

Abstract
Heterocapsa circularisquama RNA virus (HcRNAV) has at least two ecotypes (types UA and CY) that have intraspecies host specificities which are complementary to each other. We determined the complete genomic RNA sequence of two typical HcRNAV strains, HcRNAV34 and HcRNAV109, one of each ecotype. The nucleotide sequences of the viruses were 97.0% similar, and each had two open reading frames (ORFs), ORF-1 coding for a putative polyprotein having protease and RNA-dependent RNA polymerase (RdRp) domains and ORF-2 encoding a single major capsid protein. Phylogenetic analysis of the RdRp amino acid sequence suggested that HcRNAV belongs to a new previously unrecognized virus group. Four regions in ORF-2 had amino acid substitutions when HcRNAV34 was compared to HcRNAV109. We used a reverse transcription-nested PCR system to amplify the corresponding regions and also examined RNAs purified from six other HcRNAV strains with known host ranges. We also looked at natural marine sediment samples. Phylogenetic dendrograms for the amplicons correlated with the intraspecies host specificities of the test virus strains. The cloned sequences found in sediment also exhibited considerable similarities to either the UA-type or CY-type sequence. The tertiary structure of the capsid proteins predicted using computer modeling indicated that many of the amino acid substitutions were located in regions on the outside of the viral capsid proteins. This strongly suggests that the intraspecies host specificity of HcRNAV is determined by nanostructures on the virus surface that may affect binding to suitable host cells. Our study shows that capsid alterations can change the phytoplankton-virus (host-parasite) interactions in marine systems.
Only five RNA viruses are known to infect marine eukaryotic microorganisms. The following four viruses are single-stranded RNA (ssRNA) viruses: Heterosigma akashiwo RNA virus (HaRNAV) infects the noxious bloom-forming raphidophyte Heterosigma akashiwo (Raphidophyceae) (26); Rhizosolenia setigera RNA virus infects the bloom-forming diatom Rhizosolenia setigera (20); Heterocapsa circularisquama RNA virus (HcRNAV) infects the bivalve-killing bloom-forming dinoflagellate Heterocapsa circularisquama (30); and Schizochytrium sp. single-stranded RNA virus (SssRNAV) infects the marine fungoid protist Schizochytrium sp. (Labyrinthulae, Thraustochytriaceae) (27). One of the five viruses is a double-stranded RNA (dsRNA) virus (Micromonas pusilla RNA virus) that infects the cosmopolitan phytoplankton Micromonas pusilla (1). Detailed genomic analysis has been performed for two of these viruses, HaRNAV (18) and SssRNAV (Takao et al., unpublished data). Hence, genomic studies of the three other marine RNA viruses are required. In this paper we describe the genome of HcRNAV, which infects H. circularisquama.
HcRNAV infection is strain specific rather than species specific because about 6,000 combinations of cross-infection tests between H. circularisquama strains and HcRNAV strains showed that HcRNAV strains are divided roughly into two ecotypes (types UA and CY) that have complementary strain-specific infectivity (30). Fundamental characterization of HcRNAV34 and HcRNAV109, which are typical type UA and CY virus strains, respectively, has shown that these viruses are similar in terms of morphology and genome features; they are polyhedral and ca. 30 nm in diameter, lack an outer membrane and tail structure, and contain a single molecule of linear ssRNA that is approximately 4.4 kb long. A field survey carried out in Ago Bay, Japan, in 2001 revealed the intimate ecological relationship between HcRNAV and H. circularisquama blooms; the results indicated that there are at least two distinct virus ecotypes that have complementary distinct host specificities in natural water populations and dynamics of intraspecies host-parasite relationships that are independent of one another (21). Hence, the dynamics of each virus ecotype may reflect the dynamics of its host ecotype, and the two ecotypes of HcRNAV play a significant role in regulating the intraspecies diversity, as well as the biomass, of H. circularisquama populations in the natural environment.
The genome structure and the phylogeny of HcRNAV are poorly understood, and we decided to study the molecular mechanism underlying the intraspecies host specificity. In this study, we (i) compared the genome structures of two typical HcRNAV strains having different intraspecies host specificities, (ii) constructed a dendrogram describing the phylogeny of HcRNAV, and (iii) predicted the chemistry that determines the intraspecies host range of HcRNAV. Below we also discuss the ecological implications of our findings.
MATERIALS AND METHODS
Hosts and viruses.
Eight HcRNAV strains and 14 H. circularisquama strains were used (Table 1). All of the virus strains were free of bacterial contamination. The origin of each strain has been reported previously (30). Algal cultures were grown in modified SWM3 medium enriched with 2 nM Na2SeO3 (4, 10, 12) and were incubated using a cycle consisting of 12 h of light and 12 h of darkness; the light (130 to 150 μmol photons m−2 s−1) was provided by cool white fluorescent illumination at 20°C. Because HcRNAV has a strain-specific host range (30), two typical HcRNAV strains, HcRNAV34 (type UA) and HcRNAV109 (type CY), were grown using suitable hosts, H. circularisquama strains HU9433-P and HCLG-1, respectively (Table 1). The differences in intraspecies host specificity of HcRNAV strains are shown in Table 1. Four type UA HcRNAV strains and four type CY HcRNAV strains were used.
TABLE 1.
Intraspecies host specificities of HcRNAV strains tested in this study
 |
Virus strain that was fully sequenced.
Nucleotide sequence and open reading frame (ORF) analysis.
HcRNAV34 and HcRNAV109 particles were collected by the method described by Tomaru et al. (30). Viral RNAs were isolated and purified using an RNeasy Mini total RNA purification kit (QIAGEN). The purified RNA was reverse transcribed, forming cDNA, using a Time Saver cDNA synthesis kit (Amersham Biosciences Corp.) and random primers according to the manufacturer's recommendations. NotI/EcoRI adaptors (Amersham Biosciences Corp.) were ligated to the resultant dsDNA fragments, and the 5′ ends were phosphorylated using T4 polynucleotide kinase (Amersham Biosciences Corp.) The fragments were ligated to EcoRI-cleaved dephosphorylated DNA and labeled with pBlueScript SK(+) (Stratagene) using a Ligation High kit (Toyobo Co., Ltd.). The ligated dsDNA fragments were transformed into Escherichia coli DH5α (Toyobo Co., Ltd.) and were sequenced using the dideoxy method with an ABI PRISM 3100 genetic analyzer (Applied Biosystems). The fragment sequences were reassembled using the DNASIS-Mac software (Hitachi Software Engineering).
The 5′ end of the viral genome was cloned by 5′ rapid amplification of cDNA ends (5′-RACE) using the 5′-RACE System for Rapid Amplification of cDNA Ends, version 2.0 (Invitrogen); the 5′-terminal nucleotides were determined by comparing the sequences of the four clones (data not shown). To reduce the secondary structure of the isolated RNA, first-strand cDNA synthesis was done in the presence of 5% dimethyl sulfoxide (Wako Pure Chemical Industries, Ltd).
3′-RACE analysis was performed to determine the 3′ end sequence of the viral genome lacking a poly(A) tail. Fractionated viral genomic RNAs were first polyadenylated using a poly(A) polymerase (Ambion, Inc.) for 1 h at 37°C. Then the first-strand cDNAs were synthesized using the SuperscriptIII First-Strand synthesis system for reverse transcription (RT)-PCR (Invitrogen) with a poly(T) adaptor primer. The PCR was carried out using the corresponding adaptor primer and the HcRNAV-specific primer MCPF3 (5′-TAC GGA GGT GAT TTC TTT TAT G-3′). The PCR products were size selected, TA cloned using a TOPO TA cloning kit (Invitrogen), and sequenced with an ABI PRISM 3100 genetic analyzer (Applied Biosystems). The 3′-terminal nucleotides were also determined by comparison to the sequences of six clones (data not shown). A possible secondary structure for the 3′-terminal 49 nucleotides of each virus strain was predicted by the computer algorism given by Mathews et al. (19).
Automated comparisons of the complete sequences of HcRNAV34 and HcRNAV109 were performed using BLAST (Basic Local Alignment Search Tool).
Phylogenetic analysis of HcRNAV RdRp region.
A phylogenetic analysis was performed with the deduced amino acid sequence of the putative RNA-dependent RNA polymerase (RdRp) region in ORF-1. Multiple alignments were constructed using the CLUSTALW analysis system (29) and were manually corrected. The positions with gaps were removed for phylogenetic analysis. Phylogenetic trees were constructed using MEGA 3 (17) with the Jones-Taylor-Thormton matrix neighbor-joining methods. The following sequences were used (database accession numbers [from the NCBI database unless indicated otherwise] are shown in parentheses): Aichi virus (AIV) (AB010145), barley yellow dwarf virus (BYDV) (BAA01054), bean pod mottle virus (BPMV) (NC_003496), beet chlorosis virus (BChV) (AAK49964), bovine enteric calicivirus (BoCV) (AJ011099), cowpea severe mosaic virus (CPSMV) (M83830), cricket paralysis virus (CrPV) (NC_003924), cucurbit aphid-borne yellows virus (CABYV) (CAA54251), deformed wing virus (DWV) (NC_004830), Drosophila C virus (DCV) (NC_001834), H. akashiwo RNA virus (HaRNAV) (NC_005281), human poliovirus (PV) (V01149), lucerne transient streak virus (LTSV) (NP_736596), mushroom bacilliform virus (MBV) (AAA53090), Norwalk virus (NV) (M87661), parsnip yellow fleck virus (PYFV) (D14066), pea enation mosaic virus (PEMV) (AAA72297), Poinsettia latent virus (PnLV) (CAI34771), rabbit hemorrhagic disease virus (RHDV) (1KHVA), rice tungro spherical virus (RTSV) (M95497), rice yellow mottle virus (RYMV) (CAE81345), ryegrass mottle virus (RGMoV) (NP_736587), sacbrood virus (SBV) (NC_002066), Taura syndrome virus (TSV) (NC_003005), Schizochitrium single-stranded RNA virus (SssRNAV) (DDBJ accession no. AB193726), and the RdRp sequences of HcRNAV34 and HcRNAV109 (identical to each other at the amino acid sequence level; DDBJ accession numbers AB218608 and AB218609, respectively).
RT-nested PCR.
A nested PCR was designed to amplify the fragments of ORF-2 in which there were substitutions that were found to result in amino acid differences between HcRNAV34 and HcRNAV109. Primers MCPF1 (5′-CGG GAT CCA TGA CCC GTC CCC TAG CTC TTA CC-3′; an added BamHI restriction site is underlined), MCPF2 (5′-CAC GAC CAG CTT GAT CAC GTC CAG-3′), and MCPR1 (5′-CGG AAT TCT TAA GCA GCC ATC AAT GCT GGC AT-3′; an added EcoRI restriction site is underlined) were designed based on the nucleotide sequences of HcRNAV34 and HcRNAV109 (Fig. 1A). RNAs of six HcRNAV strains with known intraspecies host ranges (Table 1) and natural sediment samples collected from Ago Bay, Mie Prefecture, Japan, on 6 August 2001 and 12 August 2002 (stored at 10°C in the dark) were extracted with TRIzol LS reagent (Invitrogen); for each purification, 0.25 ml of fresh virus suspension or natural sediment were used. The resulting crude RNAs were reverse transcribed to obtain cDNAs with a High Fidelity RNA PCR kit (Takara Bio Inc.) using random primers according to the manufacturer's recommendations. The cDNAs were used as templates for the first PCR with a High Fidelity RNA PCR kit. The first PCR amplification was performed using a 100-μl mixture containing 20 μl of cDNA solution (prepared as described above), 10 μl of 10× Probest buffer, 2 μl of a deoxynucleoside triphosphate mixture (each deoxynucleoside triphosphate at a concentration of 10 mM), 2.5U of Probest DNA polymerase, and 20 pmol of primers MCPF1 and MCPR1. The PCR was performed using a GeneAmp PCR System 9700 (Applied Biosystems) with the following cycles: 5 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 3 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 59°C for 30 s, and extension at 72°C for 3 min. In the second PCR, we diluted each first PCR product 50-fold with ultrapure water, and these products were used as the templates. The second PCR amplification was performed using a 50-μl mixture containing 1.2 μl of diluted product from the first PCR, 1× Z-Taq buffer (Takara Bio Inc.), each deoxynucleoside triphosphate at a concentration of 200 μM, 4 pmol of primers MCPF2 and MCPR1, and 1.25 U of Z-Taq DNA polymerase (Takara Bio Inc.) and the GeneAmp PCR System 9700 (Applied Biosystems); it consisted of 35 cycles of denaturation at 98°C for 1 s, annealing at 58°C for 2 s, and extension at 72°C for 10 s. The resultant products were electrophoresed in 2% (wt/vol) agarose S gels (Nippon Gene Co., Ltd.). The nucleic acids were visualized by ethidium bromide staining. The bands were excised from the gel and purified using GenElute agarose spin columns (Supelco, Inc.) according to the manufacturer's recommendations. The resultant PCR products were ligated into the pGEM-T Easy vector (Promega). DNA sequencing was performed using an ABI PRISM 3100 genetic analyzer (Applied Biosystems), and the resulting fragment sequences were aligned using GENETYX-WIN, version 5.1.1 (Software Development Co., Ltd.). For samples derived from natural sediment collected on 6 August 2001 and 12 August 2002, two clones (S1-01 and S1-02) and 12 clones (S2-01 to S2-12) were ligated with target inserts and analyzed. A phylogenetic analysis was performed by using the deduced amino acid sequence of a corresponding region and the neighbor-joining method (25).
FIG. 1.

Genome of HcRNAV. (A) Schematic genome structure of HcRNAV34 and HcRNAV109 and the positions of the primers. Note the variable regions in ORF-2 correspond to those shown in Fig. 5. (B) Possible secondary structure for the 3′-terminal 49 nucleotides of HcRNAV34 and HcRNAV109.
Protein sequencing.
Purified HcRNAV virions were electrophoresed using denaturing sodium dodecyl sulfate-polyacrylamide gels stained with a Reverse-Staining kit (ATTO). The single major structural protein band was excised and purified using ATTOPREP spin columns (AB-1170; ATTO) according to the manufacturer's recommendations. The N-terminal amino acid residues were sequenced using Edman degradation (6).
Protein tertiary structure prediction.
The tertiary structure of the ORF-2 products (capsid proteins of HcRNAV34 and HcRNAV109) was partially predicted based on the amino acid sequence by using a protein discovery full automatic modeling system (PDFAMS; In-Silico Sciences, Inc.) (http://www.pd-fams.com/) and the black beetle virus (Nodaviridae) capsid protein as a reference protein (13, 22, 28). The results were manually modified so that amino acid residues that were different in HcRNAV34 and HcRNAV109 were red, and the data were viewed using the RasMol 2.6 Beta-2a program (http://www.umass.edu/microbio/rasmol/getras.htm).
Nucleotide sequence accession numbers.
The complete nucleotide sequences of the HcRNAV34 and HcRNAV109 genomes have been deposited in the DDBJ database under accession numbers AB218608 and AB218609, respectively.
RESULTS AND DISCUSSION
Features of the HcRNAV genomes.
The complete nucleotide sequences of the ssRNA virus HcRNAV34 and HcRNAV109 genomes were determined. These genomes are 4,375 and 4,391 nucleotides (nt) long, respectively, values which are in close agreement with the 4.4-kb genome size predicted using denaturing gel electrophoresis (30). The viruses were not retained by a poly(A) tail purification column (30), and reproducible 3′-RACE results were obtained only when the viral genomes were enzymatically tailed with poly(A); we concluded that the RNA genomes of both HcRNAV34 and HcRNAV109 lack a poly(A) tail. The two viral genomes are very similar to each other (∼97.0% similarity). Assuming that the initiation codon for viral replication is universal (AUG), both HcRNAV genomes were predicted to contain two major open reading frames, ORF-1 and ORF-2 (Fig. 1A).
ORF-1 of HcRNAV34 and ORF-1 of HcRNAV109 are 3,000 and 3,015 nt long, respectively; the size difference reflects deletion of 15 nt in the former strain (Fig. 1A). There are two possible initiation codons for ORF-1, which are 19 and 313 nt downstream of the 5′ end. Generally, the length of 5′ untranslated regions (5′-UTRs) of eukaryote mRNAs is 3 to 742 nt (14), and there are two consensus features for the sequence around functional AUG in eukaryotic mRNAs (15, 16), (i) a purine at position −3 (assuming that the A of initiation codon [AUG] is at position 1) and (ii) a G at position 4. Based on this information, the initiation codon is most likely AUG 19 nt downstream of the 5′ end for ORF-1. The protein sequence predicted by the ORF-1 nucleotide sequence contains two conserved domains from the RNA viruses; one is a serine protease domain, and the other is the RdRp domain. The best-fit BLAST comparison sequences were the serine protease domain of lucerne transient streak virus (e value, 2e-4) and the RdRp domain from mushroom bacilliform virus and Poinsettia latent virus (e value, 2e-11). Significant BLAST correlations with polyproteins that included both domains were found for lucerne transient streak virus, cocksfoot mottle virus, and rice yellow mottle virus. This suggests that ORF-1 codes for a polyprotein which is translated and then cleaved into smaller functional proteins, including RdRp (7).
The length of ORF-2 for both virus strains is 1,080 nt. We determined that the 5′ seven deduced amino acids of the ORF-2 product coincided completely with sequences found at the N terminus of HcRNAV's single major structural protein (TRPLALT), as shown by Edman degradation and the molecular weights predicted from the deduced amino acid sequences. The molecular masses of HcRNAV34 and HcRNAV109 ORF-2 are 38.2 and 38.3 kDa, respectively, and the molecular masses of the major structural proteins were ∼38 kDa, as predicted by denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (30). We concluded that ORF-2 codes for the single major structural protein of HcRNAV. In addition, the ORF-2 products of the two HcRNAV strains exhibited low levels of homology (∼10%) to the black beetle virus capsid protein (data not shown).
Besides the major ORFs, four and six minor ORFs (three and four ORFs on the complementary minus-sense RNA) that could code for proteins consisting of >100 amino acids and that overlap ORF-1 in HcRNAV34 and HcRNAV109, respectively, were also predicted, but none of them showed significant BLAST similarity to any known proteins (data not shown). Work needs to be done to determine the functions of these ORFs.
If the predictions described above are correct, the 5′-UTRs of HcRNAV34 and HcRNAV109 are 18 nt long and are followed by a 162-nt intercistronic region between ORF-1 and ORF-2, and the 3′-UTRs of HcRNAV34 and HcRNAV109 are 115 and 116 nt long, respectively (Fig. 1A). This reflects the difference in the predicted stem-loop structure at the 3′ termini (Fig. 1B). The stem-loop structures are considered to be essential in the replication process and may prevent enzymatic digestion of the genomic RNAs (2). The UTRs account for ∼6.7% of the HcRNAV genome.
Phylogeny of HcRNAV RdRp.
A phylogenetic analysis of the deduced amino acid sequence in the RdRp domain encoded by HcRNAV ORF-1 was performed, and this analysis showed that the amino acid sequences of HcRNAV34 and HcRNAV109 were identical. The positive-sense ssRNA viruses used in this study were roughly divided into two phylogenetic clusters, one containing the Tetraviridae, Barnaviridae, and Leteoviridae and the other containing the Picornaviridae, Caliciviridae, Dicstroviridae, Iflavirus, Comoviridae, Sequiviridae, and two marine plankton viruses, HaRNAV and SssRNAV. HcRNAV forms a sister group with the former cluster at a bootstrap value of 99% (Fig. 2). However, HcRNAV was deeply branched and apparently distinct from the cluster containing the Tetraviridae, Barnaviridae, and Leteoviridae at a bootstrap value of 97% (Fig. 2). We concluded that HcRNAV belongs to a new unrecognized positive-sense single-stranded RNA virus group.
FIG. 2.

Phylogenetic tree calculated from confidently aligned regions of amino acid sequences of RNA-dependent RNA polymerase alleles. The numbers at the nodes are bootstrap values (percentages) based on 1,000 samples. Nodes with bootstrap values less than 70% were collapsed. Scale bar = 0.5 fixed mutation per amino acid position. CABYV, cucurbit aphid-borne yellows virus; BChV, beet chlorosis virus; PEMV-1, pea enation mosaic virus 1; BYDV, barley yellow dwarf virus; PnLV, Poinsettia latent virus; MBV, mushroom bacilliform virus; RGMoV, ryegrass mottle virus; RYMV, rice yellow mottle virus; LTSV, lucerne transient streak virus; BoCV, bovine enteric calicivirus; NV, Norwalk virus; RHDV, rabbit hemorrhagic disease virus; PV, human poliovirus; BPMV, bean pod mottle virus; CPSMV, cowpea severe mosaic virus; RTSV, rice tungro spherical virus; PYFV, parsnip yellow fleck virus; SBV, sacbrood virus; DWV, deformed wing virus; TSV, Taura syndrome virus; DCV, Drosophila C virus; CrPV, cricket paralysis virus.
The first BLAST analysis showed that proteins similar to HcRNAV RdRp were proteins of land viruses infecting fungi or plants, including mushroom bacilliform virus (e value, 2e-11), Poinsettia latent virus (e value, 2e-11), ryegrass mottle virus (e value, 5e-11), lucerne transient streak virus (e value, 3e-10), and others. These data indicate that there are a great number of unrecognized viruses in the deep blanch of HcRNAV, and it may be possible to predict the evolutionary history of land viruses and marine viruses from the ancestral viruses.
Comparison of genome structure and intraspecies host specificity.
The most noticeable difference between the HcRNAV34 and HcRNAV109 genomes was the four regions (regions I to IV) in ORF-2 that resulted in high frequencies of amino acid substitutions (Fig. 1). We used an RT-nested PCR system so the corresponding variable regions were specifically amplified from the RNAs. We also purified this region from other HcRNAV strains tested with known host ranges and natural marine sediment samples. The results of the phylogenetic analysis of the amino acid sequences of the variable regions completely coincided with the intraspecies host specificities of the virus strains tested; i.e., type CY virus strains and type UA virus strains were divided into distinct clusters, which was supported at a bootstrap value of 100% (Fig. 3). In addition, sediment clonal sequences also exhibited considerable similarities to either the type UA or the type CY sequence (Fig. 3). Prediction of these capsid proteins' tertiary structures using computer modeling indicated that the three ORF-2 products form a tightly combined trimer capsid plate (i.e., T = 1 icosahedral virus) and that the variable regions (with a number of amino acid substitutions) are specifically located on the outside, on the hydrophilic side of the capsid (Fig. 4). Among the 14 amino acid residues that were distinctly different in the two ecotypes in regions I to IV (Fig. 5), one, two, and six residues were predicted to be on the outside of the capsid proteins (exposed to ambient water) only in type UA viruses, only in type CY viruses, and in both types, respectively (Fig. 5). In contrast, no significant amino acid substitution was found on the interior of the capsid proteins of HcRNAV34 and HcRNAV109 (Fig. 4). The results suggest that the intraspecies host specificity of HcRNAV is most likely determined by the exterior structures on the major capsid protein that may affect the ability of the virus particles to bind to their host ecotype, and the changes in the capsid structure in the four separate sites likely control the affinity for the host's receptors. Viral entry into the host cell through the endosomal pathway also can be a barrier for virus infection because it requires an uncoating step (11). This requires a considerable amount of surface structural change on the virion to adapt to a new entry process.
FIG. 3.
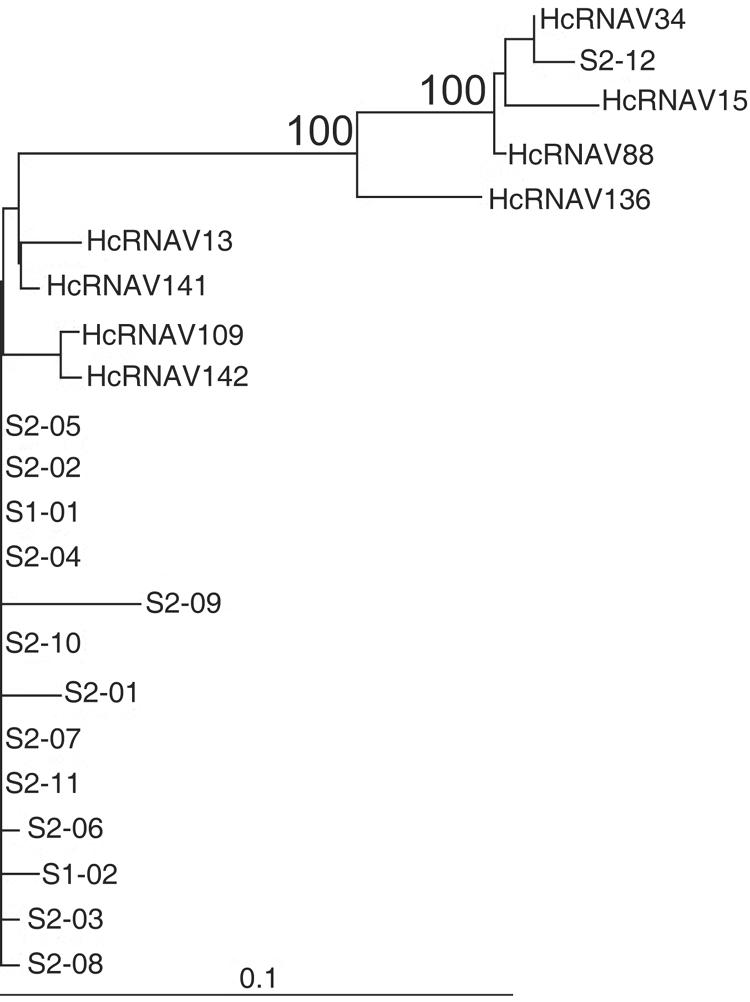
Phylogenetic tree calculated from confidently aligned regions of amino acid sequences encoded by the major capsid protein gene (ORF-2) fragments. A total of 245 amino acid sites were used to construct the tree. The numbers at the nodes are bootstrap values (percentages) based on 1,000 samples. Nodes with bootstrap values less than 70% were collapsed. Scale bar = 0.1 fixed mutation per amino acid position.
FIG. 4.

Tertiary structures of the major capsid proteins of HcRNAV34 (A to C) and HcRNAV109 (D to F) predicted by computer modeling. Three monomers that make up the capsid trimers are indicated by different colors (green, blue, and yellow), and the amino acid molecules where complete substitution was observed between the UA type and the CY type are indicated by red. (A and D) Surface side view; (B and E) reverse side view; (C and F) side view.
FIG. 5.

Amino acid alignment of the major capsid protein gene fragments of virus strains (four type UA HcRNAV strains and four type CY HcRNAV strains). Highly variable regions (regions I to IV) are highlighted. The asterisks indicate the amino acid positions at which complete substitution was observed between the UA type and the CY type for the eight strains tested.
There are two additional differences in the genomes of HcRNAV34 and HcRNAV109. One is the 15-nt deletion in HcRNAV34 ORF-1. This deletion is highly conserved, and evidently the intraspecies host specificity is not determined by the 15-nt deletion located in ORF-1 (Tomaru et al., unpublished data). The other structural difference involves the 3′ end stem-loop structure (the length of the stem and the size and sequences of the loop) (Fig. 1B). Further study is needed to determine the relationship between the stem-loop structure and the intraspecies host specificity.
Ecological implications.
Previous field surveys and laboratory experiments have shown that there are at least two distinct (and independent) host-virus systems for H. circularisquama and HcRNAV (21, 30). In other words, multiple ecotypes of the host and virus coexist within natural blooms of H. circularisquama. Each virus ecotype has its own concentration fluctuation pattern that presumably reflects the changes in abundance of its host ecotype (21). It is interesting that these two virus ecotypes are ∼97% identical despite the presence of complementary host-parasite populations. The amino acid products of ORF-1, the polyprotein gene, and ORF-2, the capsid protein gene, are 98.5% and 91.9% identical in ecotype strains HcRNAV34 and HcRNAV109, respectively. Virus strains having common replication mechanisms that are differentiated evolutionarily may exhibit structural differences in the capsid protein determining specific host ranges. Presumably due to the dominance of the two distinct ecotypes of H. circularisquama, the two virus ecotypes might have been ecologically selected and propagated. However, at present, we have not established practical methods for identifying the two host ecotypes because their morphologies are very similar and their rRNA gene nucleotide sequences, including internal transcribed spacer regions, are almost completely identical (data not shown).
The emergence of host range variants due to mutation has also been reported for canine parvovirus and feline parvovirus, which have single-stranded DNA genomes. Feline parvovirus naturally infects cats and some carnivores but not dogs (24), but a canine parvovirus type 2 strain that infected both dogs and feline cells in vitro did emerge; however, this strain did not infect cats (8, 31). Subsequently, the canine parvovirus type 2strain was replaced in nature by an antigenically variant strain, canine parvovirus type 2a, that had gained the ability to infect cats (23). These studies show that substitution of several amino acid residues in the viral structural proteins by mutation plays an important role in determining the host specificity (3, 9, 31). If an RNA virus's replication is accompanied by a considerable increase in diversity because there is no RdRp proofreading mechanism (5), this would allow the emergence of host range variants. Because of this, such RNA virus variants may occur much more frequently. We found that a relationship between HcRNAV and H. circularisquama must be one of the ordinary examples resulting from the emergence of host range variants and ecological selection for viruses in natural waters. We concluded that this study suggests that alterations in the viral surface structure can change the host-virus interaction in marine systems, similar to what happens in canine parvovirus.
Acknowledgments
We are grateful to T. Okuno, K. Mise (Kyoto University, Kyoto, Japan), N. Kamiya, and H. Naitow (RIKEN Harima Institute, Hyogo, Japan) for their helpful advice.
This work was partially supported by funds from the Industrial Technology Research Grant Program in 2004 from the New Energy and Industrial Technology Development Organization (NEDO) of Japan.
REFERENCES
- 1.Brussaard, C. P., A. A. Noordeloos, R. A. Sandaa, M. Heldal, and G. Bratbak. 2004. Discovery of a dsRNA virus infecting the marine photosynthetic protist Micromonas pusilla. Virology 319:280-291. [DOI] [PubMed] [Google Scholar]
- 2.Buck, K. W. 1996. Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv. Virus Res. 47:159-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang, S. F., J. Y. Sgro, and C. R. Parrish. 1992. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J. Virol. 66:6858-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, L. C. -M., T. Edelstein, and J. B. McLachlan. 1969. Bonnemaisonia hamifera Hariot in nature and in culture. J. Phycol. 5:211-220. [DOI] [PubMed] [Google Scholar]
- 5.Domingo, E., C. Escarmis, N. Sevilla, A. Moya, S. F. Elena, J. Quer, I. S. Novella, and J. J. Holland. 1996. Basic concepts in RNA virus evolution. FASEB J. 10:859-864. [DOI] [PubMed] [Google Scholar]
- 6.Edman, P. 1950. Method for determination of the amino acid sequence in peptides. Acta Chem. Scand. 4:283-293. [Google Scholar]
- 7.Goldbach, R. 1990. Plant viral proteinases. Semin. Virol. 1:335-346. [Google Scholar]
- 8.Horiuchi, H., M. Goto, N. Ishiguro, and M. Shinagawa. 1994. Mapping of determinants of the host range for canine cells in the genome of canine parvovirus using canine parvovirus/mink enteritis virus chimeric viruses. J. Gen. Virol. 75:1319-1328. [DOI] [PubMed] [Google Scholar]
- 9.Hueffer, K., J. S. L. Parker, W. S. Weichert, R. E. Geisel, J.-Y. Sgro, and C. R. Parrish. 2003. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 77:1718-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imai, I., S. Itakura, Y. Matsuyama, and M. Yamaguchi. 1996. Selenium requirement for growth of a novel red tide flagellate, Chattonella verruculosa (Raphidophyceae), in culture. Fish. Sci. (Tokyo) 62:834-835. [Google Scholar]
- 11.Imajoh, M., K.-I. Yagyu, and S.-I. Oshima. 2003. Early interactions of marine birnavirus infection in several fish cell lines. J. Gen. Virol. 84:1809-1816. [DOI] [PubMed] [Google Scholar]
- 12.Itoh, K., and I. Imai. 1987. Raphidophyceae, p. 122-130. In Japan Fisheries Resource Conservation Association (ed.), A guide for studies of red tide organisms. Shuwa, Tokyo, Japan.
- 13.Iwadate, M., K. Ebisawa, and H. Umeyama. 2001. Comparative modeling of CAFASP2 competition. Chem. Bio. Info. J. 1:136-148. [Google Scholar]
- 14.Kozak, M. 1983. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles,. Microbiol. Rev. 47:1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozak, M. 1989. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol. Cell. Biol. 9:5073-5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozak, M. 1986. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 44:283-292. [DOI] [PubMed] [Google Scholar]
- 17.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci. 10:189-191. [DOI] [PubMed] [Google Scholar]
- 18.Lang, A. S., A. I. Culley, and C. A. Suttle. 2004. Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology 320:206-217. [DOI] [PubMed] [Google Scholar]
- 19.Mathews, D. H., M. D. Disney, J. L. Childs, S. J. Schroeder, M. Zuker, and D. H. Turner. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for predicgtion of RNA secondary structure. Proc. Natl. Acad. Sci. USA 101:7287-7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagasaki, K., Y. Tomaru, N. Katanozaka, Y. Shirai, K. Nishida, S. Itakura, and M. Yamaguchi. 2004. Isolation and characterization of a novel single-stranded RNA virus infecting the bloom-forming diatom Rhizosolenia setigera. Appl. Environ. Microbiol. 70:704-711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagasaki, K., Y. Tomaru, K. Nakanishi, N. Hata, N. Katanozaka, and M. Yamaguchi. 2004. Dynamics of Heterocapsa circularisquama (Dinophyceae) and its viruses in Ago Bay, Japan. Aquat. Microb. Ecol. 34:219-226. [Google Scholar]
- 22.Ogata, K., and H. Umeyama. 2000. An automatic homology modeling method consisting of database searches and simulated annealing. J. Mol. Graph. Model. 18:258-272, 305-306. [DOI] [PubMed] [Google Scholar]
- 23.Parrish, C. R., C. Aquadro, M. L. Strassheim, J. F. Evermann, J.-Y. Sgro, and H. Mohammed. 1991. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 65:6544-6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parrish, C. R., C. F. Aquadro, and L. E. Carmichael. 1988. Canine host range and a specific epitope map along with variant sequences in the capsid protein gene of canine parvovirus and related feline, mink and raccoon parvoviruses. Virology 166:293-307. [DOI] [PubMed] [Google Scholar]
- 25.Saitou, N., and M. Nei. 1987. The neighbor joining method. A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 26.Tai, V., J. E. Lawrence, A. S. Lang, A. M. Chan, A. I. Culley, and C. A. Suttle. 2003. Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma akashiwo (Raphidophyceae). J. Phycol. 39:343-352. [Google Scholar]
- 27.Takao, Y., K. Nagasaki, K. Mise, T. Okuno, and D. Honda. 2005. Isolation and characterization of a novel single-stranded RNA virus infectious to a marine fungoid protist, Schizochytrium sp. (Thraustochytriaceae, Labyrinthulae). Appl. Environ. Microbiol. 71:4516-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda-Shitaka, M., D. Takaya, C. Chiba, H. Tanaka, and H. Umeyama. 2004. Protein structure prediction in structure based drug design. Curr. Med. Chem. 11:551-558. [DOI] [PubMed] [Google Scholar]
- 29.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomaru, Y., N. Katanozaka, K. Nishida, Y. Shirai, K. Tarutani, M. Yamaguchi, and K. Nagasaki. 2004. Isolation and characterization of two distinct types of HcRNAV, a single-stranded RNA virus infecting the bivalve-killing microalga Heterocapsa circularisquama. Aquat. Microb. Ecol. 34:207-218. [Google Scholar]
- 31.Truyen, U., J. F. Evermann, E. Vieler, and C. R. Parrish. 1996. Evolution of canine parvovirus involved loss and gain of feline host range. Virology 215:186-189. [DOI] [PubMed] [Google Scholar]