

Abstract
The goal of this field study was to provide insight into three distinct populations of microorganisms involved in in situ metabolism of phenol. Our approach measured 13CO2 respired from [13C]phenol and stable isotope probing (SIP) of soil DNA at an agricultural field site. Traditionally, SIP-based investigations have been subject to the uncertainties posed by carbon cross-feeding. By altering our field-based, substrate-dosing methodologies, experiments were designed to look beyond primary degraders to detect trophically related populations in the food chain. Using gas chromatography-mass spectrometry (GC/MS), it was shown that 13C-labeled biomass, derived from primary phenol degraders in soil, was a suitable growth substrate for other members of the soil microbial community. Next, three dosing regimes were designed to examine active members of the microbial community involved in phenol metabolism in situ: (i) 1 dose of [13C]phenol, (ii) 11 daily doses of unlabeled phenol followed by 1 dose of [13C]phenol, and (iii) 12 daily doses of [13C]phenol. GC/MS analysis demonstrated that prior exposure to phenol boosted 13CO2 evolution by a factor of 10. Furthermore, imaging of 13C-treated soil using secondary ion mass spectrometry (SIMS) verified that individual bacteria incorporated 13C into their biomass. PCR amplification and 16S rRNA gene sequencing of 13C-labeled soil DNA from the 3 dosing regimes revealed three distinct clone libraries: (i) unenriched, primary phenol degraders were most diverse, consisting of α-, β-, and γ-proteobacteria and high-G+C-content gram-positive bacteria, (ii) enriched primary phenol degraders were dominated by members of the genera Kocuria and Staphylococcus, and (iii) trophically related (carbon cross-feeders) were dominated by members of the genus Pseudomonas. These data show that SIP has the potential to document population shifts caused by substrate preexposure and to follow the flow of carbon through terrestrial microbial food chains.
Documentation of in situ biogeochemical processes and discovery of microbial populations responsible for such processes are long-standing challenges for microbial ecologists (27). Stable isotope probing (SIP) is a procedure that has led to recent progress in this area. The approach incorporates a stable isotope (e.g., 13C) into cellular biomarkers of organisms actively involved in metabolism of a 13C-labeled substrate (39). Biomarkers that have been used in these types of studies include phospholipid fatty acids, DNA, and RNA (6, 28, 29, 36, 37). Following 13C enrichment in nucleic acid-based studies, density gradient ultracentrifugation, and a series of molecular methods (PCR, molecular cloning, terminal restriction fragment length polymorphism [T-RFLP], and sequencing) are employed to separate and analyze the isotopically labeled biomarker, thus providing insight into the microbial populations actively involved in substrate-specific metabolism.
One of the limitations to SIP has been the ambiguities posed by carbon cross-feeding effects (37, 38, 39, 44). In addition to biomarker labeling of targeted primary degraders, the stable isotope can be incorporated indirectly into the biomass of trophically related microorganisms (28). This can occur either by uptake of labeled metabolites released by primary degraders or by microbial scavenging of labeled biomass. The uncertainties associated with carbon cross-feeding increase as incubation time with the labeled substrate increases. In some cases, SIP studies have involved incubation times of >40 days (33, 37). Longer incubations increase the potential for the isotopic label to be passed down the food chain into the biomass of nontarget organisms.
The investigative strategy developed in this study sought to differentiate between enriched and unenriched primary degraders of phenol and trophically related populations. Phenol was selected as a model organic chemical pollutant due to its ubiquitous distribution in the environment and its documented biodegradability (28, 35, 41, 44). By modifying the field-based DNA-SIP methodologies developed by Padmanabhan et al. (35), with key variables being substrate isotope and prior exposure to the substrate, we were able to gain insight into three distinct populations involved in phenol degradation in situ: (i) unenriched primary degraders, (ii) enriched primary degraders, and (iii) trophically related organisms (carbon cross-feeders).
MATERIALS AND METHODS
Chemicals and standards.
Uniformly labeled [13C6]phenol was purchased from Isotec (Miamisburg, OH). Phenol was purchased from Fisher Scientific (Fair Lawn, NJ). A carbon dioxide standard (1.0%) was purchased from Scott Specialty Gases (Plumsteadville, PA). High-purity helium was supplied by Airgas (Elmira, NY).
Field study site.
This study was conducted at the Cornell University Agricultural Experiment Station, Ithaca, NY. The soil plot (Collamer silt loam) was level and free of vegetation. A table was placed over the plot (0.8 m high) to protect the experiment from rain and direct exposure to sunlight.
Carbon cross-feeding field assay.
One gram of Collamer silt loam was added to 5 ml phosphate-buffered solution (1.44 g Na2HPO4, 0.24 g KH2PO4, 0.2 g KCl, 8 g NaCl [per liter], pH 7.4), vortexed, and allowed to settle for ∼0.5 min. The upper suspension (1 ml) was added as an inoculum to 1 liter minimal salts medium (42) with 2.5 mM [13C6]phenol and shaken at room temperature for 14 days. A similar culture was prepared using unlabeled phenol. High-performance liquid chromatography (HPLC) analysis was used to confirm phenol metabolism in the cell cultures (see below). The cells were pelleted, washed three times with phosphate-buffered saline, resuspended in 1 ml deionized water, and autoclaved. The resulting 13C- and 12C-labeled cell preparations were mixed vigorously, and 100 μl was added dropwise to the center of a 5.2-cm2 circle of soil (in triplicate) in the field as previously described (35). Septum-fitted stainless steel chambers (20) were inserted into the soil, enclosing the dosed surface. Using gastight syringes (Hamilton, Reno, NV) 100 μl of the 14.6 cm3 headspace was removed, shuttled immediately to the laboratory (∼0.5 km), and analyzed within 30 min by gas chromatography-mass spectrometry (GC/MS) for both 13CO2 and 12CO2, as previously described (35). Total carbon in the cell preparations was determined using a ThermoQuest Italia S.p.A. EA/NA 1110 automated elemental analyzer (Milan, Italy) operated by the Department of Crop and Soil Science Analytical Laboratory, Cornell University.
HPLC analysis of phenol.
Phenol was analyzed by HPLC. Samples (1.0 ml) of culture medium were collected at various time points, immediately diluted with an equal volume of methanol, sealed, and stored at 4°C until analyzed. Samples were filtered through nylon Acrodisc filters (0.2 μm, 25-mm syringe filter; Gelman, Ann Arbor, MI). Phenol was separated using a Varian Microsorb-MV 100-5 C18 HPLC column (250 by 4.6 mm). A Waters model 590 HPLC pump was used to pump a mobile phase of methanol-40 mM acetic acid (60:40) at a flow rate of 1.0 ml/min. Eluents were monitored by UV-visible light detection (ABI analytical absorbance detector, Spectroflow 757) at a wavelength of 270 nm and quantified using external standard calibration curves.
Soil field treatments.
Five soil treatments (Table 1) were designed to probe three distinct communities of phenol degraders in situ. Key variables for each treatment were the carbon isotope of phenol (unlabeled [12C] or 13C) and the number of daily doses (0 or 11) prior to a final dose of phenol. Each 20-μl dose contained 200 μg of phenol. Immediately after the final dose of [13C]phenol, the plots were covered with septum-fitted chambers, followed by headspace analysis, as described above.
TABLE 1.
Five soil treatments applied to field plots that varied the type (13C-labeled or unlabeled) of phenol and number of doses to soil
| Treatment designation | No. of prior doses | Prior isotope | Final isotope | 13C probed microbial population |
|---|---|---|---|---|
| N/12 | 0 | None | 12C | None |
| N/13 | 0 | None | 13C | Unenriched primary phenol degraders |
| 12/12 | 11 | 12C | 12C | None |
| 12/13 | 11 | 12C | 13C | Enriched primary phenol degraders |
| 13/13 | 11 | 13C | 13C | Mixed, trophically related (13C cross-feeders) |
GC/MS analysis of CO2.
A Hewlett-Packard HP5890 gas chromatograph (Wilmington, DE) equipped with an HP5971A mass-selective detector was used for the respiration analyses. With high-purity helium as the carrier gas, a Hewlett-Packard Pora Plot Q column (25 m by 0.32 mm, 10-μm film thickness) was used to separate carbon dioxide from other gaseous components. The detector was operated at an electron energy of 70 eV and a detector voltage of 2,000 V. The ion source pressure was maintained at 1 × 10−5 torr. A splitless injection was used, and the GC oven was isothermal at 60°C. CO2 eluted at 2.5 min. Single-ion monitoring allowed simultaneous quantification of both 12CO2 (m/z = 44) and 13CO2 (m/z = 45). The concentration of 13CO2 was quantified using calibration curves prepared using external standards (Scott Specialty Gases, Plumsteadville, PA). The net 13CO2 produced from metabolism of the [13C]phenol was calculated by subtracting background 13CO2 produced by the native microbial community from soil organic matter. Background 13CO2 was inferred from direct measurement of 12CO2 adjusted to the known fixed ratio of 12C to 13C in naturally occurring carbon (1.11%) (17, 35). This ratio was confirmed analytically. Net 13CO2 values from replicate chambers were averaged at each time point and compared with Student's t tests.
SIMS imaging of soil.
One-tenth of a gram of surface soil was aseptically collected from the field treatments receiving 12 doses of [13C]phenol and unlabeled phenol. The soil was fixed in 4% formalin (1 ml) and stored in screw-cap glass vials. Soil smears were prepared after dilution (1:50) in filter-sterilized deionized water by spreading 1 μl onto sterile, clean high-purity silicon wafers (∼1 cm2; Silicon Quest International, Inc.) supported by a glass microscope slide. After air drying and heat fixation by passing rapidly over a flame, secondary ion mass spectrometric (SIMS) analysis was performed. A CAMECA IMS-3f SIMS ion microscope (Paris, France) operated with a positive oxygen beam was used, and negative secondary masses were monitored in the imaging mode for the detection of 12C, 13C, 12C14N, and 13C14N signals as previously described (10). SIMS images were recorded on a charge-coupled device camera and digitized to 14 bits per pixel (Photomatrix, Tucson, AZ). Images were processed using a Macintosh computer and DIP Station image processing software (Hayden Image Processing, Inc.).
DNA extraction and isopycnic fractionation of 13C-DNA.
After headspace sampling had been completed (∼30 h), the chambers were removed from the soil, keeping a 2-cm-thick, intact soil core in the bottom. These were immediately transported to the laboratory. Using a sterile spatula, approximately 0.125 g was removed from the upper 1-mm layer of soil. Four replicates from each of the five treatments were pooled to a final weight of 0.5 g. Such composite samples, commonly used by soil scientists, minimize the potential influence of spatial heterogeneity. DNA extraction was carried out using the Fast DNASPIN kit with a bead-beating procedure (Qbiogene, Carlsbad, CA).
As positive controls for 13C- and 12C-DNA, Pseudomonas putida strain G7 and Bacillus subtilis were grown in two mineral salts media: one with 0.4% [13C6]glucose and one with 0.4% unlabeled glucose. DNA was extracted as described above. One hundred microliters of both the heavy (13C labeled) and light (12C labeled) DNA solutions from both P. putida and B. subtilis were combined and brought to a final volume of 1 ml with TE buffer (10 mM Tris-1 mM EDTA, pH 8). To examine the influence of G+C content of DNA on its migration during ultracentrifugation, we compared band locations of the pseudomonad (62% G+C) with Bacillus subtilis (43% G+C).
One milliliter of the DNA solution from the standard and each field treatment was diluted to 4.5 ml with TE buffer, and 4.5 g CsCl was added and shaken gently until dissolved. Ethidium bromide (100 μl, 10 mg/ml) was added to each ultracentrifuge tube, which was then sealed. Tubes were centrifuged at 140,000 × g (41,900 rpm; Vti 80 rotor) for 66 h at 20°C (20, 35). Resultant bands in the standard were clearly separated. The 13C band in field treatments was not visible; we used the standard to guide DNA removal. Using an 18-gauge needle to puncture ∼2 mm below each band, 0.5 ml of CsCl solution containing DNA was withdrawn. Ethidium bromide was extracted from the DNA by the addition of 10 volumes of TE-saturated 1-butanol and gentle mixing. The organic layer was discarded, and the extraction was repeated five times. The DNA was brought to a final volume of 3 ml in TE. DNA precipitation occurred overnight at −20°C by addition of 300 μl of 3 M sodium acetate (pH 4.6) and a 2× volume of ethanol. After pelleting at 13,000 to 15,000 × g for 30 min, the DNA was washed twice with 70% ethanol, centrifuged at the same speed for 10 min, resuspended in 100 μl of distilled water, and stored at −20°C.
PCR cloning, restriction digestion, and sequencing.
PCR amplification of 16S rRNA genes in the 13C-DNA fraction from soil used universal eubacterial primers (27f and 1492r) by methods described previously (4, 20, 35). Cloning from the 13C-DNA derived from [13C]phenol-treated soil only proceeded when the corresponding band from [12C]phenol-treated soil failed to yield a PCR amplicon. The product was ligated into the vector pCR2.1 (TA cloning; Invitrogen) by following the manufacturer's recommended protocol. Following transformation of plasmids into host cells and blue/white screening, colonies with inserts were verified by PCR with vector-specific primers (5′-GTAACGGCCGCCAGTGTGCT and 5′-CAGTGTGATGGATATCTGCA) that flanked the cloning region. The amplicons were digested with HaeIII and HhaI. RFLP patterns were analyzed on 3% MetaPhore agarose gels (BioWhittaker; Molecular Applications, Rockland, Maine) with a 100-bp ladder (Promega) as a marker. Clones containing unique RFLP patterns were selected for sequencing, grown overnight in 5 ml of Luria-Bertani broth with kanamycin (50 μg/μl), and pelleted, and plasmids were purified (QiaPrep spin miniprep kit; QIAGEN, Santa Clarita, Calif.). Sequencing (Cornell University DNA Sequencing Facility) was conducted with 6 primers: M13 forward (5′-TGTAAAACGACGGCCAGT-3′), M13 reverse (5′-AACAGCTATGACCATG-3′), 1114 forward (5′-GCAACGAGCGCAACCC-3′), 907 reverse (5′-CCGTCAATTCATTTGAGTTT-3′), 531 reverse (5′-TACCGCGGCTGCTGGCAC-3′), and 533 forward (5′-GTGCCAGCMGCCGCGG-3′). Raw sequence data from both strands were assembled into full-length sequences with at least 2× coverage using the program SEQMAN II (DNASTAR,Inc.). After assembly, the consensus sequence was verified manually by referring to the corresponding ABI chromatograms of the sequencing reaction mixtures. The computational tools of the Ribosomal Database II project (http://rdp@cme.msu.edu) were used to check chimeras and to calculate the similarity values for individual rRNA gene sequences by using the sequence_match program. A BLAST search (http://ncbi.nlm.nih.gov/BLAST) was also used to identify the additional related sequences. The closest relatives identified from both searches were included in dendrograms.
Nucleotide sequence accession numbers.
The nucleotide sequence data reported here have been submitted to GenBank under accession no. DQ158099 to 158132.
RESULTS
We set out to demonstrate that 13C-labeled biomass, derived from primary phenol degraders in soil, is a suitable growth substrate for other members of the soil microbial community. To accomplish this, a site-derived soil inoculum was used to prepare two mixed cultures of phenol degraders. These were grown in the laboratory in minimal salts medium supplemented with either [13C]phenol or unlabeled (12C) phenol as the sole carbon source. After a 14-day incubation, HPLC analysis showed that all initial phenol was below detection (data not shown). The resulting labeled and unlabeled cells were harvested, washed three times, and autoclaved, and the resulting cell preparations were used as the substrate in a field respiration assay. Net 13CO2 over the background was measured by GC/MS over the course of 24 h (Fig. 1). Treatments with 13C-labeled biomass as the added carbon source produced a significant increase in net 13CO2, while treatments with unlabeled cell substrate displayed no detectable increase in net 13CO2 production. The mass of labeled carbon recovered as 13CO2 amounted to 21% of the added labeled substrate within 10 h (Fig. 1). This confirmed that microorganisms native to our field site can rapidly metabolize labeled biomass derived from primary degraders of phenol. Thus, carbon in added phenol can be expected to flow through the microbial community.
FIG. 1.

Evolution of 13CO2 from unlabeled (12C) and 13C-labeled biomass added to field soil plots. GC/MS analysis monitored both 12CO2 and 13CO2 concentrations. Net 13CO2 reflects total 13CO2 minus inferred background 13CO2 (35). The percentage in parentheses shows the maximum proportion of the total added [13C]carbon recovered as 13CO2. Data points are the averages of the results from three replicate treatments. Error bars indicate standard deviations. The field chambers were not sealed beneath the soil surface. The late drop in 13CO2 concentration was caused by diffusion of gas into the soil.
In our prior field-based SIP studies (20, 35), our goal was to identify members of the soil community responsible for initial catabolism of the added 13C-labeled substrate. Thus, in prior studies, we intentionally minimized the time interval between substrate dosing, 13CO2 determination, and DNA extraction and analysis. In the experimental design implemented here, the three treatments sought to identify three distinct microbial populations involved in in situ metabolism of phenol (Table 1). In one of these treatments (N/13), soil microorganisms in the field plots received no prior exposure to the substrate but simply received one dose of [13C]phenol on day 12. A net increase in 13CO2 production was observed in the headspace of the chamber covering the soil. This amounted to 1.3% of the added labeled carbon within 24 h (Fig. 2). Respiration in soil plots in control treatments receiving a single dose of unlabeled (12C) phenol produced no net increase in 13CO2 over the background, as expected. The second key treatment (12/13) received 11 doses of [12C]phenol prior to a final [13C]phenol dose. This dosing regime undoubtedly caused an in situ enrichment of phenol degraders before the 13C label was introduced to the system. The resulting respiration data (Fig. 2) showed that prior exposure to the substrate boosted 13CO2 production by a factor of 10, as 18% of the added 13C label was recovered as 13CO2. The third key treatment (13/13) received 11 prior doses of [13C]phenol prior to a final dose of the labeled substrate. While this treatment allowed for enrichment of phenol degraders, it also delivered (in repeated pulses) a substantial mass of [13C]phenol to the microbial community. We hypothesize that this delivery regimen allowed the [13C]carbon to pass through the food chain into the biomass of nondegraders of phenol. As expected, the net 13CO2 produced was greatest in the 13/13 treatment because of the cumulative dose of 13C. The respective control treatment 12/12, which received multiple doses of unlabeled phenol, showed no net increase in 13CO2 over the background (Fig. 2).
FIG. 2.
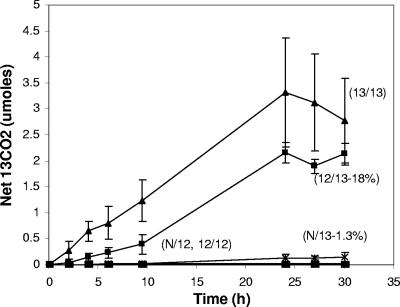
Evolution of 13CO2 from [13C]phenol added in the field to 5 experimental treatments (N/12, N/13, 12/12, 12/13, and 13/13) (Table 1). N/12 and 12/12 were control treatments receiving only unlabeled (12C)phenol. Net 13CO2 reflects total 13CO2 minus inferred background 13CO2. The percentages in parentheses in the N/13 and 12/13 treatment populations show the proportions of the total added [13C]carbon recovered as 13CO2. Data points are the averages of the results from three replicate treatments. Error bars indicate standard deviations.
To microscopically verify that the soil microbial community grew in situ in the treatment receiving [13C]phenol, samples were fixed in formalin and diluted, and then the soil smear was analyzed using SIMS. The instrument was adjusted to focus on 4 key masses: 12C and 13C (whose emissivity and, hence, sensitivity and resolution are relatively low) and 12C14N and 13C14N (whose emissivity and, hence, sensitivity and resolution are relatively high). The four images on the left of Fig. 3 (panels a to d) were derived from the treatment that received 12 doses of [12C]phenol in the field plots. Only the mass 26 signal, indicative of 12C-labeled bacteria growing on native soil carbon or [12C]phenol, showed strong signals from individual cells above background levels of ∼1% 13C. However, when the same analyses were performed on the soil smear from the treatment receiving 12 doses of [13C]phenol, very strong signals were detected from mass 27, indicative of 13C in combination with 14N (Fig. 3h). Clearly, the 13C-treated cells showed enrichment of 13C and brighter signals for 13C14N (see arrows in Fig. 3 comparing 13C and 13C14N images of the same cells). Digital image analysis of ratios of 13C and 12C signals in individual cells revealed 10- to 40-fold enhancement of 13C signals in the 13C treatment. As expected, signals from unlabeled cells (mass 26) were also detected in the [13C]phenol treatment, and the unlabeled cells were both more numerous and largely distinctive from those found in [12C]phenol-treated soil.
FIG. 3.

Dynamic SIMS ion microscopic images of soil bacteria from 12/12 and 13/13 field treatments receiving unlabeled phenol (a to d) and [13C]phenol (e to h), respectively. The upper row of images shows signals from 12C and 13C. The lower row shows higher resolution, high-emissivity images combining signals from 12C and 13C with 14N (see the text).
Following the field respiration and SIMS assays, total DNA was extracted from each of the five soil treatments (N/12, N/13, 12/12, 12/13, 13/13) (Table 1) and density gradient ultracentrifugation was performed to separate the heavy (13C enriched) from light (unenriched or 12C) DNA. Although only the 12C-DNA band was visible, the 13C-DNA band's location was determined using known 13C-DNA standards (35) prepared from P. putida and B. subtilis. In these, the difference in G+C content (19%, B. subtilis versus P. putida) caused a splitting of the 12C- and 13C-DNA bands by 6 mm. Furthermore, the high-percent G+C 12C-DNA from P. putida (lower member of the light pair) was separated from the low-percent G+C 13C-DNA from B. subtilis (upper member of the heavy pair) by 5 mm. Thus, despite potential complications posed by DNA composition, we feel that the separation of heavy and light DNA fractions was adequate. As found in previous studies (13, 20, 35, 40), when PCR primers designed to amplify the 16S rRNA gene were applied to DNA recovered from the location in the CsCl gradient that corresponded with a 13C-DNA standard, a robust amplicon was obtained from the [13C]phenol-treated soils but not from the corresponding dilution from the [12C]phenol-treated soils (Fig. 4). These results provided confidence that 16S rRNA gene sequences amplified and cloned from heavy DNA fractions reveal the identity of populations involved in [13C]phenol metabolism. Following cloning of the 16S rRNA gene amplicons, 100 white colonies from each [13C]phenol-dosed treatment were screened for the properly sized (1,500 bp) insert. Transformants potentially containing the 16S rRNA gene insert were screened by RFLP, and those with unique RFLP patterns were sequenced (Table 2). After chimeras and other sequences of questionable quality were discarded, a dendrogram was constructed from 34 soil-derived sequences and 11 reference sequences (Fig. 5). Sequences from the N/13 treatment (those which received no prior exposure to phenol) were scattered throughout the dendrogram. These were diverse, containing representatives from the α-, β-, and γ-proteobacteria and high-G+C-content gram-positive bacteria. These findings support the RFLP data which showed that 34 of the 38 clones analyzed produced unique RFLP patterns.
FIG. 4.

PCR amplification of 16S rRNA genes in 13C-DNA fractions from 12/12 (lanes 1 to 3), 12/13 (lanes 4 to 6), and 13/13 (lanes 7 to 9) treatments. Each cluster of 3 lanes shows amplicons from 102-, 103-, and 104-fold DNA dilutions. Lane − was a negative PCR control using sterile water. Lane + was a positive PCR control using DNA obtained from the 12C band of the 12/12 treatment. Lane M was a 1-kb ladder. Note that amplicons were only obtained from treatments receiving [13C]phenol. Lack of amplification at low dilutions can be attributed to soil-derived inhibitors of the PCR.
TABLE 2.
Description and status of 16S rRNA gene clone libraries obtained from the three soil treatments that received [13C]phenol
| Parameter | Result for soil treatment:
|
||
|---|---|---|---|
| N/13 | 12/13 | 13/13 | |
| Initial no. of clones picked | 100 | 100 | 100 |
| No. of clones containing ∼1,500 bp insert | 38 | 86 | 100 |
| No. of unique RFLPs | 34 | 11 | 9 |
| No. sequenced | 31 | 29 | 24 |
| No. of chimeras/questionable sequences | 24 | 9 | 10 |
| No. of sequences entered in Fig. 4 dendrogram | 7 | 13 | 14 |
FIG. 5.

Phylogenetic analysis of clone libraries derived from three soil treatments: N/13, 12/13, and 13/13 (see treatment designations in Table 1). 16S rRNA genes were cloned from the soil-derived 13C-DNA. Clones were screened by RFLP, and 84 clones were sequenced. After chimeras and other sequences of questionable quality were discarded, 34 sequences were aligned and phylogenetic relationships were completed using the computational tools of the Ribosomal Database II Project (http://rdp.cme.msu.edu). Percentages are percentages of clones analyzed with identical RFLPs for a given treatment (e.g., 70% of the 12/13 clones had identical RFLPs and are most closely identified with the genus Kocuria). Sequences from the N/13 treatment are distributed broadly throughout the tree.
Interestingly, sequences from the 12/13 and 13/13 treatments (which received multiple doses of phenol) were far less diverse than those found from the N/13 treatment. Furthermore, the clones from the 12/13 and 13/13 treatments clustered in distinct clades on the dendrogram (Fig. 5). Sixty of the 86 clones from the 12/13 treatment yielded identical RFLP patterns; these had high sequence similarity (99.5%) with Kocuria kristinae. Another 21 clones from the same treatment also produced a common RFLP pattern and had high sequence similarity (99.8%) with a member of the genus Staphylococcus. The 13/13 treatment yielded sequences that grouped in a distinct cluster on the dendrogram, one with high similarity values (99.9%) to the genus Pseudomonas. Low diversity and distinctiveness in the 16S rRNA gene sequences found in the 13/13 treatment were expected, as RFLP analysis prior to cloning found that 70 of the 100 clones were virtually identical.
DISCUSSION
SIP is a means of ecological inquiry that has been applied to both model systems (7, 15, 16, 19, 28, 32, 37, 38, 40) and to field sites (5, 20, 23, 31, 35, 36). The goal of SIP is to identify which microbial populations within complex communities are responsible for a probed process. Like all measurement procedures in microbial ecology, data must be interpreted with an awareness of potential pitfalls and artifacts. Results of SIP studies may suffer from ambiguities posed by (i) the addition of substrates at unrealistic concentrations, (ii) transfer of the labeled atom(s) through food chains, and (iii) isotopic fractionation during metabolism. The first two points were addressed experimentally in the present study and are discussed below. We feel the latter point is a negligible concern when highly isotopically enriched substrates are utilized. Carbon isotope ratios in bacterial biomass have been found to generally match the isotopic ratios of their food sources (1, 2, 5, 8, 11, 12, 43).
This investigation sought to identify three distinct groups of microorganisms involved in phenol degradation at an agricultural field site: (i) unenriched, primary degraders, (ii) enriched primary degraders, and (iii) trophically related organisms (carbon cross-feeders). The field-based assay, established by Padmanabhan et al. (35), was employed to minimize experimental artifacts that may develop during laboratory incubations. The first group (unenriched, primary degraders [N/13]) (Table 1) received only one dose of labeled phenol. The brief exposure (30 h) to the labeled substrate was designed to minimize carbon cross-feeding and reveal the 16S rRNA gene sequences of organisms directly involved in phenol metabolism prior to any enrichment. This treatment identified a relatively diverse group of microorganisms (Fig. 5), containing representatives from the α-, β-, and γ-proteobacteria and high-G+C-content gram-positive bacteria. Given the reduced cloning efficiency of the DNA recovered from the N/13 treatment (Table2), the diversity of sequences representing the unenriched, primary phenol degraders was probably conservative. To our knowledge, two other studies to date have used SIP to identify microorganisms actively involved in phenol degradation. Using DNA-SIP, Padmanabhan et al. identified Pseudomonas, Acinetobacter, and Variovorax spp. as active phenol degraders at the same field site as the current study (35). Manefield et al. used RNA-SIP to show that a member of the genus Thauera dominated phenol degradation within a bioreactor (28).
The second group of populations (enriched, primary degraders [12/13]) (Table 1) probed in this study received the same amount of labeled substrate as the N/13 treatment. Multiple daily doses of unlabeled phenol, prior to the respiration assay, resulted in an in situ enrichment of phenol-metabolizing organisms. The 10-fold increase in net 13CO2 over the background (Fig. 2) compared to the N/13 treatment confirmed that soil populations had been enhanced because substrate metabolism occurred at a faster rate. Because the exposure time to the single dose of 13C-substrate was identical to that of the N/13 treatment, we again are reasonably confident that minimal carbon cross-feeding of 13C atoms occurred. The 13C-enriched 16S rRNA genes obtained from this treatment represented a much less diverse group of bacteria than the N/13 treatment. Low sequence diversity was illustrated by small variability in RFLPs (Table 2) and by the localized clusters in the 16S rRNA gene dendrogram (Fig. 5). The two dominant genera in the 12/13 treatment, Kocuria and Staphylococcus, have routinely been isolated from soil habitats (3, 22, 30). One might expect some common sequences to be revealed in the N/13 and 12/13 treatments. We hypothesize that the absence of commonalities between the two retrieved populations can be explained by the relatively small number of clones analyzed (100 from each treatment), by dilution effects associated with microbial enrichment in the 12/13 treatment, and/or by small-scale heterogeneity between soil samples.
The third group of populations (trophically related carbon cross-feeders [13/13]) (Table 1) received the same total mass of phenol as the 12/13 treatment population prior to the respiration assay. However, unlike the 12/13 treatment population, the 13/13 treatment population uniformly received only the 13C-labeled substrate over the 14-day dosing period. The resulting net 13CO2 produced in this treatment was greater than that of the other two key treatments (Fig. 2). This rapid rate of 13CO2 production can be attributed to microbial enrichment (as in the 12/13 treatment) in combination with the larger mass of labeled carbon administered to this treatment population. We validated our notion that 13C-labeled, phenol-derived biomass is a suitable substrate for the soil microbial community (Fig. 1). The addition of phenol-free, sterile biomass was essential for data interpretation. Because autoclaved cell preparations are almost surely a more readily utilizable carbon source than intact cells grown in situ, the rates of 13C transfer shown in Fig. 1 are probably unrealistically high. Interestingly, sequencing data indicate that the 12/13 and 13/13 treatment populations were dominated by completely different organisms (Fig. 5). While the 12/13 treatment population featured representatives of the genera Kocuria and Staphylococcus, the 13/13 treatment population was dominated by members of the genus Pseudomonas. We hypothesize that the 13C label was incorporated into the biomass of the pseudomonads as a direct result of microbial scavenging of labeled biomass and metabolites derived from primary phenol degraders. This theory is supported by the fact that pseudomonads can use a wide range of carbon sources for energy (14). Although 13C enrichment of Pseudomonas DNA could also have resulted from direct metabolism of phenol, the contrast in clone libraries retrieved from the 12/13 and 13/13 treatments suggests that labeling of the pseudomonads was more likely the result of carbon cross-feeding.
The sequences retrieved via SIP provide clues about the ecological role of active microbial populations. Our data show that enriched, Kocuria-related, phenol-degrading soil populations (repeatedly exposed to high concentrations) are distinctive and less diverse than unenriched populations. Such changes in community composition in response to long-term exposure to pollutants has been documented previously (21, 24). Our data also indicate that, contrary to expectation, pseudomonads in the agricultural study site can be indirectly involved in in situ phenol metabolism. This implies that the long-recognized metabolic versatility of pseudomonads may be a manifestation of their role in catalyzing carbon flow within the microbial community at large.
Other recent SIP-based studies have aimed to document carbon flow in microbial communities. Using a mixed culture (containing a phenol-degrading- and a non-phenol-degrading pseudomonad) grown on [13C]phenol, Manefield et al. showed that labeling of nondegrader RNA occurred later in the incubation period (28). The authors attribute this finding to 13C cross-feeding between species. Other groups have used 13CO2 in an attempt to trace 13C-labeled plant photosynthate into the biomass of microbial populations in the rhizosphere (7, 9, 16, 25). Similarly, Middelburg et al. used [13C]bicarbonate to study the flow of carbon from microphytobenthos to benthic consumers (31) We feel that the potential is enormous for the combination of SIP and SIMS (as supported by other analytical, molecular, and microscopic procedures) to probe detailed complexities of carbon flow through microbial communities. To our knowledge, microscopic images documenting the isotopic composition of bacteria in environmental samples has appeared in only two prior reports (18, 34).
Without a doubt, the weakest methodological step in our procedures is choosing the 13C-labeled DNA that represents the active members of the microbial community. In the procedures described here, we were careful to be sure that identically processed 12C-treated negative controls were implemented. Only when our negative controls (invisible bands from the location where the 13C-DNA is expected) were successful did we trust that the amplicons from the 13C treatments represented the sought microorganisms. But clearly, resolving labeled (13C) from unlabeled (12C) DNA can be confounded by the percent G+C content of microorganisms (see Results) as well as by heterotrophs or predators whose biomass is derived from mixtures of labeled and unlabeled carbon. In following the flow of 13C from the added substrate to primary degraders and to secondary and tertiary consumers, the likelihood of mixotrophic feeding increases. Eventually, the added 13C atoms are lost in a blur of trophic interactions and DNA composition. To improve resolution of labeled from unlabeled populations, molecular fingerprinting (e.g., T-RFLP, denaturing gradient gel electrophoresis, etc.) procedures have been implemented (13, 20, 26, 28). This and related quality control procedures are likely to ensure continued application of SIP to microbial ecological inquiries.
Acknowledgments
We thank Jane Yagi and Daniella Bocioaga for carrying out the experiments on the influence of G+C content on DNA recovery. We thank Subhash Chandra, Cornell SIMS ion microscopy laboratory in the Department of Earth and Atmospheric Sciences, for SIMS analysis. We also acknowledge constructive criticism from two anonymous reviewers.
The research was supported by NIEHS grant no. 1-R21-ES012834.
REFERENCES
- 1.Abelson, P., and T. Hoering. 1961. Carbon isotope fractionation in formation of amino acids by photosynthetic organisms. Proc. Natl. Acad. Sci. USA 47:623-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham, W.-R., C. Hesse, and O. Pelz. 1998. Ratios of carbon isotopes in microbial lipids as an indicator of substrate usage. Appl. Environ. Microbiol. 64:4202-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajaz, M., N. Noor, S. A. Rasool, and S. A. Khan. 2004. Phenol resistant-bacteria from soil: identification-characterization and genetical studies. Pak. J. Bot. 36:415-424. [Google Scholar]
- 4.Bakermans, C., and E. L. Madsen. 2002. Diversity of 16S rDNA and naphthalene dioxygenase genes from coal-tar-waste-contaminated aquifer waters. Microb. Ecol. 44:95-106. [DOI] [PubMed] [Google Scholar]
- 5.Blair, N. E., L. A. Levin, D. J. DeMaster, and G. Plaia. 1996. The short-term fate of fresh algal carbon in continental slope sediments. Limnol. Oceanogr. 41:1208-1219. [Google Scholar]
- 6.Boschker, H. T. S., S. C. Nold, P. Wellsbury, D. Bos, W. de Graff, R. Pel, R. J. Parkes, and T. E. Cappenberg. 1998. Direct linking of microbial populations to specific biogeochemical processes by 13C-labelling of biomarkers. Nature 392:801-805. [Google Scholar]
- 7.Boschker, H. T. S., A. Wielemaker, B. E. M Schaub, and M. Holmer. 2000. Limited coupling of macrophyte production and bacterial carbon cycling in the sediments of Zostera spp. meadows. Mar. Ecol. Prog. Ser. 203:181-189. [Google Scholar]
- 8.Boschker, H. T. S., and J. J. Middelburg. 2002. Stable isotopes and biomarkers in microbial ecology. FEMS Microbiol. Ecol. 40:85-95. [DOI] [PubMed] [Google Scholar]
- 9.Butler, J. L., M. A. Williams, P. J. Bottomley, and D. D. Myrold. 2003. Microbial community dynamics associated with rhizosphere carbon flow. Appl. Environ. Microbiol. 69:6793-6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandra, S., D. R. Smith, and G. H. Morrison. 2000. Subcellular imaging by dynamic SIMS ion microscopy. Anal. Chem. 72:104A-114A. [DOI] [PubMed] [Google Scholar]
- 11.Coffin, R., B. Fry, B. Peterson, and R. Wright. 1989. Carbon isotopic compositions of estuarine bacteria. Limnol. Oceanogr. 34:1305-1310. [Google Scholar]
- 12.Coffin, R., R. Devereux, W. Price, and L. Cifuentes. 1990. Stable carbon isotope analysis of nucleic acids to trace sources of dissolved substrates used by estuarine bacteria. Appl. Environ. Microbiol. 56:2012-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallagher, E. M., L. McGuinness, C. Phelps, L. Y. Young, and L. J. Kerkhof. 2005. 13C-carrier DNA shortens the incubation time needed to detect benzoate-utilizing bacteria by stable-isotope probing. Appl. Environ. Microbiol. 71:5192-5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galli, E., S. Silver, and B. Witholt. 1992. Pseudomonas: molecular biology and biotechnology. American Society for Microbiology, Washington, D.C.
- 15.Ginige, M. P., P. Hugenholtz, H. Daims, M. Wagner, J. Keller, and L. L. Blackall. 2004. Use of stable-isotope probing, full-cycle rRNA analysis, and fluorescence in situ hybridization-microautoradiography to study a methanol-fed denitrifying microbial community. Appl. Environ. Microbiol. 70:588-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffiths, R. I., M. Mafefield, N. Ostle, N. McNamara, A. G. O'Donnell, M. J. Bailey, and A. S. Whiteley. 2004. 13CO2 pulse labeling of plants in tandem with stable isotope probing: methodological considerations for examining microbial function in the rhizosphere. J. Microbiol. Methods 58:119-129. [DOI] [PubMed] [Google Scholar]
- 17.Hoefs, J. 2004. Stable isotope geochemistry, 5th ed. Springer, New York, N.Y.
- 18.Huang, W. E., R. I. Griffiths, I. P. Thompson, M. J. Bailey, and A. S. Whiteley. 2004. Raman microscopic analysis of single microbial cells. Anal. Chem. 76:4452-4458. [DOI] [PubMed] [Google Scholar]
- 19.Hutchens, E., S. Radajewski, M. G. Dumont, I. McDonald, and J. C. Murrell.2004. Analysis of methanotrophic bacteria in Movile Cave by stable isotope probing. Environ. Microbiol. 6:111-120. [DOI] [PubMed] [Google Scholar]
- 20.Jeon, C. O., W. Park, P. Padmanabhan, C. DeRito, J. R. Snape, and E. L. Madsen. 2003. Discovery of a bacterium, with distinctive dioxygenase, that is responsible for in situ biodegreadation in contaminated sediment. Proc. Natl. Acad. Sci. USA 100:13591-13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ka, J. O., P. Burauel, J. A. Bronson, W. E. Holben, and J. M. Tiedje. 1995. DNA probe analysis of microbial community selected in field by long-term 2,4-D application. Soil Sci. Soc. Am. J. 59:1581-1587. [Google Scholar]
- 22.Katayama, A., H. Y. Hu, M. Nozawa, H. Yamakawa, and K. Fujie. 1998. Long-term changes in microbial community structure in soils subjected to different fertilizing practices revealed by quinone profile analysis. Soil Sci. Plant Nutr. 44:559-569. [Google Scholar]
- 23.Kritzberg, E. S., J. J. Cole, M. L. Pace, W. Graneli, and D. L. Bade. 2004. Autochthonous versus allochthonous carbon sources of bacteria: results from whole-lake 13C addition experiments. Limnol. Oceanogr. 49:588-596. [Google Scholar]
- 24.Langworthy, D. E., R. D. Stapelton, G. S. Sayler, and R. H. Findlay. 1998. Genotypic and phenotypic responses of a riverine microbial community to polycyclic aromatic hydrocarbon contamination. Appl. Environ. Microbiol. 64:3422-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu, Y., J. Murase, A. Watanabe, A. Sugimoto, and M. Kimura. 2004. Linking microbial community dynamics to rhizosphere carbon flow in a wetland rice soil. FEMS Microbiol. Ecol. 48:179-186. [DOI] [PubMed] [Google Scholar]
- 26.Lueders, T., M. Manfield, and M. W. Friedrich. 2004. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ. Microbiol. 61:73-78. [DOI] [PubMed] [Google Scholar]
- 27.Madsen, E. L. 2005. Identifying microorganisms responsible for ecologically significant biogeochemical processes. Nat. Rev. Microbiol. 3:439-446. [DOI] [PubMed] [Google Scholar]
- 28.Manefield, M., A. S. Whiteley, R. I. Griffiths, and M. J. Bailey. 2002. RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl. Environ. Microbiol. 68:5367-5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manefield, M., A. S. Whitely, N. Ostle, P. Ineson, and M. J. Bailey. 2002. Technical considerations for RNA-based stable isotope probing: an approach to associating microbial diversity with microbial community function. Rapid Commun. Mass Spectrom. 16:2179-2183. [DOI] [PubMed] [Google Scholar]
- 30.Michaud, M., C. Martinez, A. M. Simao-Beaunoir, R. R. Belanger, and R. J. Tweddell. 2002. Selection of antagonist microorganisms against Helminthosporium solani, causal agent of potato silver scurf. Plant Dis. 86:717-720. [DOI] [PubMed] [Google Scholar]
- 31.Middelburg, J. J., C. Barranguet, H. T. S. Boschker, P. M. J. Herman, T. Moens, and C. H. R. Heip. 2000. The fate of intertidal microphytobenthos carbon: an in situ 13C-labeling study. Limnol. Oceanogr. 45:1224-1234. [Google Scholar]
- 32.Moodley, L., H. T. S. Boschker, J. J. Middelburg, R. Pel, P. M. J. Herman, E. de Deckere, and C. H. R. Heip. 2000. Ecological significance of benthic foraminifera: 13C labelling experiments. Mar. Ecol. Prog. Ser. 202:289-295. [Google Scholar]
- 33.Morris, S. A., S. Radajewski, T. W. Willison, and J. C. Murrell. 2002. Identification of the functionally active methanotroph population in a peat soil microcosm by stable-isotope probing. Appl. Environ. Microbiol. 68:1446-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orphan, V. J., C. H. House, K.-U. Hinrich, K. D. McKeegan, and E. F. DeLong. 2001. Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science 293:484-487. [DOI] [PubMed] [Google Scholar]
- 35.Padmanabhan, P., S. Padmanabhan, C. DeRito, A. Gray, D. Gannon, J. R. Snape, C. S. Tsai, W. Park, C. Jeon, and E. L. Madsen. 2003. Respiration of 13C-labeled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13C-labeled soil DNA. Appl. Environ. Microbiol. 69:1614-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pombo, S. A., O. Pelz, M. H. Schroth, and J. Zeyer. 2002. Field-scale 13C-labeling of phospholipid fatty acids (PLFA) and dissolved inorganic carbon: tracing acetate assimilation and mineralization in a petroleum hydrocarbon-contaminated aquifer. FEMS Microbiol. Ecol. 41:259-267. [DOI] [PubMed] [Google Scholar]
- 37.Radajewski, S., P. Ineson, N. R. Parekh, and J. C. Murrell. 2000. Stable-isotope probing as a tool in microbial ecology. Nature 403:646-649. [DOI] [PubMed] [Google Scholar]
- 38.Radajewski, S., G. Webster, D. S. Reay, S. A. Morris, P. Ineson, D. B. Nedwell, J. I. Prosser, and J. C. Murrell. 2002. Identification of active methylotroph populations in an acidic forest soil by stable-isotope probing. Microbiology 148:2331-2342. [DOI] [PubMed] [Google Scholar]
- 39.Radajewski, S., I. R. McDonald, and J. C. Murrell. 2003. Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr. Opin. Biotechnol. 14:296-302. [DOI] [PubMed] [Google Scholar]
- 40.Singleton, D. R., S. N. Powell, R. Sangaiah, A. Gold, L. M. Ball, and M. D. Aitken. 2005. Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene, or phenanthrene in a bioreactor treating contaminated soil. Appl. Environ. Microbiol. 71:1202-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sparling, G. P., B. G. Ord, and D. Vaughan. 1981. Changes in microbial biomass and activity in soils amended with phenolic acids. Soil Biol. Biochem. 13:455-460. [Google Scholar]
- 42.Stanier, R. Y., N. J. Palleroni, and M. Douderoff. 1966. The aerobic pseudomonas: a taxonomic study. J. Gen. Microbiol. 43:159-271. [DOI] [PubMed] [Google Scholar]
- 43.Sun, M. Y., R. C. Aller, C. Lee, and S. G. Wakeham. 1999. Enhanced degradation of algal lipids by benthic macrofaunal activity: effect of Yoldia limatula. J. Mar. Res. 57:775-804. [Google Scholar]
- 44.Watanabe, K., M. Teramoto, H. Futamata, and S. Harayama. 1998. Molecular detection, isolation, and physiological characterization of functionally dominant phenol-degrading bacteria in activated sludge. Appl. Environ. Microbiol. 64:4396-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wellington, E. M. H., A. Berry, and M. Krsek. 2003. Resolving functional diversity in relation to microbial community structure in soil: exploiting genomics and stable isotope probing. Curr. Opin. Microbiol. 6:295-301. [DOI] [PubMed] [Google Scholar]