

Abstract
Fluorescence in situ hybridization (FISH) is a widely used method to detect environmental microorganisms. The standard protocol is typically conducted at a temperature of 46°C and a hybridization time of 2 or 3 h, using the fluorescence signal intensity as the sole parameter to evaluate the performance of FISH. This paper reports our results for optimizing the conditions of FISH using rRNA-targeted oligonucleotide probes and flow cytometry and the application of these protocols to the detection of Escherichia coli in seawater spiked with E.coli culture. We obtained two types of optimized protocols for FISH, which showed rapid results with a hybridization time of less than 30 min, with performance equivalent to or better than the standard protocol in terms of the fluorescence signal intensity and the FISH hybridization efficiency (i.e., the percentage of hybridized cells giving satisfactory fluorescence intensity): (i) one-step FISH (hybridization is conducted at 60 to 75°C for 30 min) and (ii) two-step FISH (pretreatment in a 90°C water bath for 5 min and a hybridizing step at 50 to 55°C for 15 to 20 min). We also found that satisfactory fluorescence signal intensity does not necessarily guarantee satisfactory hybridization efficiency and the tightness of the targeted population when analyzed with a flow cytometer. We subsequently successfully applied the optimized protocols to E. coli-spiked seawater samples, i.e., obtained flow cytometric signatures where the E. coli population was well separated from other particles carrying fluorescence from nonspecific binding to probes or from autofluorescence, and had a good recovery rate of the spiked E. coli cells (90%).
The detection of environmentally important bacteria generally relied upon the conventional method of bacterial cultivation on selective media until the introduction of fluorescence in situ hybridization (FISH) using rRNA-targeted oligonucleotide probes by DeLong et al. (6). Some of the problems with the method of bacterial cultivation are that many bacteria are difficult to culture or are unculturable; media are not specific enough or, conversely, are too selective for some bacteria; and the overall procedures of cultivation are very time-consuming. The FISH approach using rRNA-targeted oligonucleotide probes has advantage as a fast and effective technique, and in the last decade, many attempts have been made to develop and optimize the protocol and its application (2, 3, 8, 9, 14, 15, 20; A. Oppedahl, K. R. Harkins, B. G. Smith, and K. A. Harrigan, Abstr. Gen. Meet. Am. Soc. Microbiol., abstr. 103, p. 485, 2003). FISH of oligonucleotide probes, when combined with flow cytometry (FCM), offers the advantages of both high resolution for taxonomic identification and rapid, automated cell counting (3, 20) and has been recommended as a standard method (1).
Most of the studies applying FISH of rRNA-targeted oligonucleotide probes and flow cytometry to environmental samples use a FISH protocol in which hybridization is typically carried out at 40 to 46°C for 2 to 3 h (2, 9, 10, 13, 17, 18, 20, 21). Some studies have applied hybridization of DNA probes at lower temperatures but for longer times to preserve the morphology of the targeted chromosome and obtain higher fluorescence. For instance, Winkler et al. (22) applied a hybridization time of 15 h at 37°C, and Buno et al. (4) used 4 to 5 h at 37°C. It has generally been accepted that low-stringency hybridization (i.e., low temperature) corresponds to a stronger binding of probes to the targeted rRNA sites (9). Therefore, higher temperatures appear to be less suitable for FISH in terms of obtaining a satisfactory fluorescence signal.
However, this hypothesis does not always hold true, as in some situations an opposite trend has been observed. For example, Fuchs et al. (9) observed that two out of nine probes exhibited lower signal intensities at lower temperatures. A few studies have also tried higher temperatures for FISH: Prescott and Fricker (15), using a peptide nucleic acid probe targeting the rRNA of E. coli and microscopy, conducted FISH at 50°C for 30 min; Oppedahl et al. (Abstr. Gen. Meet. Am. Soc. Microbiol., 2003) applied FISH at 55°C for 30 min using rRNA-targeted peptide nucleic acid probes and flow cytometry; da Silva and da Cruz (5) applied FISH at 60°C for 4 h with DNA-targeted probes on animal cells; and Durm et al. (7) conducted FISH at 40 to 75°C for 15 to 120 min using DNA probes and animal cells for microscopic discrimination of the hybridization stringency. These studies led us to speculate that there may be a complicated relationship between the hybridization temperature and fluorescence intensity, because temperature does not only affect the dissociation of a probe, but also affects the conformation of the targeted rRNA or DNA. In view of these studies and speculations, we undertook a study to systematically optimize the FISH protocols using higher temperatures and shorter hybridization times, in order to obtain a more efficient protocol for the combined techniques of FISH (using rRNA-targeted probes) and flow cytometry for the detection of environmental microorganisms. After reviewing the procedure of in situ PCR (11, 16), we speculated that applying a pretreatment step at a temperature of 90°C, before the FISH took place at a temperature below the melting point of the 20-mer probe, might be helpful in increasing the accessibility of the rRNA sites to the probes, such as through dissociation of selected helices and changing the permeability of the bacterial cells.
In reviewing the literature, it was also noted that in most of the studies involving optimization or application of whole-cell FISH using rRNA-targeted oligonucleotide probes and flow cytometry (or microscopy), the fluorescence signal intensity was adopted as the sole parameter in evaluating the performance of different protocols (2, 7, 9, 10, 17, 18, 20). However, high fluorescence signal intensity does not necessarily ensure satisfactory hybridization efficiency (i.e., the percentage of cells that were hybridized well with satisfactory fluorescence intensity). As long as binding of a probe to a target cell occurs, the fluorescence intensity of an individual cell mainly depends on the labeling reagent used (i.e., species, storage time, and buffering solutions) and the number of copies of rRNA (or targeted DNA), while the hybridization efficiency mainly depends on the sample processing and FISH conditions applied, such as the fixative used, the permeabilizing reagent, FISH temperature, hybridization time, probe concentration, and mixing efficiency. In cases where both enumeration and identification of the targeted microorganisms in the environmental samples are major goals, the hybridization efficiency becomes a parameter of importance equal to that of the signal intensity. Therefore, both the signal intensity and FISH efficiency should be taken into account when developing a protocol for FISH with rRNA-targeted probes and flow cytometry.
In this paper, we report the results of optimizing FISH procedures using rRNA-targeted probes and flow cytometry for the detection of E. coli in seawater samples. E. coli was chosen as a model microorganism because enumeration of these indicator microorganisms is a routine practice for microbial-quality monitoring of aquatic environments. Our emphasis was to develop a rapid protocol so that the time taken to detect and quantify organisms could be minimized. Shorter analytical times are needed to prevent outbreaks of waterborne diseases, and hence, the speed and accuracy with which these microbes can be identified is of paramount importance for protecting human and ecosystem health.
MATERIALS AND METHODS
16S rRNA-targeted oligonucleotide probes.
The probes used in the study, Eco541 and Eco1482, were adopted from Fuchs et al. (9) and have the following sequences: 5′-CCG ATT AAC GCT TGC ACC-3′ and 5′-TAC GAC TTC ACC CCA GTC-3′. The probes were monolabeled with fluorescein isothiocyanate at the 5′ end. They reportedly have high fluorescence intensity and thus high accessibility to the targeted sites on rRNA of Escherichia coli (9) but are not highly specific to E. coli. As the major objective of this study was to optimize the FISH protocol using pure cultures, the nonspecificity of the probes to E. coli was not a major concern. The stock solutions of probes were stored in TE buffer (10 mmol/liter Tris-HCl, 1.0 mmol/liter EDTA-Na2, pH 7.6) at −20°C and diluted to the working concentrations with nonformamide hybridization buffer (0.9 M NaCl, 0.1% sodium dodecyl sulfate, 20 mM Tris-HCl, pH 7.2) immediately before hybridization.
Culture of E. coli and sample pretreatment.
A pure culture of Escherichia coli (ATCC 700891) was grown in Luria Broth (Difco Laboratories) and prepared according to the manufacturer's instructions. Two hundred fifty milliliters of culture was placed in 500-ml flasks and grown at 37°C (shaken at 150 rpm) for 3 to 4 h. Aliquots (20 or 40 ml) of culture broth were centrifuged for 5 min at 4,000 × g at 4°C (Jouan BR4). After the supernatant was removed, the pellet of E. coli cells was resuspended in 10 ml 1× phosphate-buffered saline (PBS) (130 mM NaCl, 10 mM Na2HPO4, 10 mM NaH2PO4, pH 7.2) and 30 ml of 4% (wt/vol) cold, freshly prepared paraformaldehyde (PFA) solution (in PBS), and the suspension was mixed and incubated overnight (16 h) at 4°C. The samples were then centrifuged, the pellet was washed with 10 ml PBS, and an appropriate volume of the mixture of 1:1 1× PBS-absolute ethanol was added. The samples were then aliquoted into 2-ml Eppendorf microcentrifuge tubes (1ml for each) and stored at −20°C for several weeks until they were used. In addition to the tests designed to optimize the FISH temperature and duration, another set of tests was arranged to use fixatives other than PFA. After being harvested and washed, the E. coli cells were fixed with 1% glutaraldehyde (GTA) in 1× PBS (pH 7.2) at room temperature for 20 min in darkness or fixed with 50% cold ethanol in 1× PBS (pH 7.2). The GTA-fixed samples were then processed similarly to those fixed by PFA, while the 50% ethanol-fixed samples were stored at −20C° before FISH (and analyzed within a week).
Processing of seawater samples.
In order to enhance the sensitivity of analyzing seawater for coliform bacteria, it is sometimes necessary to preconcentrate seawater samples so that sufficient bacteria can be enumerated. For this reason, we applied our FISH protocols to seawater that had been concentrated and spiked with E. coli before analysis by flow cytometry. A volume of 10 liters of seawater was collected from the coast of St John's Island, Singapore. Prior to the analysis, the background concentration of the general bacterial population was measured with FCM after SYBR Green 1 staining. Half of the seawater (5 liters) was spiked with 200 μl of fresh culture of E. coli (the E. coli cell concentration was later measured to be 1.93 × 104 cells/ml) and exposed to seawater for 3 h to allow the E. coli cells to equilibrate with the ambient environmental conditions. Both the nonspiked (5 liters) and the spiked seawater samples were then filtered through a 15-μm nylon membrane (to avoid clogging the flow cell of the flow cytometer). The filtrates were centrifuged at 4,000 × g and 4°C for 15 min with a Jouan BR4 centrifuge; the pellets were resuspended in 10 ml 1× PBS, vortexed, and centrifuged again; and the resulting pellet was resuspended in 5 ml PBS. The suspension was then added to a volume of 15 ml freshly prepared 4% cold paraformaldehyde solution, vortexed, and incubated overnight at 4°C (for 16 to 18 h). After fixation, the procedure for the remaining treatment was the same as that for the E. coli pure culture. (Note that the sample aliquots were concentrated 250 times.)
Conditions of FISH.
The samples stored in the microcentrifuge tubes with 1:1 PBS-ethanol were centrifuged at 10,000 × g for 2 min with a microcentrifuge (Eppendorf centrifuge 5415C; Germany). One hundred microliters of the formamide-free hybridization buffer with or without 4.0 ng/μl probe was then added to the pellets. After being mixed, the samples were subjected to one of the following three types of FISH on a rotating incubator: (i) standard FISH (46°C for 3 h) (9), (ii) one-step FISH at temperatures from 46°C to 75°C for 10 min to 30 min, or (iii) two-step FISH with prewarming at 90°C for 5 min in a water bath tank and FISH at 50 or 55°C for 10 min to 30 min on a rotating incubator (the samples were shifted from the water bath to the incubator within a minute using a small tub containing 90°C water). The negative controls consisted of hybridization buffer without the addition of E. coli cells and probes or E. coli cells (or seawater sample) without probes subjected to standard FISH conditions. After FISH, the samples were centrifuged at 10,000 × g for 2 min, the pellets were resuspended in 100 μl of hybridization buffer containing no probe, and the samples were then washed at 46°C for 30 min (or 50°C for 20 min for two-step FISH) on a rotating incubator. After being centrifuged at 10,000 × g for 2 min, the pellets were resuspended in 1,000 μl 1× PBS (pH 8.4) and put on ice until flow cytometric analysis was performed.
Flow cytometry.
A Coulter EPICS Elite ESP flow cytometer was used for all sample analyses, and all acquired data were analyzed with the software WinMDI version 2.8. The flow cytometer was equipped with an argon ion laser (model 621; Coherent Innova Enterprise) capable of producing 488-nm light emission at a power of 200 mW. The flow cell used was a SortSense Enhanced Quartz flow cell with a 100-μm orifice (Coulter Corp.). Spherical polystyrene Flow-Check Fluorospheres (Coulter) beads, 10-μm diameter, were used for basic alignment of the laser. Generally, 20 μl of 0.75-μm- or 2-μm-diameter blue-excitable beads (Fluoresbrite YG; Polysciences, Inc.) was added to 1,000 μl sample for general optical alignment and for standardization of the fluorescence intensities of probes and concentration calibration. Each sample was run in triplicate. The green-fluorescence-versus-forward-scatter dot plots were used for the determination of fluorescence intensity (the mean of the gated population) and cell counts. Background noise was removed by adjusting the value of the discriminator on the green fluorescence. However, for the nonspiked and E. coli-spiked seawater samples, the acquisition mode was set as “gated” to exclude the “unwanted” signals, because there were high concentrations of interfering particles (e.g., phytoplankton, other bacteria, or debris) and a relatively low concentration of E. coli. In addition, the preconcentrated seawater samples were diluted 5- to 20-fold and run for different durations to enhance the performance of flow cytometry.
The concentrations of E coli cells in the sample were calculated as follows: concentration of E. coli cells = (number of cells counted/number of beads counted) × concentration of beads in the sample. Fluorescence intensity was determined as the mean green fluorescence (relative to standard calibration beads) of the E. coli population defined in the green-fluorescence-versus-forward-scatter dot plot. SYBR Green 1 (a nucleic acid stain used to detect DNA) was used as a reference for the hybridization efficiency of FISH, in addition to the standard FISH conditions. The SYBR Green 1 staining was conducted at 80°C for 10 min in darkness using the reagent diluted 5,000-times from the stock solution.
In addition to fluorescence intensity and hybridization efficiency, we define another parameter, the tightness of population (Tp), as a quantitative measure to evaluate the tightness (compactness) of the detected population on the cytogram, as follows: 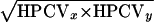 (0 < Tp < 100%), where HPCVx and HPCVy are the half-peak coefficients of variation for the values on the x axis (e.g., forward scatter) and y axis (e.g., green fluorescence), respectively. Note that the HPCV is derived from the fixed mathematical relationship between the standard deviation (SD) and the full-width half-max (FWHM) value of a normal or Gaussian peak (SD = FWHM/2.354); thus, HPCV is usually smaller than the CV calculation normally used in statistical analysis (cytometry data acquisition and analysis software, Expo32 version 1.2B; Beckman Coulter Inc.). In general, the smaller the value of Tp, the tighter the population on the cytogram, and thus, the better the performance of the overall experimental procedures (FISH and flow cytometric analysis). A Tp of less than 10% is generally recommended as acceptable.
(0 < Tp < 100%), where HPCVx and HPCVy are the half-peak coefficients of variation for the values on the x axis (e.g., forward scatter) and y axis (e.g., green fluorescence), respectively. Note that the HPCV is derived from the fixed mathematical relationship between the standard deviation (SD) and the full-width half-max (FWHM) value of a normal or Gaussian peak (SD = FWHM/2.354); thus, HPCV is usually smaller than the CV calculation normally used in statistical analysis (cytometry data acquisition and analysis software, Expo32 version 1.2B; Beckman Coulter Inc.). In general, the smaller the value of Tp, the tighter the population on the cytogram, and thus, the better the performance of the overall experimental procedures (FISH and flow cytometric analysis). A Tp of less than 10% is generally recommended as acceptable.
RESULTS
Performance of different fixatives.
In addition to the optimization of FISH temperature and duration, the performances of different fixatives were evaluated, i.e., PFA, GTA, and 50% ethanol. It was seen that the GTA-fixed samples showed significant green autofluorescence before hybridization, although the E. coli cells were well separated from background noise and their signature showed a tight or compact population (Tp= 7.97%) (Fig. 1A). In fact, the observed autofluorescence has also been pointed out by Vives-Rego et al. (18), and hence, GTA appeared to be a poor fixative for the purpose of this study. As for the ethanol-fixed samples, the results showed a good separation of the hybridized E. coli population from the noise and the nonhybridized E. coli population (Fig. 1B). However, the FISH efficiency (i.e., the percentage of hybridized cells in the total targeted cells) was only 0.26% of the FISH sample fixed with PFA (Fig. 1C and D), presumably due to the dehydration effect of ethanol. As expected, the best performance was obtained with the samples fixed with PFA, in terms of both the fluorescence signal intensity and the FISH efficiency.
FIG. 1.

Flow cytometric results for (A) E. coli fixed with GTA, showing autofluorescence (region R1 shows the position for GTA-fixed E. coli after FISH at 50°C for 20 min, following a pretreatment at 90°C for 5 min using Eco541); (B) 50% ethanol-fixed E. coli hybridized at 50°C for 20 min, following pretreatment at 90°C for 5 min using Eco541 (note that FISH efficiency was only 0.26% of that for a sample fixed with PFA and FISH under the same conditions); (C) PFA-fixed E. coli hybridized under conditions similar to those in panel B, showing much higher hybridization efficiency; and (D) a one-parameter histogram of green fluorescence for the same sample shown in panel C. Standard calibration beads (0.75 μm) were used for reference.
Performance of one-step FISH.
In general, we were able to obtain FISH performance, in terms of both the green fluorescence intensity and the hybridization efficiency, equivalent or superior to the standard protocol using temperatures higher than 46°C and with a hybridization time of less than 30 min (Fig. 2 and 3). Figure 2 shows a typical cytogram and histogram obtained from an E. coli pure culture hybridized at 75°C for 30 min. The population of E. coli cells is well defined (Tp = 3.85%) and shows satisfactory green fluorescence intensity, demonstrating that a temperature even higher than the melting point of the 18-mer probe can be adopted in the process of FISH using rRNA-targeted probes. From the different treatments tested (Fig. 3 and Table 1), the following three observations were made. First, the trends for the fluorescence signal intensity with temperature were not very consistent with those for the hybridization efficiency, in that higher signal intensity could correspond to low hybridization efficiency and vice versa (e.g., see the results for 60°C and 65°C in Fig. 3). Second, when the hybridization was conducted at temperatures higher than 46°C, it appeared that 30 min is required to allow the FISH reaction to complete or to reach an extent equivalent to that with the standard conditions (Fig. 3B). For example, in Table 1(also in Fig. 3A), while satisfactory signal intensity was obtained from FISH at 75°C for 10 min, the hybridization efficiency was lower (by about 15%) than with FISH at 46°C for 3 h and FISH at 75°C for 20 min (6%) and 30 min (12%). Similar results were also obtained at temperatures of 60, 65, and 70°C. Third, for certain combinations of high temperature and hybridization time, both satisfactory signal intensity and hybridization efficiency could be obtained, which were equivalent to or better than with the standard conditions (i.e., 46°C for 2 to 3 h). This demonstrates that FISH can be conducted at temperatures higher than 46°C without loss of hybridization efficiency, and the hybridization time requirement at these higher temperatures can be significantly shorter than that for standard FISH. From our experimental results, it can be seen that, in general, FISH conducted at 65 to 75°C for 30 min will give satisfactory results comparable to the results from the standard FISH protocol (at 46°C for 3 h).
FIG. 2.

Typical flow cytometric results for one-step FISH conducted at 75°C for 30 min. (A) Cytogram of green fluorescence versus forward scatter. (B) One-parameter histogram of green fluorescence.
FIG. 3.

Comparison of (A) fluorescence intensities and (B) hybridization efficiencies for one-step FISH at different temperatures and hybridization times. The error bars indicate the standard deviations of triplicates. The mean fluorescence intensities of hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to the cell counts for the standard conditions (Std) (46°C; 3 h). The FISH conditions for each sample are expressed in terms of the FISH temperature (°C), followed by the hybridization time (min).
TABLE 1.
Comparison of fluorescence intensities (mean value of triplicates) and hybridization efficiencies (mean value of triplicates) for one-step FISH and two-step FISH at different temperatures and hybridization timesa
| FISH conditions | Fluorescence intensity | SD | FISH efficiency (%) | SD |
|---|---|---|---|---|
| Std | 225.7 | 1.3 | 100.0 | 0.7 |
| 50°C, 30 min | 205.6 | 0.8 | 82.1 | 1.6 |
| 55°C, 30 min | 215.1 | 1.5 | 91.3 | 2.7 |
| 60°C, 20 min | 213.4 | 0.7 | 64.7 | 0.1 |
| 60°C, 30 min | 215.7 | 0.8 | 83.2 | 1.9 |
| 65°C, 20 min | 196.2 | 0.2 | 86.1 | 4.1 |
| 65°C, 30 min | 161.8 | 0.6 | 100.1 | 1.7 |
| 70°C, 15 min | 199.5 | 3.5 | 83.9 | 4.0 |
| 70°C, 20 min | 195.4 | 8.1 | 94.9 | 1.9 |
| 70°C, 30 min | 192.0 | 11.7 | 98.3 | 6.5 |
| 75°C, 10 min | 201.7 | 0.6 | 85.5 | 0.1 |
| 75°C, 20 min | 209.9 | 0.2 | 91.3 | 2.7 |
| 75°C, 30 min | 219.8 | 2.9 | 97.1 | 3.8 |
| 20°C, 20 minb | 207.2 | 2.2 | 73.2 | 2.8 |
| 50°C, 15 minb | 212.8 | 3.2 | 100.5 | 0.9 |
| 50°C, 20 minb | 211.8 | 2.2 | 99.5 | 2.6 |
| 50°C, 25 minb | 211.0 | 1.8 | 80.0 | 2.2 |
| 55°C, 20 minb | 198.5 | 1.4 | 100.8 | 3.9 |
Data are illustrated in Fig. 3 and 5. The fluorescence intensities of hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to the cell counts for the standard conditions (Std) (46°C; 3 h).
Indicates pretreatment of sample at 90°C for 5 min.
Performance of two-step FISH.
In addition to one-step FISH, satisfactory performance was also obtained with the two-step FISH protocol, which consisted of a pretreatment step in which the sample was incubated in a water bath at 90°C for 5 min before FISH hybridization at 50 or 55°C for 10 to 30 min in a rotating incubator. A representative dot plot and histogram, which were obtained from a test in which FISH took place at 55°C for 20 min after the 90°C pretreatment, are presented in Fig. 4. It can be seen that the fluorescence of the hybridized population was generally high and the signature of the cell population was compact (Tp = 3.93%) (Fig. 4). A compilation of the results for relative signal intensity and hybridization efficiency under different test conditions is shown in Fig. 5 and Table 1; this demonstrates that, after pretreatment at 90°C in a water bath, FISH conducted at a temperature slightly higher than 46°C (i.e., 50 to 55°C) can achieve performance equivalent (in terms of both the fluorescence intensity and the hybridization efficiency) to that with the standard conditions but in a much shorter time, between 15 and 20 min. In fact, if the hybridization time was longer than 20 min, it was observed that the hybridization efficiency of FISH at 50 to 55°C after pretreatment at 90°C would decrease, as shown in Fig. 5B and additional results (data not shown). However, by comparing Fig. 5A and B, it can be seen that although FISH can take place at room temperature (20 ± 2°C) within 20 min after the pretreatment (90°C; 5 min) to give a fluorescence intensity similar to that of samples hybridized at 50 or 55°C, the hybridization efficiency was much lower. This indicates that it is necessary to apply the second step of FISH at a temperature between 50 and 55°C after the 90°C pretreatment to allow the FISH reaction to reach completion (since the pretreatment at 90°C is not the step in which hybridization takes place). Thus, the recommended procedure would be to pretreat samples in a 90°C water bath for 5 min, followed by FISH at 50 to 55°C for no more than 20 min.
FIG. 4.

Typical flow cytometric results for two-step FISH conducted at 55°C for 20 min after pretreatment at 90°C for 5 min. (A) Cytogram of green fluorescence versus forward scatter. (B) One-parameter histogram of green fluorescence.
FIG. 5.

Comparison of (A) fluorescence intensities and (B) hybridization efficiencies for two-step FISH at different temperatures and hybridization times. The error bars indicate the standard deviations of triplicates. The mean fluorescence intensities of the hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to cell counts for the standard conditions (Std) (46°C; 3 h). The FISH conditions for each sample are expressed in terms of the FISH temperature (°C), followed by the hybridization time (min). The asterisks indicate that pretreatment at 90°C for 5 min was carried out.
Comparison of performances of one-step and two-step FISH.
Given that both one-step and two-step FISH could provide satisfactory fluorescence intensity and hybridization efficiency, we decided to compare their performances based on the same FISH temperature and duration to understand whether the pretreatment step at 90°C was helpful in increasing the overall performance. From the results of one batch of tests, it can be seen that FISH at 50°C for 20 min after the 90°C pretreatment had the highest fluorescence intensity and had a hybridization efficiency higher than FISH at 55°C for 30 min after pretreatment. This was equivalent to one-step FISH at 60°C for 30 min and the SYBR Green-stained sample but was slightly lower than FISH at 65°C for 30 min (Fig. 6 and Table 2). For the same temperature, our earlier results (Fig. 3 and 5) also showed that two-step FISH at 50 to 55°C for 20 min had a better hybridization efficiency than one-step FISH at 50 to 55°C for 30 min, while they had equivalent fluorescence intensities. Thus, it appears that the pretreatment step at 90°C is beneficial, in that a higher hybridization efficiency can be obtained in a shorter hybridization time than for one-step FISH at the same temperature. In general, the overall performance of one-step FISH was enhanced with increasing FISH temperature over the range of temperatures applied (46 to 65°C) for the same hybridization time (30 min) (Fig. 6 and Table 2). Note that in Table 2, while the hybridization efficiency for FISH at 46°C for 30 min is comparable to that for standard FISH, its fluorescence intensity is much lower than those for the other FISH conditions at higher temperatures.
FIG. 6.

Comparison of (A) fluorescence intensities and (B) hybridization efficiencies between one-step and two-step FISH at different temperatures and hybridization times. The error bars indicate the standard deviations of six replicates. The mean fluorescence intensities of the hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to cell counts obtained from SYBR Green 1 (SYBR-Grn) staining. The FISH conditions for each sample are expressed in terms of the FISH temperature (°C), followed by the hybridization time (min). The asterisks indicate that pretreatment at 90°C for 5 min was carried out.
TABLE 2.
Comparison of fluorescence intensities and hybridization efficiencies between one-step and two-step FISH and between Eco541 and Eco1482 probes at different temperatures and hybridization timesa
| FISH conditions | Fluorescence intensity | SD | FISH efficiency (%) | SD |
|---|---|---|---|---|
| Std | 31.6 | 1.9 | 83.9 | 9.0 |
| 46°C, 30 min | 27.8 | 10.4 | 88.7 | 7.9 |
| 55°C, 30 min | 59.2 | 4.5 | 87.5 | 4.3 |
| 60°C, 30 min | 59.2 | 26.2 | 95.7 | 8.3 |
| 65°C, 30 min | 68.3 | 5.6 | 106.2 | 19.5 |
| 50°C, 20 minb | 73.7 | 3.1 | 100.1 | 6.3 |
| SYBR Green | 37.9 | 3.1 | 100.0 | 8.8 |
| Eco541-Std | 203.6 | 5.1 | 58.7 | 1.6 |
| Eco1482-Std | 159.0 | 1.5 | 66.1 | 5.1 |
| Eco541-55°C, 30 min | 199.7 | 0.8 | 82.6 | 4.7 |
| Eco1482-55°C, 30 min | 150.6 | 0.4 | 105.5 | 2.7 |
Data are also illustrated in Fig. 6 and 7. The fluorescence intensities of hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to the cell counts for the standard conditions (Std) (46°C; 3 h) (for comparison of one-step and two-step FISH) or normalized to the counts of reference beads (for comparison of the two probes).
Indicates pretreatment of sample at 90°C for 5 min.
Comparison of two probes used.
The two probes used in the study, Eco541 and Eco1482, both targeting 16S rRNA, were adopted from Fuchs et al. (9). It was reported in the original paper that Eco1482 had higher fluorescence intensity than Eco541 when they were hybridized with E. coli K-12 DSM 30083T (9). However, in the present study, we found that Eco1482 had lower fluorescence intensity but higher hybridization efficiency than Eco541 when they were hybridized with E. coli Famp ATCC 700891 under FISH conditions of 46°C for 3 h and 55°C for 30 min (Fig. 7 and Table 2). This indicates that the accessibility of rRNA to a certain probe may depend on the strain of E. coli and the FISH protocol used or other unknown factors, such as the species of labeling reagent. For example, Fuchs et al. (10) found that different labeling reagents reversed the order of the relative fluorescence for probes.
FIG. 7.

Comparison of the Eco541 and Eco1482 probes for E. coli based on their (A) fluorescence intensities and (B) hybridization efficiencies under different FISH conditions. The mean fluorescence intensities of the hybridized E. coli cells were normalized to 0.75-μm beads, while the hybridization efficiencies were normalized to the counts of reference beads. FISH conditions are expressed as standard conditions (Std) (46°C; 3 h) or FISH temperature (°C), followed by the hybridization time (min).
Application of the two FISH protocols to E. coli-spiked seawater samples.
Initially, the concentrated seawater samples (see Materials and Methods) were directly subjected to the standard and optimized FISH protocols as used for the pure culture. However, this resulted in a poor separation of the hybridized E. coli population from the noise, although most of the population could be discriminated on the cytogram (Fig. 8A). From the positions of the cells in the cytogram, it was postulated that some of the hybridized E. coli cells probably adhered to other particles present (e.g., phytoplankton and debris) because of the very high concentration of particles in the sample. (Note that the background concentration of the general bacterial population was about 3 × 105 to 4 × 105 cells/ml when measured with SYBR Green 1 staining and FCM). This led to a displacement of the signals for the hybridized E. coli cells, i.e., E. coli cells appeared to be larger and to have relatively lower green fluorescence. For the next round of analysis, a satisfactory separation of the hybridized E. coli population from the “unwanted” signals (representing other particles carrying fluorescence from nonspecific binding to probes or from autofluorescence) was obtained (Fig. 8B and C; Tp = 2.0% and 2.5%, respectively) by diluting the concentrated samples before and after FISH by different amounts (5 to 20×) and vortexing them immediately before running them on the flow cytometer. Furthermore, it was calculated that the recovery rate of the spiked E. coli cells in the seawater sample was as high as 90% (n = 2) for the samples with the highest dilution before flow cytometric analysis (20× dilution from the sample concentrated 250 times from seawater initially), based on enumeration of the pure culture (n = 6). However, for those samples with lower dilution (i.e., 5×), the recovery rate was only 29% (n = 3), although there was still a good separation of the E. coli population from “unwanted” signals. Thus, the presence of high concentrations of particulates can affect the enumeration of E. coli cells, and samples would have to be adjusted according to the preconcentration factor used. In spite of this, however, the results still showed that the two FISH protocols using higher temperatures could be applied to environmental samples successfully.
FIG. 8.

Cytograms of green fluorescence versus forward scatter for flow cytometric analysis of E. coli-spiked seawater samples after concentration. (A) FISH conducted at 70°C for 30 min. (B) Concentrated sample was diluted before FISH at 50°C for 20 min, following pretreatment at 90°C. (C) Concentrated sample was diluted before FISH at 70°C for 30 min. Note that noise and “unwanted signals” were gated out from the data collection.
DISCUSSION
The results presented in this paper demonstrate that a FISH protocol using temperatures higher than the widely used 46°C can be successfully applied to the flow cytometric detection of bacteria in environmental samples, using oligonucleotide probes targeting rRNA. The new FISH protocols resulted in a much shorter analytical time than for standard procedures (i.e., 25 or 30 min versus 2 to 3 h) and yet also gave higher hybridization efficiencies without compromising the fluorescence signal intensities. FISH protocols using higher temperatures have been considered to be able to effectively decrease nonspecific binding (2), and temperatures higher than 46°C have been tried by different researchers (5, 7, 15; Oppedahl et al., Abstr. Gen. Meet. Am. Soc. Microbiol., 2003), although it appears that these conditions have been applied randomly. In most cases, the general theoretical basis for selecting the FISH temperature depends only upon the melting point of the probe (2, 9). However, our results have indicated that application of higher temperatures for FISH can increase the accessibility of the targeted rRNA to oligonucleotide probes. We assumed that the higher temperatures applied to FISH (60 to 70°C for one-step FISH) or a pretreatment step (90°C for two-step FISH) could potentially serve the following multiple functions: (i) change the conformation of the secondary and tertiary structures of rRNA (e.g., dissociation of some local helices) and consequently increase the possibility of exposing the targeted sites to probes, (ii) increase the permeability of cells by changing the structure of cell walls or membranes, (iii) ensure that (or help) both oligonucleotide probes and the targeted fragments of rRNA strands maintain a linear conformation and thus help the orientation (i.e., one-to-one match) and alignment of the probes, and (iv) decrease the possibility of nonspecific binding of probes to rRNA because of the assumed linear conformation of both probes and targeted sequences on rRNA. In fact, the successful application of in situ PCR within cells (11, 16) provides strong support for the above assumptions, in particular, for the functions of the pretreatment step in a 90°C water bath. This step presumably enhances the accessibility of the rRNA sites to the probes, such as through dissociation of selected double-stranded helices in the rRNA structure. However, the actual in situ hybridization reaction was assumed to occur during the cooling phase of the sample, if the applied temperature was higher than the melting point of the probes for one-step FISH or during the second step for the two-step protocol. It may be further anticipated that the last hybridization step could take place on a time scale of seconds. In addition, the fact that the FISH efficiency was not decreased for high-temperature protocols but even increased (in comparison with that of the standard protocol) suggests that nonspecific binding was minimal, since we did not observe higher noise signals for the high-temperature protocols when we applied these protocols to nonspiked and spiked seawater samples (data not shown).
Previous studies have generally evaluated the performance of FISH by assessing the fluorescence intensity (2, 7, 9, 10, 17, 18, 20). However, as seen from the results presented above, high fluorescence signal intensity does not ensure satisfactory hybridization efficiency, possibly because of the reasons discussed in the introduction. In cases where both the enumeration and identification of the targeted microorganisms in the environmental samples are major goals, the FISH efficiency becomes a parameter as important as the signal intensity, because complete hybridization would be a primary requirement. Therefore, it is strongly suggested that both the fluorescence signal intensity and the FISH efficiency be taken into account when developing a protocol for using FISH with rRNA-targeted probes and flow cytometry to quantify target species in a natural sample.
Among the methods that have been used to quantify specific environmental microorganisms, FISH combined with flow cytometry has offered the advantages of both high resolution for taxonomic identification and automated cell counting. However, since targeted microorganisms, such as E. coli, in natural environments are likely to be at much lower concentrations than the assemblage of nontargeted microorganisms, it is necessary to concentrate the original samples before FISH and flow cytometric analysis. Clumps of cells from different taxonomic groups in concentrated environmental samples, together with cell losses and low signal-to-background noise, have been pointed out as major problems for routine application of this combination of techniques (3). Inherent difficulties also arise from the presence of autofluorescent particles, such as minerals and algae, in environmental samples and nonspecific binding of fluorescent probes to detritus particulates (8). In applying our protocols to E. coli-spiked seawater samples, we also encountered significant “unwanted” signals coming from both autofluorescence and nonspecific binding that interfered with the signals of the hybridized E. coli. This problem was addressed by using dilution, mixing, and gating out the “unwanted” signals, and satisfactory separation of the E. coli population for both samples hybridized with one-step FISH and two-step FISH was obtained. We also obtained a recovery rate of 90% for spiked E. coli cells. With the consideration that some microorganisms (e.g., dinoflagellates) of potential interest may not be able to withstand high-temperature treatments, the two protocols are provided as alternatives which may be applied to further studies. Nevertheless, these results demonstrate that the two types of high-temperature FISH protocols can be successfully applied for the detection and enumeration of specific microorganisms in environmental samples.
Acknowledgments
This study was funded by the Agency of Science and Technology Research, Singapore (R347-000-027-305).
We thank Sun Xiaofei for technical assistance and Xie Shuang and Bharti Dewangan for helping to analyze some of the flow cytometry samples.
REFERENCES
- 1.Amann, R., and W. Ludwig. 2000. Ribosomal RNA-targeted nucleic acid probes for studies in microbial ecology. FEMS Microbiol. Rev. 24:555-565. [DOI] [PubMed] [Google Scholar]
- 2.Amann, R. I., L. Krumholz, and D. A. Stahl. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J. Bacteriobiol. 172:762-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biegala, I. C., F. Not, D. Vaulot, and N. Simon. 2003. Quantitative assessment of picoeukaryotes in the natural environment by using taxon-specific oligonucleotide probes in association with tyramide signal amplification-fluorescence in situ hybridization and flow cytometry. Appl. Environ. Microbiol. 69:5519-5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buno, I., E. Moreno-Lopez, M. Jimenez-Mahillo, P. Balsalobre, D. Serrano, R. Carrion, A. Gomez-Pineda, and J. L. Diez-Martin. 2002. Post-BMT chimerism quantification by FISH on routine smears: easier, faster and more sensitive than STR-PCR. Blood 100:5271. [Google Scholar]
- 5.da Silva, C. C., and A. D. da Cruz. 2002. An easy procedure for cytogenetic analysis of aged chromospme preparation using FISH-WCP probes. Chromosome Res. 10:233-238. [DOI] [PubMed] [Google Scholar]
- 6.DeLong, E. F., G. S. Wickham, and N. R. Pace. 1989. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science 243:1360-1363. [DOI] [PubMed] [Google Scholar]
- 7.Durm, M., F. M. Haar, M. Hausmann, H. Ludwig, and C. Cremer. 1996. Optimization of fast-fluorescence in situ hybridization with repetitive alpha-satellite probes. Z. Naturforsch. Teil C 51:253-261. [DOI] [PubMed] [Google Scholar]
- 8.Ferrari, B. C., G. Vesey, K. A. Davis, M. Gauci, and D. Veal. 2000. A novel two-color flow cytometric assay for the detection of Cryptosporidium in environmental water samples. Cytometry 41:216-222. [PubMed] [Google Scholar]
- 9.Fuchs, B. M., G. Wallner, W. Beisker, I. Schwippl, W. Ludwig, and R. Amann. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs, B. M., K. Syutsubo, W. Ludwig, and R. Amann. 2001. In situ accessibility of Escherichia coli 23S rRNA to fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 67:961-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoshino, T., N. Noda, F. Tsuneda, A. Hirata, and Y. Inamori. 2001. Direct detection by in situ PCR of the amoA gene in biofilm resulting from a nitrogen removal process. Appl. Environ. Microbiol. 67:5261-5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reference deleted.
- 13.Lebaron, P., P. Catala, C. Fajon, F. Joux, J. Baudart, and L. Bernard. 1997. A new sensitive, whole-cell hybridization technique for detection of bacteria involving a biotinylated oligonucleotide probe targeting rRNA and tyramide signal amplification. Appl. Environ. Microbiol. 63:3274-3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemarchand, K., N. Parthuisot, P. Catala, and P. Lebaron. 2001. Comparative assessment of epifluorescence microscopy, flow cytometry and solid-phase cytometry used in the enumeration of specific bacteria in water. Aquat. Microb.Ecol. 25:302-309. [Google Scholar]
- 15.Prescott, A. M., and C. R. Fricker. 1999. Use of PNA oligonucleotides for the in situ detection of Escherichia coli in water. Mol. Cell. Probes 13:261-268. [DOI] [PubMed] [Google Scholar]
- 16.Sachidanandham, R., and K. Y. H. Gin. 2003. Flow cytometric detection of β-d-glucuronidase gene in wild-type bacterial cells using in-situ PCR. Biotechnol. Bioeng. 82:127-133. [DOI] [PubMed] [Google Scholar]
- 17.Schönhuber, W., B. Fuchs, S. Juretschko, and R. Amann. 1997. Improved sensitivity of whole-cell hybridization by the combination of horseradish peroxidase-labeled oligonucleotides and tyramide signal amplification. Appl. Environ. Microbiol. 63:3268-3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon, N., N. Lebot, D. Marie, F. Partensky, and D. Vaulot. 1995. Fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes to identify small phytoplankton by flow cytometry. Appl. Environ. Microbiol. 61:2506-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vives-Rego, J., P. Lebaron, and G. Nebe-von Caron. 2000. Current and future applications of flow cytometry in aquatic microbiology. FEMS Microbiol. Rev. 24:429-448. [DOI] [PubMed] [Google Scholar]
- 20.Wallner, G., R. Amann, and W. Beisker. 1993. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14:136-143. [DOI] [PubMed] [Google Scholar]
- 21.Wallner, G., R. Erhart, and R. Amann. 1995. Flow cytometric analysis of activated sludge with rRNA-targeted probes. Appl. Environ. Microbiol. 61:1859-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winkler, R., B. Perner, A. Rapp, M. Durm, C. Cremer, K. O. Greulich, and M. Hausmann. 2003. Labelling quality and chromosome morphology after low temperature FISH analyzed by scanning far-field and near-field optical microscopy. J. Microsc. 209:23-33. [DOI] [PubMed] [Google Scholar]