

INTRODUCTION
This discussion attempts to present a simple explanation of the current theory of error catastrophe and why it does not herald a paradigm shift in antiviral strategy. In the main text of the paper, the workings of a simple model of error catastrophe are examined to demonstrate what actually causes error catastrophe. The Appendix contains a more detailed discussion of the original error threshold model of Eigen and Schuster and how it applies to a viral quasispecies.
RNA viruses are said to replicate at the edge of “error catastrophe” (18). Error catastrophe is a term coined to describe the supposed inability of a genetic element to be maintained in a population as the fidelity of its replication machinery decreases beyond a certain threshold value. Error catastrophe has been invoked as a theoretical basis for treatment of viral infection with drugs that would push the error rate for copying of the viral genome beyond this threshold (1, 4, 5, 6, 7, 9, 10, 14, 15, 16, 17, 19, 22, 25, 26, 28, 29, 40). Numerous publications aimed at the detection of virus extinction by error catastrophe induced by viral mutagens have appeared in recent years (8, 11, 12, 13, 24, 27, 33, 34, 35, 36, 38, 39, 46, 47).
The catastrophic effect of high error rates was originally predicted in a mathematical model by Eigen and Schuster (20), in which a master genetic sequence replicated in competition with a collection of variants generated by errors in replication of the master sequence. In the simplest versions of the model (41), the variants all typically have a lower replication rate than the master sequence, and the effect of their replication errors is to convert one variant into another. When the distribution of genomes in such a replicating system was calculated to steady state, it was found that beyond a threshold error rate the master sequence effectively disappeared, becoming no more frequent than any single variant sequence. Eigen and Schuster referred to this hypothetical redistribution of the genetic information of the system as an error catastrophe (not to be confused with the theory of ageing that is also called error catastrophe [30, 31, 32]). Various treatments of the basic model have appeared in the literature since publication of Eigen and Schuster's original paper (2, 3, 21, 42, 42, 44, 45).
We present here an examination of the theoretical basis for error catastrophe as predicted by the accepted mathematical simulations. For this purpose, we have constructed our own relatively simple model, based on ordinary differential equations, that reproduces error catastrophe. Using this model, we show that an error threshold is predicted to occur solely because of the implausible proposition that all progeny genomes that are not the master sequence continue to replicate at a finite rate no matter how many replication errors they contain, whereas replication of the master sequence is disqualified by a single error in the progeny genome. The disappearance of the master sequence at the error threshold is predicated on competition between the progeny and the master sequence to infinite time. We will show that, without the assumption that all mutants, no matter what their sequences, continue to replicate, mathematical models do not predict error catastrophe.
MODELS THAT PRODUCE ERROR CATASTROPHE
Our model is patterned after that of Swetina and Schuster (41) but is highly simplified to make its workings more transparent to a wider audience. In it (see Appendix) v0 represents any genome of unspecified length with the exact master sequence with no errors, and v>0 is any genome with one or more errors. Together, these two classes of genomes make up the viral quasispecies that in Swetina and Schuster's model was explicitly defined as classes of genomes 50 nucleotides in length, differing from the master sequence by 0, 1, 2, or 50 nucleotides. We shall refer to the sequence of v0 as the wild-type genome and the various sequences of v>0 as mutant genomes. When the wild-type genome is copied to produce a progeny molecule, an average number (m) of errors is randomly introduced into the progeny. Since errors are random, some progeny of wild-type genomes receive more than m errors, some receive fewer than m errors, and some receive no errors. From the Poisson distribution, we calculate the fraction of progeny genomes receiving no errors as e−m, and only those genomes correspond to the wild-type sequence. All other genomes are mutant.
The effect of errors on the replication rates of wild-type and mutant genomes.
In our model, k0 is the replication rate constant of v0, and k1 is the replication rate constant of all v>0 genomes. According to the conditions assumed in the original model of error catastrophe, all mutant genomes replicate at one-tenth the wild-type rate, so k1 is equal to 0.1 · k0. However, the apparent rate constant of the wild type, k0a, is reduced directly in proportion to the fraction of master sequences produced and therefore varies with the error rate such that k0a = e−m · k0; that is, the higher the mutation frequency, the lower the rate of production of wild-type genomes. In contrast, the replication rate constant of the mutants is not affected by the error rate, no matter how high, because errors simply convert one mutant into another with the same rate of growth (except for rare errors that cause reversion to the wild type). The effect of the error rate m on k0a and k1 is illustrated in Fig. 1A. Swetina and Schuster's model and ours both predict that at values of m < −ln 0.1, or 2.303, the growth rate of the wild type is greater than that of the mutants, while for m > 2.303, the mutant growth rate is greater. Note that the model does not predict a sudden or catastrophic drop in k0a at a “threshold” error rate but produces a continuous first-order decline (Fig. 1A, inset).
FIG. 1.

Simplified error catastrophe model. (A) Apparent rate constants as a function of error rate m. The apparent rate constant k0a for wild-type replication (black) was calculated as k0a is equal to e−m · k0, where k0 is equal to 1. The rate constant for mutant replication (blue) is constant when k1 is equal to 0.1. The vertical red arrow represents the value of m beyond which replication of mutant genomes is more rapid than that of the wild type. (B) Virus extinction by error catastrophe. Wild-type virus (black) is completely replaced with mutants (blue) when k0 is equal to 1 and k1 is equal to 0.1, i.e., when mutants can replicate. The probability that the consensus sequence is identical to the wild-type sequence (red) switches from 1 to 0 at the error threshold. (C) Virus extinction does not occur when mutants cannot replicate. When k0 is equal to 1 and k1 is equal to 0, wild-type virus (black) steadily decreases with the increasing error rate and mutants (blue) increase, but mutants never completely replace the wild type. Because the replication rate of wild-type virus is always higher that that of the mutants, the probability that the consensus sequence remains identical to the wild-type sequence (red) remains at 1 for all error rates. Steady-state conditions are satisfied at t → ∞ (see Appendix, equations 2 and 3). Inserts in the right of each panel are the same curves on a semilog plot.
Wild-type virus extinction at steady state.
In Eigen's model of error catastrophe, competition between the wild type and mutants in a population of fixed size continues to steady state conditions, a requirement that is truly satisfied only at infinite times. Although the replication rate of the wild-type virus decreases in a continuous fashion with the error rate, as shown in Fig. 1A, it is easy to see why the wild-type sequence will disappear from the population when it replicates even marginally slower than the mutants if competition occurs over a sufficient number of generations. This disappearance with increasing mutation rate is shown in Fig. 1B, which plots the fraction of the wild type (F0) as a function of the error rate at t → ∞. As in Swetina and Schuster's model, the switch to extinction occurs when m is equal to 2.302. Because the consensus sequence of the quasispecies (the most prevalent nucleotide at each position) is identical to the sequence with the highest replication advantage (20), elimination of the replication advantage of the wild type results in the loss of that consensus sequence. This loss is represented in Fig. 1B by the red dotted line, which is the probability that the consensus sequence is identical to the wild-type sequence. This probability goes discontinuously from 1 to 0 at the error threshold. In fact, above the error threshold there is no preferred consensus sequence. All sequences are equally probable among the competing mutant viral genomes because there is no replication penalty for mutations caused by errors. The loss of any consensus sequence for the quasispecies population has been referred to as error catastrophe, virus extinction, lethal mutagenesis, and mutational meltdown. That is, the population is predicted to consist entirely of replicating genomes with randomized sequences.
Shown in Fig. 1C is the steady state fraction of v0 (F0) when competition with mutants is eliminated by setting k1 equal to 0, i.e., assuming that mutants are produced solely during replication of the wild-type genomes and that the mutants do not replicate. In this case, there is no error catastrophe, and the steady-state fraction of v0 simply decreases as a first-order function of the error rate, in parallel with the decreasing replication rate (Fig. 1A, B, and C, compare insets). This example serves to illustrate that the extinction of the wild type in models of error catastrophe is a result of competition with variants that replicate. In reality, variants that result from replication errors will not, of course, conform to either of these two extreme properties, i.e., constant replication versus no replication, but will consist of a spectrum of phenotypes ranging from enhanced replication to lethal mutation. However, we may assume that higher rates of errors in the variant population will render an increasing proportion of variants replication incompetent; i.e., the only viable genomes will be those that have not received a lethal mutation. Under no conditions can we imagine that the replication of any variant genome would be insensitive to the deleterious effects of any and all additional mutations, as specified in models of error catastrophe.
It is a common misconception that the model of error catastrophe is about the loss of viability of a genome when it accumulates a number of errors greater than the error threshold. However, many experiments have shown that the relationship between mutagen exposure and virus viability follows single-hit kinetics, as does the k0a curve in Fig. 1A, and the interpretation of this observation has been that genome inactivation events are, on average, the result of single lethal events rather than accumulating effects of multiple sublethal events (23). More-recent studies have shown that approximately 40% of random single nucleotide substitutions in the genome of vesicular stomatitis virus were lethal, while only 30% were deleterious but not lethal, with an average reduction in growth rate of 19% (37). These results demonstrate directly that the major effects of errors on virus viability occur through single-hit lethal mutations. The reduction in k0a due to mutation in models of error catastrophe likewise follows single-hit kinetics with no error threshold and, in this regard, resembles the real-life effects of mutations on viability. However, unlike real life, mutagenesis in the error catastrophe model does not produce any lethal mutations but only limited deleterious mutations. This assumption of the model is essential for generating error catastrophe at a threshold error rate.
DISCUSSION
Error catastrophe simulations.
In the first numerical simulation of error catastrophe (41), genomes were classified as being either identical to or distinct from the master sequence according to the number of mutations. To simplify the mathematical treatment, only two fitness levels were assumed: that of the master sequence (superior) and that of all other sequences (inferior but finite). That a genome could acquire a property of zero fitness through the degeneration of its sequence was not provided for in the model. Thus, only one outcome of errors introduced during replication was possible: a one-time reduction in replication rate. The inevitable outcome of this assumption was that all mutant nucleic acid sequences would be replication competent. Thus, it was possible through this assumption to postulate the existence of a quasispecies consisting entirely of a population of replication-competent genomes with randomized mutant sequences. It is this population of replicating mutants that displaces the wild-type genome at the error threshold in the process of mutational meltdown.
Extinction at the error threshold does not occur because the wild-type genome suddenly fails to replicate but because the mutant genome replication rate is invariant while the yield of wild-type genomes continuously declines with error rate. Subsequent elaborations on the model have retained the basic premise that further errors do not affect the replication of mutants (2, 3, 21, 42, 43, 44, 45). It can be shown, however, that if the deleterious effects of mutations on the population of variants is taken into account, Eigen and Schuster's model of the quasispecies does not predict error catastrophe (see Appendix).
Empirical evidence for error catastrophe.
Despite the absence of any realistic theoretical underpinning, error catastrophe claims experimental support from two general types of observation in the literature: (i) loss of virus infectivity from cell cultures after serial passage in the presence of a mutagen and (ii) an apparent threshold mutation frequency for infectivity of viruses or viral RNAs. While a detailed critique of the literature in this field is beyond the scope of this commentary, we find that, in general, experimental support for error catastrophe is marred by the failure to propose or test alternative explanations for the results and by inadequate precision in the data.
For example, in Fig. 1A it is clear that the effect of mutagens on v0 replication rates is no different from the effect of other antiviral agents (nucleoside chain terminators, for example) in that both reduce the yield after replication of the viral genome in a continuous manner. In a virus infection in real life or in an experimental setting, all viruses may be eliminated from a system if the amount of virus consumed in the process of initiating an infectious cycle exceeds the amount of infectious virus produced in that cycle. For example, if a “conventional” inhibitor or a mutagen reduced the yield of progeny such that, on average, ten cells have to become infected to produce one infectious particle, then each cycle of infection would reduce the virus population by a factor of ten and a virus population of 1010 particles would then become extinct after approximately 11 such cycles of infection. Therefore, extinction of a virus population caused by the action of a mutagen is not, by itself, evidence of error catastrophe because extinction, in theory, can occur by simple inhibition of the production of infective particles.
Of equal concern is that published specific infectivity and mutation frequency measurements entail large errors and few data points, and attempts to determine whether the results fit the error catastrophe model better than other, simpler models have not been reported. In fact, it would be somewhat difficult to detect error catastrophe by such an analysis. Wild type “infectivity” for both the error catastrophe model (Fig. 1B) and the conventional model without error catastrophe (Fig. 1C) decreases smoothly with increasing average error rate. The curves are very similar, except that wild-type infectivity goes to zero at the error threshold, whereas without error catastrophe the wild-type infectivity approaches zero asymptotically. Detection of this kind of difference would require accurate measurements of specific infectivity over several orders of magnitude. The reported data are simply insufficient to demonstrate an error threshold for viability.
Even if such data existed, there could be other interpretations. For example, the direct effect of mutagens on the yield of viable progeny genomes might be potentiated if mutations resulted in the production of defective interfering proteins, as suggested recently (24). It is possible that such interference could magnify the decreases of specific infectivity caused by high mutation frequencies, creating the appearance of a threshold.
A third type of observation in the literature has been interpreted as providing support for error catastrophe but actually argues against it. It has been noted for several RNA viruses that it is not possible to increase the frequency of mutations more than a fewfold without large losses in viability. This observation has been interpreted as indicating that RNA viruses, indeed, replicate at the edge of error catastrophe. There are obvious reasons why substantially increased mutation rates cannot be detected. Genomes with high numbers of mutations may not be readily recovered because they are selected against due to the presence of lethal mutations; RNA viruses already have a high rate of mutation, and a fewfold increase in mutations is a large number of additional mutations. But the prediction of the theory of error catastrophe is that the accumulation of mutations by RNA genomes should be unlimited, allowing a mutational meltdown to occur. The products of such a meltdown, according to theory, should be hypermutated genomes with random sequences. At least one study specifically designed to detect such genomes was unsuccessful (27). Objectively speaking, the failure to find such products might actually be considered a disproof of error catastrophe.
In our opinion, the theoretical model predicting error catastrophe cannot realistically represent any virus infection in the biological world as we know it and by itself cannot serve as a new paradigm for antiviral therapy. Explanations for the antiviral behavior of mutagens other than error catastrophe need to be considered and tested, with the possibility that other, more significant mechanisms might be discovered.
Acknowledgments
We thank David Peabody, Brian Hjelle, Bill Mason, Adam Zlotnick, Eugene Toll, and Glenn Rall for helpful suggestions.
This work was supported by a grant from the National Institutes of Health.
APPENDIX
Our model of error catastrophe.
Treatments of error catastrophe have been concerned with the fate of the “wild-type” quasispecies, the collection of variants of a master wild-type sequence that is generated by replication errors. The consensus sequence of the quasispecies at steady state, i.e., the most prevalent nucleotide at each position, is exactly that of the master sequence molecule as long as the master sequence molecule maintains any replication advantage over other members of the quasispecies (20). Molecules with the master sequence may be a minority or a majority of all sequences in the quasispecies, depending on the relative fitness of the master sequence. If the master sequence molecule loses its replication advantage, then the consensus sequence assumes that of a different most-fit sequence (20). The replication efficiency of the master sequence molecule may be reduced by high error rates, and error catastrophe occurs when the error rate destroys its replication advantage. Because maintenance of a wild-type quasi species depends on the presence of the master sequence molecule, this model will consider the dynamics of that molecule itself relative to all other members of the quasispecies.
A genome with the master (wild-type) sequence, whose population size is indicated by ν0, replicates through a conservative nucleic acid synthesis pathway. During replication it incurs an error rate in progeny genomes of m mutations per daughter genome to produce a mixture of wild-type and non-wild-type genomes whose yield is indicated by ν0 and ν>0. A single replication event is represented by the pathway
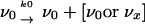 |
where the index, x, represents any one of the collection of various mutants, a, b… x, all of which replicate according to the pathway
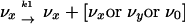 |
Although reversion of any mutant to the master sequence, 0, occurs at a finite rate, the probability that an x genome will produce a 0 genome during one replication cycle is the probability that every mutation in the genome will be reverted and that no other mutation will occur. If errors are random, the reversion rate is 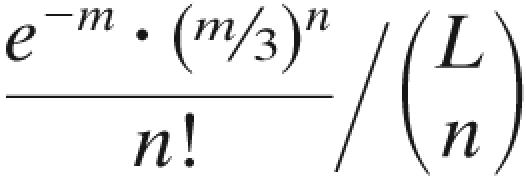 for mutants with n errors, where L is the length of the genome in nucleotides and
for mutants with n errors, where L is the length of the genome in nucleotides and 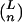 is the binomial coefficient. Since this rate is very small compared with the error rate, we have ignored the contribution of reversion to the number of v0 genomes. If all mutants replicate at the same rate, k1, the collection of mutants can be absorbed into one term, v>0. The result of this simplification is that the replication of the >0 population is not affected by the error rate, whereas that of the 0 population is diminished by the term e−m, the fraction of 0 progeny that do not incur errors during replication. The time evolution of the populations v0 and v>0 is then given by
is the binomial coefficient. Since this rate is very small compared with the error rate, we have ignored the contribution of reversion to the number of v0 genomes. If all mutants replicate at the same rate, k1, the collection of mutants can be absorbed into one term, v>0. The result of this simplification is that the replication of the >0 population is not affected by the error rate, whereas that of the 0 population is diminished by the term e−m, the fraction of 0 progeny that do not incur errors during replication. The time evolution of the populations v0 and v>0 is then given by
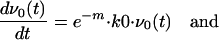 |
(1) |
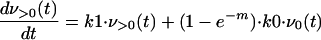 |
(2) |
Virus extinction.
Equations 1 and 2 can be solved to derive the fraction F0 of v0 in the virus population:
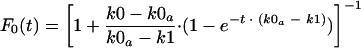 |
where the apparent rate constant k0a is equal to k0 · e−m. The value of F0 at steady-state conditions, achieved at t → ∞, depends on the relative values of k0a and k1. For k0a > k1,
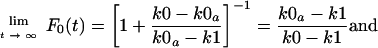 |
(3) |
for k0a < k1,
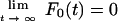 |
(4) |
Conditions for extinction of 0 (F0 → 0) occur only when k0a ≤ k1. k0a decreases with an increasing mutation rate, but since k1 is not affected by errors, the value of k1 eventually exceeds that of k0a and virus extinction occurs. The crucial assumption in the model that generates virus extinction by error catastrophe is that k1 is not sensitive to errors.
Comparison with Eigen and Schuster's model of the error threshold.
The theory of Eigen and Schuster (20) has been used to postulate a threshold error rate by the following argument. On page 555 (equation 27), Eigen and Schuster define the condition that guarantees the stable conservation of master sequence information in the quasispecies at steady state and state that if this condition is not met the information will be lost. It is summarized as
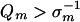 |
(5) |
Qm,the “quality factor,” is defined on page 548 (equation 1) as the probability that the master sequence genome (designated by subscript m) will be reproduced without error. σm is the “superiority” of the master sequence over the (weighted) average of all its mutants at steady state, as defined on page 552 (equation 17; equation 6 below):
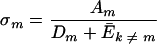 |
(6) |
Am is a term that determines the replication rate of the master sequence, and Dm determines its decomposition rate (Eigen and Schuster, page 548). 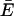 k≠m is defined on page 252 (equation 16) as the (weighted) average “excess productivity,” Ek, of the mutants at steady state, where Ek is equal to Ak − Dk, corresponding roughly to the replication rate constant minus the decomposition rate constant for each mutant. Assuming that the decomposition rate constants are equal for all genomes, then the following is true:
k≠m is defined on page 252 (equation 16) as the (weighted) average “excess productivity,” Ek, of the mutants at steady state, where Ek is equal to Ak − Dk, corresponding roughly to the replication rate constant minus the decomposition rate constant for each mutant. Assuming that the decomposition rate constants are equal for all genomes, then the following is true:
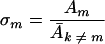 |
(7) |
Combining equation 7 with Eigen and Schuster's inequality (page 555, equation 27) above, we get
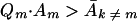 |
(8) |
The inequality expressed in equation 8 is the same as the constraint for maintenance of the wild type in our model,
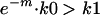 |
(9) |
Moreover, Eigen and Schuster's equation 18 (equation 10 below),
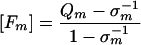 |
where Fm is the fraction of the master sequence, is equivalent to equation 3 in our model. Therefore, our model reproduces the essential features of the error threshold theory of Eigen and Schuster.
Effect of error rate on the mutant population.
In both equations 8 and 9, it can be seen that an error threshold can be reached only if (i) Ak≠m (or k1) is greater than 0, that is, if there are replicating mutants and if (ii) errors reduce the apparent replication rate of the wild type (left side) to or below the average replication rate of the mutants (right side). The form of equations 8 and 9 does not indicate explicitly that either 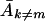 or k1 is affected by the error rate, but in reality, the average replication capacity of the mutant population,
or k1 is affected by the error rate, but in reality, the average replication capacity of the mutant population, 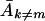 , or k1, diminishes with an increased error rate if the deleterious effects of mutations are taken into account.
, or k1, diminishes with an increased error rate if the deleterious effects of mutations are taken into account.
Eigen and Schuster's selection equation (equation 10; equation 11 below) describes the competition among all sequences of a defined length over time. This vast population is grouped into closely related sequences (quasispecies), and the selection criterion for each quasispecies is expressed by the equation
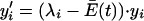 |
(11) |
where yi is the concentration of each quasispecies i, λi is a measure of the rate of its production, and 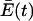 is the average excess productivity (net replication) of the entire collection of quasispecies evolving over time (this term was constructed to maintain a constant population of genomes). The selection equation shows that with time any quasispecies that is characterized by a λi that is less than the average replication rate of the population as a whole is lost because yi′ is negative. This process causes
is the average excess productivity (net replication) of the entire collection of quasispecies evolving over time (this term was constructed to maintain a constant population of genomes). The selection equation shows that with time any quasispecies that is characterized by a λi that is less than the average replication rate of the population as a whole is lost because yi′ is negative. This process causes 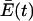 to increase until a steady state is reached when
to increase until a steady state is reached when
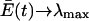 |
(12) |
Thus, only the quasispecies with the maximum λi is retained. This example, as explained by Eigen and Schuster, produces an extremum principle, expressed in equation 12, that says that the weighted average of replication rate constants of the entire population, 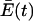 , evolves toward the replication rate constant of the most-fit quasispecies, λi, and that all other quasispecies are eliminated. The model describes how the quasispecies acts as the target of selection, based on its fitness and its ability to replicate itself effectively. For our purposes, each quasispecies is, in effect, a different virus species with its own fidelity of replication, and the sequence space that exists between two different viruses consists of replication-incompetent space that is unlikely to be traversed by simple errors in replication.
, evolves toward the replication rate constant of the most-fit quasispecies, λi, and that all other quasispecies are eliminated. The model describes how the quasispecies acts as the target of selection, based on its fitness and its ability to replicate itself effectively. For our purposes, each quasispecies is, in effect, a different virus species with its own fidelity of replication, and the sequence space that exists between two different viruses consists of replication-incompetent space that is unlikely to be traversed by simple errors in replication.
Let us examine, however, what happens when the error rate in a population already at steady state, i.e., consisting of one quasispecies, is increased (by the addition of a mutagenic nucleoside analog, for example) so that the accuracy (Q) of copying of all templates in the quasispecies is reduced. Initially, the excess productivity, 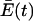 , of the entire population is unaffected, but the production of more highly mutated copies of every member is increased at the expense of exact copies. This process will result in a remodeling of the quasispecies in the direction of a greater proportion of more highly mutated error copies. Change in the concentrations of the various species, xi, is described by the selection equation (Eigen and Schuster equation 6; equation 13 below), simplifiedby setting the decomposition term, Di, to 0,
, of the entire population is unaffected, but the production of more highly mutated copies of every member is increased at the expense of exact copies. This process will result in a remodeling of the quasispecies in the direction of a greater proportion of more highly mutated error copies. Change in the concentrations of the various species, xi, is described by the selection equation (Eigen and Schuster equation 6; equation 13 below), simplifiedby setting the decomposition term, Di, to 0,
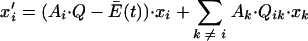 |
(13) |
where Qik is the probability that species k will produce species i as a result of replication errors. At steady state, before addition of a mutagen, all xi are equal to 0, so that for all species the net production of accurate copies is less than the average excess productivity and the deficit is made up by production of copies of i through replication errors; i.e.,
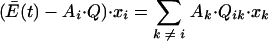 |
(14) |
When Q is decreased in a quasispecies at steady state, both sides of equation 14 are increased. However, the right side of the equation is a very small number compared with 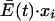 and Ai · Q · xi, so that the magnitude of changes in the latter parameters determines primarily whether any particular species expands or contracts. For example, for rapidly replicating genomes that are present in high amounts, the left side of the equation is increased by decreasing Q to a greater extent than the right side is increased by Qik, which results in
and Ai · Q · xi, so that the magnitude of changes in the latter parameters determines primarily whether any particular species expands or contracts. For example, for rapidly replicating genomes that are present in high amounts, the left side of the equation is increased by decreasing Q to a greater extent than the right side is increased by Qik, which results in
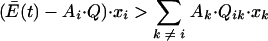 |
at the new value of Q. These species will acquire a negative value of xi and will be depleted over time. Conversely, species with lower replication rates and concentrations will be enriched.
Depletion of the species with higher replication rates cannot continue indefinitely because the value of 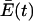 decreases over time as species with higher replication rates are diminished. Steady state will be reached for each species when equation 14 is again satisfied at the new value of Q.
decreases over time as species with higher replication rates are diminished. Steady state will be reached for each species when equation 14 is again satisfied at the new value of Q. 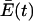 will continue to approach Ai · Q (which is invariant with time) as closely as required to achieve the condition (expressed by equation 14) for steady state because the continued accumulation of error copies over time produces increasingly higher frequencies of deleterious and lethal mutations in the quasispecies as a whole.
will continue to approach Ai · Q (which is invariant with time) as closely as required to achieve the condition (expressed by equation 14) for steady state because the continued accumulation of error copies over time produces increasingly higher frequencies of deleterious and lethal mutations in the quasispecies as a whole.
We will designate the new steady-state value of 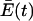 as
as 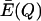 in order to recognize that the excess productivity depends on the accuracy of replication Q for the quasispecies.
in order to recognize that the excess productivity depends on the accuracy of replication Q for the quasispecies. 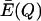 can be determined by the conditions for steady state of the master sequence concentrationxm (from Eigen and Schuster, equation 6) as follows:
can be determined by the conditions for steady state of the master sequence concentrationxm (from Eigen and Schuster, equation 6) as follows:
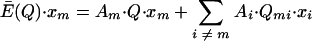 |
(15) |
The steady-state value of 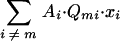 , which is an exceedingly small number (for realistic genome sizes) compared with Am·Q·xi, varies directly with Q because (i) the mutant population is enriched in mutants with more errors (xi≠m), (ii) the high-error copies have a reduced rate of replication (Ai≠m), and (iii) the high-error copies have a reduced probability of reversion per replication (Qmi). Consequently, at the new steady-state distribution, if the term
, which is an exceedingly small number (for realistic genome sizes) compared with Am·Q·xi, varies directly with Q because (i) the mutant population is enriched in mutants with more errors (xi≠m), (ii) the high-error copies have a reduced rate of replication (Ai≠m), and (iii) the high-error copies have a reduced probability of reversion per replication (Qmi). Consequently, at the new steady-state distribution, if the term 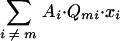 is neglected, the excess productivity of the quasispecies is expressed by
is neglected, the excess productivity of the quasispecies is expressed by 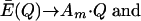
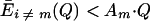 |
In other words, within a quasispecies the master sequence competes successfully with the aggregate of its error copies at all error rates. Therefore, increasing the error rate cannot result in a loss of the master sequence information.
REFERENCES
- 1.Anderson, J. P., R. Daifuku, and L. A. Loeb. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58:183-205. [DOI] [PubMed] [Google Scholar]
- 2.Bagnoli, F., and M. Bezzi. 1998. Eigen's error threshold and mutational meltdown in a quasispecies model. Int. J. Mod. Phys. C 9:1-7. [Google Scholar]
- 3.Brumer, Y., and E. I. Shakhnovich. 22. December 2004, posting date. Importance of DNA repair in tumor suppression. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70:061912. [Online.] http://link.aps.org/abstract/PRE/v70/e061912. [DOI] [PubMed] [Google Scholar]
- 4.Cameron, C. E., and C. Castro. 2001. The mechanism of action of ribavirin: lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Curr. Opin. Infect. Dis. 14:757-764. [DOI] [PubMed] [Google Scholar]
- 5.Castro, C., J. J. Arnold, and C. E. Cameron. 2005. Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective. Virus Res. 107:141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, R., M. E. Quinones-Mateu, and L. M. Mansky. 2004. Drug resistance, virus fitness and HIV-1 mutagenesis. Curr. Pharm. Des. 10:4065-4070. [DOI] [PubMed] [Google Scholar]
- 7.Clementi, M. 2004. Can modulation of viral fitness represent a target for anti-HIV-1 strategies? New Microbiol. 27:207-214. [PubMed] [Google Scholar]
- 8.Contreras, A. M., Y. Hiasa, W. He, A. Terella, E. V. Schmidt, and R. T. Chung. 2002. Viral RNA mutations are region specific and increased by ribavirin in a full-length hepatitis C virus replication system. J. Virol. 76:8505-8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crotty, S., and R. Andino. 2002. Implications of high RNA virus mutation rates: lethal mutagenesis and the antiviral drug ribavirin. Microbes Infect. 4:1301-1307. [DOI] [PubMed] [Google Scholar]
- 10.Crotty, S., C. Cameron, and R. Andino. 2002. Ribavirin's antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. 80:86-95. [DOI] [PubMed] [Google Scholar]
- 11.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 98:6895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crotty, S., D. Maag, J. J. Arnold, W. Zhong, J. Y. Lau, Z. Hong, R. Andino, and C. E. Cameron. 2000. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6:1375-1379. (Erratum, 7: 255.) [DOI] [PubMed] [Google Scholar]
- 13.de la Torre, J. C. 2005. Arenavirus extinction through lethal mutagenesis. Virus Res. 107:207-214. [DOI] [PubMed] [Google Scholar]
- 14.Domingo, E. 2003. Quasispecies and the development of new antiviral strategies. Prog. Drug Res. 60:133-158. [DOI] [PubMed] [Google Scholar]
- 15.Domingo, E., C. Escarmis, E. Lazaro, and S. C. Manrubia. 2005. Quasispecies dynamics and RNA virus extinction. Virus Res. 107:129-139. [DOI] [PubMed] [Google Scholar]
- 16.Domingo, E., A. Mas, E. Yuste, N. Pariente, S. Sierra, M. Gutierrez-Riva, and L. Menendez-Arias. 2001. Virus population dynamics, fitness variations and the control of viral disease: an update. Prog. Drug Res. 57:77-115. [DOI] [PubMed] [Google Scholar]
- 17.Domingo, E., N. Pariente, A. Airaksinen, C. Gonzalez-Lopez, S. Sierra, M. Herrera, A. Grande-Perez, P. R. Lowenstein, S. C. Manrubia, E. Lazaro, and C. Escarmis. 2005. Foot-and-mouth disease virus evolution: exploring pathways towards virus extinction. Curr. Top. Microbiol. Immunol. 288:149-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake, J. W., and J. J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eigen, M. 2002. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA 99:13374-13376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eigen, M., and P. Schuster. 1977. The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle. Naturwissenschaften 64:541-565. [DOI] [PubMed] [Google Scholar]
- 21.Eigen, M., J. McCaskill, and P. Schuster. 1988. Molecular quasi-species. J. Phys. Chem. 92:6881-6891. [Google Scholar]
- 22.Freistadt, M. S., G. D. Meades, and C. E. Cameron. 2004. Lethal mutagens: broad-spectrum antivirals with limited potential for development of resistance? Drug Resist. Updates 7:19-24. [DOI] [PubMed] [Google Scholar]
- 23.Gard, S., and O. Maaløe. 1959. Inactivation of viruses, p. 359-427. In M. F. Burnet and W. M. Stanley (ed.), The viruses. Academic Press, Orlando, Fla.
- 24.Gonzalez-Lopez, C., A. Arias, N. Pariente, G. Gomez-Mariano, and E. Domingo. 2004. Preextinction viral RNA can interfere with infectivity. J. Virol. 78:3319-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graci, J. D., and C. E. Cameron. 2002. Quasispecies, error catastrophe, and the antiviral activity of ribavirin. Virology 298:175-180. [DOI] [PubMed] [Google Scholar]
- 26.Graci, J. D., and C. E. Cameron. 2004. Challenges for the development of ribonucleoside analogues as inducers of error catastrophe. Antivir. Chem. Chemother. 15:1-13. [DOI] [PubMed] [Google Scholar]
- 27.Grande-Perez, A., S. Sierra, M. G. Castro, E. Domingo, and P. R. Lowenstein. 2002. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc. Natl. Acad. Sci. USA 99:12938-12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonsson, C. B., B. G. Milligan, and J. B. Arterburn. 2005. Potential importance of error catastrophe to the development of antiviral strategies for hantaviruses. Virus Res. 107:195-205. [DOI] [PubMed] [Google Scholar]
- 29.Loeb, L. A., and J. I. Mullins. 2000. Lethal mutagenesis of HIV by mutagenic ribonucleoside analogs. AIDS Res. Hum. Retrovir. 16:1-3. [DOI] [PubMed] [Google Scholar]
- 30.Orgel, L. E. 1963. The maintenance of the accuracy of protein synthesis and its relevance to aging. Proc. Natl. Acad. Sci. USA. 49:517-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orgel, L. E. 1970. The maintenance of the accuracy of protein synthesis and its relevance to aging: a correction. Proceedings of the National Academy of Science USA. 67:1476. [DOI] [PMC free article] [PubMed]
- 32.Orgel, L. E. 1973. Ageing of clones of mammalian cells. Nature 243:441-445. [DOI] [PubMed] [Google Scholar]
- 33.Pariente, N., A. Airaksinen, and E. Domingo. 2003. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J. Virol. 77:7131-7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pariente, N., S. Sierra, and A. Airaksinen. 2005. Action of mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 107:183-193. [DOI] [PubMed] [Google Scholar]
- 35.Pariente, N., S. Sierra, P. R. Lowenstein, and E. Domingo. 2001. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 75:9723-9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Jarabo, C. M., C. Ly, E. Domingo, and J. C. de la Torre. 2003. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV). Virology. 308:37-47. [DOI] [PubMed] [Google Scholar]
- 37.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schinkel, J., M. D. de Jong, B. Bruning, B. van Hoek, W. J. Spaan, and A. C. Kroes. 2003. The potentiating effect of ribavirin on interferon in the treatment of hepatitis C: lack of evidence for ribavirin-induced viral mutagenesis. Antivir. Ther. 8:535-540. [PubMed] [Google Scholar]
- 39.Severson, W. E., C. S. Schmaljohn, A. Javadian, and C. B. Jonsson. 2003. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 77:481-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith, R. A., L. A. Loeb, and B. D. Preston. 2005. Lethal mutagenesis of HIV. Virus Res. 107:215-228. [DOI] [PubMed] [Google Scholar]
- 41.Swetina, J., and P. Schuster. 1982. Self replication with errors, a model for polynucleotide replication. Biophys. Chem. 16:329-345. [DOI] [PubMed] [Google Scholar]
- 42.Tannenbaum, E., and E. I. Shakhnovich. 15. January 2004, posting date. Error and repair catastrophes: a two-dimensional phase diagram in the quasispecies model. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 69:011902. [Online.] http://link.aps.org/abstract/PRE/v69/e011902. [DOI] [PubMed] [Google Scholar]
- 43.Tannenbaum, E., and E. I. Shakhnovich. 11. August 2004, posting date. Solution of the quasispecies model for an arbitrary gene network. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70:021903. [Online.] http://link.aps.org/abstract/PRE/v70/e021903. [DOI] [PubMed] [Google Scholar]
- 44.Tannenbaum, E., E. J. Deeds, and E. I. Shakhnovich. 16. June 2004, posting date. Semiconservative replication in the quasispecies model. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 69:061916. [Online.] http://link.aps.org/abstract/PRE/v69/e061916. [DOI] [PubMed] [Google Scholar]
- 45.Tarazona, P. 1992. Error thresholds for molecular quasispecies as phase transitions: from simple landscapes to spin-glass models. Phys Rev. A 45:6038-6050. [DOI] [PubMed] [Google Scholar]
- 46.Vignuzzi, M., J. K. Stone, and R. Andino. 2005. Ribavirin and lethal mutagenesis of poliovirus: molecular mechanisms, resistance and biological implications. Virus Res. 107:173-181. [DOI] [PubMed] [Google Scholar]
- 47.Zhou, S., R. Liu, B. M. Baroudy, B. A. Malcolm, and G. R. Reyes. 2003. The effect of ribavirin and IMPDH inhibitors on hepatitis C virus subgenomic replicon RNA. Virology 310:333-342. [DOI] [PubMed] [Google Scholar]