the infectious cycle of Listeria using, in hindsight, what we now know to have been a ΔperRlg mutant (42). That study demonstrated that a moderate (10-fold) reduction in virulence potential was associated with the perR deletion. In the current study the ΔperRsm mutant, which we believe represents the true ΔperR phenotype, exhibited a significant and substantial reduction in virulence compared to both the wild type (100-fold reduction) and the ΔperRlgmutant (10-fold reduction). This shows that environmental adaptation through PerR is essential for complete virulence in L. monocytogenes and that the true phenotype may be underrepresented in clones that have developed a secondary mutation (ΔperRlg). It is likely that the ΔperRsm mutant examined in this study differs from the general small-colony variants that have been isolated from infections caused by a variety of gram-negative and gram-positive organisms (9, 44, 45, 48, 49). These small-colony variants actually have a greater capacity to persist intracellularly than the wild type, whereas the ΔperRsm mutant has a greatly reduced virulence potential.
In conclusion, deletion of the gene encoding the PerR repressor results in increased expression of its regulon, probably at such a level that it decreases the fitness of the cell, resulting in a slower growth rate and increased sensitivity to peroxide stress. This is, at least in part, due to the increase in catalase activity in the cell. This imposes selection pressure on the cell, which results in a secondary, unidentified mutation, which results in down-regulation of the PerR regulon to a level which restores fitness but is still greater than the wild-type level of expression. This is an important finding that should influence the design of future experiments to examine the nature of PerR-dependent gene regulation in Listeria spp. and related organisms.
Acknowledgments
We acknowledge the financial assistance of the Irish Government under National Development Plan 2000-2006 and funding of the Alimentary Pharmabiotic Centre by the Science Foundation of Ireland Centres for Science Engineering and Technology scheme.
REFERENCES
- 1.Andrews, S. C., A. K. Robinson, and F. Rodríguez-Quiñones. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215-237. [DOI] [PubMed] [Google Scholar]
- 2.Bagg, A., and J. B. Neilands. 1987. Ferric uptake regulation protein acts as a repressor, employing iron(II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 26:5471-5477. [DOI] [PubMed] [Google Scholar]
- 3.Baichoo, N., T. Wang, R. Ye, and J. D. Helmann. 2002. Global analysis of the Bacillus subtilis Fur regulon and the iron starvation stimulon. Mol. Microbiol. 45:1613-1629. [DOI] [PubMed] [Google Scholar]
- 4.Beers, R. F., Jr., and I. W. Sizer. 1951. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 196:133-140. [PubMed] [Google Scholar]
- 5.Begley, M., R. D. Sleator, C. G. M. Gahan, and C. Hill. 2005. Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect. Immun. 73:894-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenot, A., K. Y. King, and M. G. Caparon. 2004. The PerR regulon in peroxide resistance and virulence of Streptococcus pyogenes. Mol. Microbiol. 55:221-234. [DOI] [PubMed] [Google Scholar]
- 7.Bsat, N., L. Chen, and J. D. Helmann. 1996. Mutation of the Bacillus subtilis alkyl hydroperoxide reductase (ahpCF) operon reveals compensatory interactions among hydrogen peroxide stress genes. J. Bacteriol. 178:6579-6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bsat, N., A. Herbig, L. Casillas-Martinez, P. Setlow, and J. D. Helmann. 1998. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol. Microbiol. 29:189-198. [DOI] [PubMed] [Google Scholar]
- 9.Cano, A. D., M. Graciela Pucciarelli, M. Martínez-Moya, J. Casadesús, and F. García-del Portillo. 2003. Selection of small-colony variants of Salmonella enterica serovar Typhimurium in nonphagocytic eukaryotic cells. Infect. Immun. 71:3690-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casillas-Martinez, L., A. Driks, B. Setlow, and P. Setlow. 2000. Lack of a significant role for the perR regulator in Bacillus subtilis spore resistance. FEMS Microbiol. Lett. 188:203-208. [DOI] [PubMed] [Google Scholar]
- 11.Cotter, P. D., N. Emerson, C. G. M. Gahan, and C. Hill. 1999. Identification and disruption of lisRK, a genetic locus encoding a two-component signal transduction system involved in stress tolerance and virulence in Listeria monocytogenes. J. Bacteriol. 181:6840-6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cotter, P. D., C. M. Guinane, and C. Hill. 2002. The LisRK signal transduction system determines the sensitivity of Listeria monocytogenes to nisin and cephalosporins. Antimicrob. Agents Chemother. 46:2784-2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalet, K., E. Gouin, Y. Cenatiempo, P. Cossart, and Y. Hechard. 1999. Characterisation of a new operon encoding a Zur-like protein and an associated ABC zinc permease in Listeria monocytogenes. FEMS Microbiol. Lett. 174:111-116. [DOI] [PubMed] [Google Scholar]
- 14.de Ruyter, P. G., O. P. Kuipers, and W. M. de Vos. 1996. Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl. Environ. Microbiol. 62:3662-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dussurget, O., D. Cabanes, P. Dehoux, M. Lecuit, C. Buchrieser, P. Glaser, P. Cossart and the European Listeria Genome Consortium. 2002. Listeria monoytogenes bile salt hydrolase is a PrfA-regulated virulence factor involved in the intestinal and hepatic phases of listeriosis. Mol. Microbiol. 45:1095-1106. [DOI] [PubMed] [Google Scholar]
- 16.Fuangthong, M., and J. D. Helmann. 2003. Recognition of DNA by three ferric uptake regulator (Fur) homologs in Bacillus subtilis. J. Bacteriol. 185:6348-6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuangthong, M., A. F. Herbig, N. Bsat, and J. D. Helmann. 2002. Regulation of the Bacillus subtilis fur and perR genes by PerR: not all members of the PerR regulon are peroxide inducible. J. Bacteriol. 184:3276-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaballa, A., and J. D. Helmann. 1998. Identification of a zinc-specific metalloregulatory protein, Zur, controlling zinc transport operons in Bacillus subtilis. J. Bacteriol. 180:5815-5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaballa, A., T. Wang, R. W. Ye, and J. D. Helmann. 2002. Functional analysis of the Bacillus subtilis Zur regulon. J. Bacteriol. 184:6508-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaballa, A., and J. D. Helmann. 2002. A peroxide-induced zinc uptake system plays an important role in protection against oxidative stress in Bacillus subtilis. Mol. Microbiol. 45:997-1005. [DOI] [PubMed] [Google Scholar]
- 21.Goodman, A. L., B. Kulasekara, A. Rietsch, D. Boyd, R. S. Smith, and S. Lory. 2004. A signalling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 5:745-754. [DOI] [PubMed] [Google Scholar]
- 22.Helmann, J. D., M. F. W. Wu, A. Gaballa, P. A. Kobel, M. M. Morshedi, P. Fawcett, and C. Paddon. 2003. The global transcriptional response of Bacillussubtilis to peroxide stress is coordinated by three transcription factors. J. Bacteriol. 185:243-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbig, A. F., and J. D. Helmann. 2001. Roles of metal ions and hydrogen peroxide in modulating the interaction of the Bacillus subtilis PerR peroxide regulon repressor with operator DNA. Mol. Microbiol. 41:849-859. [DOI] [PubMed] [Google Scholar]
- 24.Horsburgh, M. J., M. O. Clements, H. Crossley, E. Ingham, and S. J. Foster. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 69:3744-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horsburgh, M. J., S. J. Wharton, A. G. Cox, E. Ingham, S. Peacock, and S. J. Foster. 2002. MntR modulates expression of the PerR regulon and superoxide resistance in Staphylococcus aureus through control of manganese uptake. Mol. Microbiol. 44:1269-1286. [DOI] [PubMed] [Google Scholar]
- 26.Imlay, J. A. 2002. How oxygen damages microbes: oxygen tolerance and obligate anaerobiosis. Adv. Microb. Physiol. 46:111-153. [DOI] [PubMed] [Google Scholar]
- 27.Israelsen, H., S. M. Madsen, A. Vrang, E. B. Hansen, and E. Johansen. 1995. Cloning and partial characterization of regulated promoters from Lactococcus lactis Tn917-lacZ integrants with the new promoter probe vector pAK80. Appl. Environ. Microbiol. 61:2540-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King, K. Y., A. Joshua, and M. G. Caparon. 2000. Aerotolerance and peroxide resistance in peroxidase and perR mutants of Streptococcus pyogenes. J. Bacteriol. 182:5290-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lauer, P., M. Y. Chow, M. J. Loessner, D. A. Portnoy, and R. Calender. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184:4177-4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leblond-Francillard, M., J.-L. Gaillard, and P. Berche. 1989. Loss of catalase activity in Tn1545-induced mutants does not reduce growth of Listeria monocytogenes in vivo. Infect. Immun. 57:2569-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, H., A. K. Singh, L. M. McIntyre, and L. A. Sherman. 2004. Differential gene expression in response to hydrogen peroxide and the putative PerR regulon of Synechocystis sp. strain PCC 6803. J. Bacteriol. 186:3331-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 33.Marron, L., N. Emerson, C. G. M. Gahan, and C. Hill. 1997. A mutant of Listeria monocytogenes LO28 unable to induce an acid tolerance response displays diminished virulence in a murine model. Appl. Environ. Microbiol. 63:4945-4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mongkolsuk, S., and J. D. Helmann. 2002. Regulation of inducible peroxide stress responses. Mol. Microbiol. 45:9-15. [DOI] [PubMed] [Google Scholar]
- 35.Mostertz, J., C. Scharf, M. Hecker, and G. Homuth. 2004. Transcriptome and proteome analysis of Bacillus subtilis gene expression in response to superoxide and peroxide stress. Microbiology 150:497-512. [DOI] [PubMed] [Google Scholar]
- 36.O'Driscoll, J., F. Glynn, O. Cahalane, M. O'Connell-Motherway, G. F. Fitzgerald, and D. Van Sinderen. 2004. Lactococcal plasmid pNP40 encodes a novel, temperature-sensitive restriction-modification system. Appl. Environ. Microbiol. 70:5546-5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsen, K. N., M. H. Larsen, C. G. M. Gahan, B. Kallipolitis, X. A. Wolf, R. Rea, C. Hill, and H. Ingmer. 2005. The Dps-like protein Fri of Listeria monocytogenes promotes stress tolerance and intracellular multiplication in macrophage-like cells. Microbiology 151:925-933. [DOI] [PubMed] [Google Scholar]
- 38.Park, S. F., and G. S. Stewart. 1990. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94:129-132. [DOI] [PubMed] [Google Scholar]
- 39.Que, Q., and J. D. Helmann. 2000. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol. Microbiol. 35:1454-1468. [DOI] [PubMed] [Google Scholar]
- 40.Ratledge, C., and L. G. Dover. 2000. Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 54:881-941. [DOI] [PubMed] [Google Scholar]
- 41.Raya, R., J. Bardowski, P. S. Andersen, S. D. Ehrlich, and A. Chopin. 1998. Multiple transcriptional control of the Lactococcus lactis trp operon. J. Bacteriol. 180:3174-3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rea, R. B., C. G. M. Gahan, and C. Hill. 2004. Disruption of putative regulatory loci in Listeria monocytogenes demonstrates a significant role for Fur and PerR in virulence. Infect. Immun. 72:717-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricci, S., R. Janulczyk, and L. Björck. 2002. The regulator PerR is involved in oxidative stress response and iron homeostasis and is necessary for full virulence of Streptococcus pyogenes. Infect. Immun. 70:4968-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Martínez, A., A. F. Kelly, S. F. Park, and B. M. Mackey. 2004. Emergence of variants with altered survival in stationary phase cultures of Campylobacter jejuni. Int. J. Food Microbiol. 90:321-329. [DOI] [PubMed] [Google Scholar]
- 45.Roggenkamp, A., A. Sing, M. Hornef, U. Brunner, I. B. Autenrieth, and J. Heesemann. 1998. Chronic prosthetic hip infection caused by a small-colony variant of Escherichia coli. J. Clin. Microbiol. 36:2530-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rouquette, C., M. T. Ripio, E. Pellegrini, J. M. Bolla, R. I. Tasion, J. A. Vasquez-Boland, and P. Berche. 1996. Identification of a ClpC ATPase required for stress tolerance and in vivo survival of Listeria monocytogenes. Mol. Microbiol. 21:977-987. [DOI] [PubMed] [Google Scholar]
- 47.Vázquez-Boland, J. A., M. Khun, P. Berche, T. Chakraborty, G. Domínguez-Bernal, W. Goebel, B. González-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Eiff, C., R. A. Procters, and G. Peters. 2000. Staphylococcus aureus small colony variants: formation and clinical impact. Int. J. Clin. Pract. 115:44-49. [PubMed] [Google Scholar]
- 49.von Götz, F., S. Häussler, D. Jordan, S. S. Saravanamuthu, D. Wehmhöner, A. Strübmann, J. Lauber, I. Attree, J. Buer, B. Tümmler, and I. Steinmetz. 2004. Expression analysis of a highly adherent and cytotoxic small-colony variant of Pseudomonas aeruginosa isolated from a lung of a patient with cystic fibrosis. J. Bacteriol. 186:3837-3847. [DOI] [PMC free article] [PubMed] [Google Scholar]