

Abstract
The vaccinia virus A35R gene is highly conserved among poxviruses and encodes a previously uncharacterized hydrophobic acidic protein. Western blotting with anti-A35R peptide antibodies indicated that the protein is expressed early in infection and resolved as a single sharp band of ∼23 kDa, slightly higher than the 20 kDa predicted from its sequence. The protein band appeared to be the same molecular weight on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, whether expressed in an in vitro transcription/translation system without microsomes or expressed in infected cells, suggesting that it was not glycosylated. A mutant virus with the A35R gene deleted (vA35Δ) formed wild-type-sized plaques on all cell lines tested (human, monkey, mouse, and rabbit); thus, A35R is not required for replication and does not appear to be a host range gene. Although the A35R protein is hydrophobic, it is unlikely to be an integral membrane protein, as it partitioned to the aqueous phase during TX-114 partitioning. The protein could not be detected in virus-infected cell supernatants. A35R localized intracellularly to the virus factories, where the first stages of morphogenesis occur. The vA35Δ mutant formed near-normal levels of the various morphogenic stages of infectious virus particles and supported normal acid-induced fusion of virus-infected cells. Despite normal growth and morphogenesis in vitro, the vA35Δ mutant virus was attenuated in intranasal challenge of mice compared to wild-type and A35R rescue virus. Thus, the intracellular A35R protein plays a role in virulence. The A35R has little homology to any protein outside of poxviruses, suggesting a novel virulence mechanism.
Poxviruses are large double-stranded DNA viruses with genomes that range from 130 to 379 kbp (39). Poxviruses have worldwide distribution and infect a wide variety of animals, including insects, birds, and mammals (18). While smallpox was eradicated from nature, variola virus continues to present a bioterrorism concern, and several other poxviruses infect humans, causing morbidity and mortality: molluscum contagiosum virus (MCV), monkeypox virus, Tanapox virus, Yaba-like disease virus, cowpox virus, and Cantagalo virus (evolved from a vaccinia virus [VV] vaccine strain) (5, 7, 10, 16-18, 27, 32, 33, 38). The prevalence of poxviruses in animals and humans and their propensity for recombination and gene acquisition suggest that it would be unwise to discount them as important human pathogens. This is especially true, since most emerging infectious diseases are zoonoses, crossing from animals to humans, and poxviruses are known to acquire mutations and become highly pathogenic in a new animal species (19).
The complex poxvirus replication cycle occurs in the cytoplasmic areas of dense viroplasm called virus factories. Gene expression is temporally regulated, with early, intermediate, and late gene expression. Early genes include those encoding intermediate transcription factors, components of the DNA replication machinery, inhibitors of cellular apoptosis and proteins that interfere with immune clearance of virus-infected cells. Among the last group, there are inhibitors of complement; alpha interferon (IFN-α), IFN-β, and IFN-γ; major histocompatility complex class I expression; antigen presentation; interleukin 1-converting enzyme; Fas-induced killing; chemokines; and interleukins, (2, 34, 40, 54). While a great many viral pathogenic strategies have been described, my colleagues and I have documented that of the 90 most widely conserved poxvirus genes, 25 remain uncharacterized (55). This fact underscores the large amount that is still unknown about poxviruses and highlights the need for further study of these viruses and genes.
Most poxvirus research has focused on VV, the prototypical poxvirus that was used as the live smallpox vaccine (39). Poxvirus morphogenesis is complex with multiple viral forms. The spherical immature virus particle matures into the intracellular mature virus (IMV) concomitant with proteolytic cleavage. The IMV is infectious, but to escape the cell during infection, IMV must undergo a second membrane-wrapping event, during which the IMV acquires two additional membrane layers derived from the trans-Golgi network or early endosomes, forming the intracellular enveloped virus (IEV) (23, 28, 39, 48). The IEV fuses out, losing its outermost membrane and releasing the extracellular enveloped virus (EEV) (6, 20, 26, 47, 56, 57, 59, 60). Seven proteins have been identified that are specifically incorporated into the IEV and/or EEV membranes: an 89-kDa hemagglutinin (encoded by the VV A56R gene) (44, 53), a 42 kDa protein (B5R) (14, 15, 30), a 43- to 50-kDa protein (A36R) (43), the 65-kDa (F12L) protein (56, 61), the acylated 37-kDa (F13L) protein (3, 24), and two variably glycosylated 21- to 24-kDa proteins encoded by A33R and A34R (11, 37, 46, 47). The A33R, A34R, and A36R envelope genes are contained within a small contiguous region of the genome interrupted only by the A35R open reading frame (ORF). The location of the A35R and its hydrophobic nature suggested that it might also be an envelope-associated protein. This report describes the first characterization of the A35R protein and its role in virulence.
MATERIALS AND METHODS
Cells.
HeLa cells were used for growth of virus stocks as previously described (12). BS-C-1 cells were routinely used for plaque assays, immunostaining, immunoprecipitations, and Western blotting. For titrations of VV and analysis of plaque size, monolayers were fixed and stained with 0.1% crystal violet in 20% ethanol.
RK13 cells were used to propagate virus for CsCl purification of virus particles. Cells were grown in Eagle's minimum essential medium with 10% fetal bovine serum, and infections were carried out in Eagle's minimum essential medium with 2.5% fetal bovine serum. Cell lines used for host range studies are listed with the American Type Culture Collection number in parenthesis: RK-13, normal rabbit kidney (CCL-37); CV-1, normal African Green monkey kidney (CCL-70); HeLa, human cervical adenocarcinoma (CCL-2); human, thymidine kinase-negative (TK−) (CRL-8303); African green monkey normal kidney, BS-C-1 (CCL-26); human lung carcinoma, A549 (CCL-185); normal human lung fibroblast, MRC-5 (CCL-171); and hamster kidney, BHK-21 (CRL 8544).
Antibodies.
Polyclonal rabbit sera were raised to three A35R peptides (supplied by J. Coligan, National Institute of Allergy and Infectious Diseases). Sequences were derived from hydrophilic regions of the predicted amino acid sequence of A35R. The peptide A35R-55 sequence was CQKCYFSYKGKIVPQDSND (residues 55 to 72), the A35R-102 peptide sequence was CDIEDKHQPFY (residues 102 to 111), and A35R-85 was YRSKNTIIIACDYDC (85 to 98). Cysteine residues (underlined) were included to aid in peptide coupling. Peptides were coupled to keyhole limpet hemocyanin using the Imject Activated Immunogen Conjugation kit (Pierce, Rockford, IL). A total of five injections were given, and the rabbits were bled to prepare antiserum.
Cloning and in vitro expression of A35R.
The Sal L opp plasmid (a gift from Nelson Cole) was cut with NcoI and SpeI restriction enzymes to release a DNA segment containing the A35R gene, from 1 nucleotide before the ATG translation start site to 37 nucleotides after the stop codon. The A35R segment and NcoI- and SpeI-cut pGem5Zf (Promega) and pTM1 (39) plasmids were gel purified using the Geneclean II kit, ligated, and transformed into Escherichia coli. Ampicillin-resistant colonies were picked and screened for inserts. In vitro expression was carried out using the TNT reticulolysate system (Promega) in the absence of microsomal membranes.
vA35RΔ mutant virus construction.
DNA segments containing the A35R flanking regions were joined with the E. coli xanthine guanine phosphoribosyl transferase (gpt) gene under the p7.5 promoter by recombinant PCR (47, 58). The flanking sequence that included part of the adjacent A34R gene was amplified by using the primer pair CCAGATTGCCTAGACCGGATACTAG and GCGGGTGGGTTTGGAATTAGTGTGGCGGCGTACGTTAACGACTTA. Sequences of the gpt gene (underlined) were incorporated into the primers for sequential amplification and joining of the gpt and A35R flanking regions. The flanking sequence that included part of the adjacent A36R gene was amplified using the primer pair CCTGGCACTGCCGGGCGTTCATAAAAGTTGTAAAGTAAATAATAAAAC and TCATTCCTAGAAATATTATCTACG. Primers used to amplify the gpt cassette were CACTAATTCCAAACCCACCCGCTT and AACGCCCGGCAGTGCCAGGCGT. The three DNA segments were amplified by PCR individually, purified by using Promega PCR Preps, and then joined by recombinant PCR in a stepwise fashion. The final PCR product was purified, TA cloned (Invitrogen), and sequenced using a Prism Dye Deoxy Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, Calif.) in conjunction with a model 373 DNA sequencer (Applied Biosystems). The TA plasmid containing the A35Δ construct was transfected with N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP; Roche) into cells infected with the wild-type VV strain Western Reserve (WR). Viruses were grown under semisolid agarose (GIBCO/BRL, Grand Island, N.Y.) containing mycophenolic acid (Sigma) for selection of recombinant virus. Viruses were plaque purified three times and amplified. Recombinant viruses were PCR screened for the presence of the gpt gene and the absence of the A35R gene using primers flanking the A35R locus. Protein expression was analyzed by Western blotting.
vA35-Res virus construction.
The A35R gene and flanking sequences were PCR amplified using primers CCAGATTGCCTAGACCGGATACTAG and TCATTCCTAGAAATATTATCTACG. The PCR product was cloned into the TA cloning vector (Invitrogen). Plasmids were sequenced to confirm the correct sequence and transfected into vA35Δ-infected cells. vA35-Res recombinant viruses were selected on STO cells in the presence of 6-thioguanine to select against viruses containing the gpt gene (29). Expression of A35R protein was confirmed by Western blotting.
Immunoprecipitation and Western blotting.
Immunoprecipitation and Western blots were performed essentially as described previously (46). Rabbit anti-A35R sera were used at a 1:1,000 dilution in Western blots.
Triton X-114 partitioning of integral membrane proteins.
BS-C-1 cells were infected at a multiplicity of infection (MOI) of 10 for 24 h. The medium was removed, and cells were extracted in cold buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 2% Triton X-114 (Pierce, Rockford, IL). Following a 15-min incubation, lysates were centrifuged for 7 min at 14,000 × g to remove insoluble matter. Supernatants were warmed to 37°C, underlaid twice with 6% sucrose, and centrifuged for 10 min at 1,000 × g to separate the upper aqueous phase from the lower detergent phase. Samples were analyzed by immunoprecipitation and sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Immunofluorescence microscopy.
Fluorescence microscopy was performed as previously described (47). Infected HeLa cell monolayers on coverslips were washed with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Rabbit anti-A35R sera were used at a 1:1,000 dilution. The secondary antibody was Alexa 568-labeled goat anti-rabbit immunoglobulin G (Molecular Probes, Inc.) diluted 1:400, and DNA was stained by 500-ng/ml Hoechst 33258 (Sigma). Samples were viewed and images were collected with a Zeiss IIIRS immunofluorescent microscope.
One-step growth curve.
Confluent BS-C-1 cell monolayers in six-well plates were infected with VV at an MOI of 10 for 2 h. The inocula were removed, the cells were washed, and fresh medium was added. At intervals, the medium from an individual well was harvested and centrifuged at 1,800 × g for 5 min to pellet detached cells. The resulting cell pellet was combined with infected cells that had been scraped from the plate into 1 ml of fresh medium. Cells were frozen, thawed three times, and sonicated. Viruses from the media and cells were individually titrated in duplicate on BS-C-1 cell monolayers.
Virus purification.
Virus particles were separated and purified based on differential buoyant density centrifugation in CsCl, as described previously (46). Briefly, RK-13 cells were infected at a multiplicity of infection of 10 for 48 h. For EEV, supernatants were harvested and centrifuged twice at 3,000 rpm in a Sorvall RT 6000B to remove cells. Then, supernatants were centrifuged at 22,000 rpm in a Beckman L8-70 M for 80 min to pellet virus. For IMV, cells were scraped into 10 mM Tris (pH 9.0) and Dounce homogenized. After a low-speed centrifugation, virus was pelleted through a 36% sucrose cushion. Afterwards, both the EEV and IMV were purified on a CsCl gradient (layered densities of 1.3, 1.25, and 1.2 g/ml) and centrifuged at 32,000 rpm with slow acceleration for 60 min in a Beckman L8-70 M.
Syncytium formation.
Confluent BS-C-1 cell monolayers were infected at an MOI of 10 for 2 h, washed, and incubated in medium for an additional 10 h as described previously (47). Cells were washed and treated with fusion buffer [phosphate-buffered saline with 10 mM 2-(N-morpholino)ethanesulfonic acid and 10 mM HEPES] at pH 5.5 or 7.4 for 2 min at 37°C. Afterwards, fusion buffer was replaced by medium, and the cells were incubated at 37°C and then observed by phase-contrast microscopy.
Determination of virulence in mice.
Groups of six female BALB/c 6-week-old mice were anesthetized and intranasally challenged with 104 to 106 PFU of vA35Δ, WR, vA35-Res virus, or PBS in 20 μl 10 mM Tris-HCl (pH 9.0). Virus titers were determined on the day of challenge to confirm virus dose. Individual mice were weighed every 2 days, and mice were sacrificed if they lost 30% of their body weight. All animal experiments were carried out in accordance with National Institutes of Health guidelines.
RESULTS
vA35Δ mutant virus forms normal plaques.
A35R orthologs are conserved in all mammalian-tropic poxvirus genomes, except the ORF is fragmented in variola virus (13, 55). To assess the importance of the A35R protein in the virus life cycle, a recombinant VV was constructed replacing all but four of the A35R coding nucleotides by the E. coli gpt gene (Fig. 1A). Recombinant viruses were selected, purified, and screened by PCR to confirm the absence of the A35R gene (data not shown). Several virus isolates missing the A35R gene grew normally on African green monkey normal kidney BS-C-1 cells, forming large wild-type plaques (Fig. 1B). To determine whether the A35R gene plays a role in VV host range, growth of the vA35Δ mutant was assessed on several cell lines: RK-13 normal rabbit kidney, CV-1 normal African green monkey kidney; HeLa human cervical adenocarcinoma; human TK−; A549 human lung carcinoma; MRC-5 normal human lung fibroblast, and BHK-21 hamster kidney cells. The vA35Δ mutant grew normally (data not shown) on all cell lines tested, suggesting that A35R is not involved in host range growth of VV in any of these cell lines.
FIG. 1.

vA35Δ mutant virus. (A) To construct the vA35Δ mutant virus, wild-type (WR) infected cells were transfected with a recombinant PCR product containing A34R and A36R flanking sequences with the A35R gene replaced by the E. coli gpt gene. The recombinant vA35Δ was selected in medium containing mycophenolic acid. (B) Wild-type and vA35Δ mutant viruses form similarly sized plaques on BS-C-1 cell monolayers. Monolayers were stained with crystal violet 40 h postinfection.
vA35Δ mutant virus replicates normally.
To evaluate the kinetics of virus replication, a one-step growth curve was performed. BS-C-1 cells were infected with WR or vA35Δ at an MOI of 10 for 2 h, and then inocula were removed. At various time points postinfection, virus in the medium and associated with cells were collected, and titers were determined. There was a <2-fold difference between the amount of virus produced by WR- or vA35Δ-infected cells, except at 48 h postinfection (hpi) when vA35Δ made 2.4 times as much virus as WR (Fig. 2). Virus in the supernatant varied by <1.4 fold at all time points after 8 hpi. Since virus particles begin to be released after approximately 8 hpi, virus present before that time was expected to represent input virus. These results indicate that A35R is not required for normal replication and production of intracellular or extracellular virus particles.
FIG. 2.
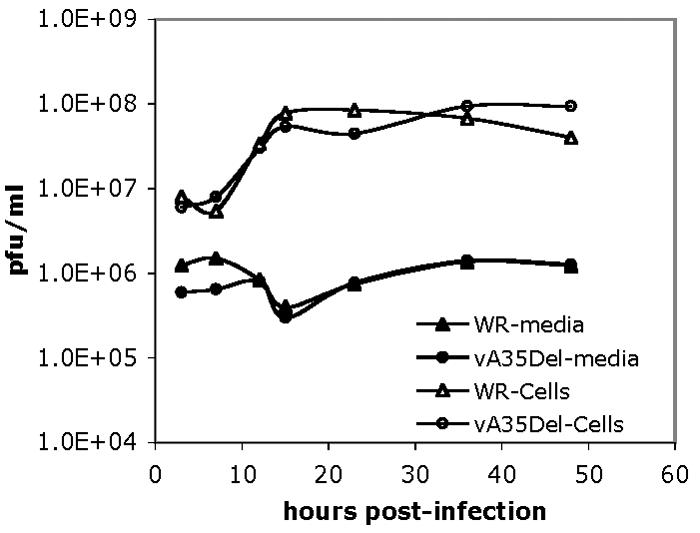
Wild-type (WR) or vA35Δ replication in a one step growth curve. BS-C-1 cells were infected with wild-type (WR) or vA35Δ at an MOI of 10, and supernatants and cells were harvested at designated time points postinfection. Cell-associated virus and virus in the medium were titrated on separate BS-C-1 monolayers. Representative data are shown.
A35R protein is expressed early.
The VV A35R ORF is predicted to encode a 20-kDa protein. Proteins from infected cells were analyzed by Western blotting for reactivity with polyclonal antisera prepared by immunization of rabbits with synthetic hydrophilic peptides chosen from the predicted A35R protein sequence. Figure 3A shows that the rabbit polyclonal sera recognized a 23-kDa band in proteins expressed in wild-type-infected cells but not in cells infected with the vA35RΔ. In Western blots, the antisera to peptides 55 to 85 and 102 to 111 gave the strongest signals (data not shown) and are used throughout this paper.
FIG. 3.

A35R protein expression during poxvirus infection. (A) Western blot of A35R protein at time points 3, 6, and 18 h postinfection with wild-type WR (W) or vA35Δ (Δ) mutant virus. The arrow points to the expressed A35R protein band. (B) A35R protein is expressed early in infection. Cells were infected with wild-type (WT) or vA35Δ mutant virus and pulse labeled with [35S]methionine for 2 h at the times shown (hours postinfection). Cytosine arabinoside (AraC) was also added 1 h prior to infection into wells containing each virus sample to inhibit DNA replication and late gene expression. At the end of the labeling period, cells were processed for immunoprecipitation with polyclonal anti-A35R sera. The arrow points to the A35R protein band expressed.
Searching upstream of the predicted A35R translation ATG start site showed the absence of a late promoter TAAAT element within 150 bp and the presence of a putative early promoter element, AAAAAATGAATTAATA, from position −24 to −39. This putative early promoter differs from the early promoter consensus sequence (8) by the presence of two T residues (underlined) in the A35R promoter region in place of two A residues present in the consensus. Since the TT dinucleotide is found in this position in known early promoters (8), it seemed likely that this was a functional early promoter. As shown in Fig. 3B, the A35R protein could be detected in wild-type-infected cells by Western blotting from 3 to 18 hpi. To determine the expression time of the A35R protein, proteins from wild-type- and vA35Δ mutant virus-infected cells were metabolically labeled at time intervals after infection. Figure 3B shows that the A35R protein is expressed in wild-type virus-infected cells during a 1- to 3-h period after infection and that no significant expression of A35R occurred between 5 to 7 h postinfection, or 18 to 20 h postinfection (data not shown). The presence of cytosine arabinoside, a DNA synthesis inhibitor that also blocks VV late gene expression, did not block the expression of A35R. These data confirm that A35R is expressed early during the infection cycle in wild-type-infected cells.
Together, these data indicate that while the A35R protein is expressed early, the A35R protein is stable and present through later stages of the virus life cycle, albeit at lower concentrations.
A35R hydrophobic nature.
Sequence analysis of the A35R open reading frame showed that the A35R protein is very hydrophobic (Fig. 4A). The multiple hydrophobic areas of the protein suggested that it might be a membrane protein. The amino terminus contains a stretch of hydrophobic amino acids suggestive of a signal peptide; however, analysis with SignalP software (41, 42) did not predict this region to encode a signal peptide. Analysis by TMpred software (25), however, reported a significant transmembrane score (556) in the region from amino acid (aa) 112 to aa 130. Any score over 500 is considered significant; however, this score was much lower than the scores for the known A33R and A34R transmembrane regions, which were near 2000. The A35R protein was predicted to encode two potential N-linked glycosylation consensus sites at aa 120 and 152.
FIG. 4.

Biochemical characterization of A35R protein. (A) The hydrophobic region predicted to be a transmembrane region by TMpred software is shown with a line above and labeled TMpred. (B) Triton X-114 partitioning. Cells were infected with wild-type WR (W) or vA35Δ (Δ) mutant virus and then lysed in either radioimmunoprecipitation buffer (Rip) or 2% TX-114 buffer. TX-114 lysates were warmed, and proteins were separated into aqueous soluble phase (Sol Aq) and detergent phase (TX-114). All samples were then immunoprecipitated with anti-A35R sera. The arrow points to the expressed A35R protein band visible in the Rip sample and in the aqueous phase. (C) Autoradiogram of proteins transcribed and translated in vitro in the absence of microsomal membranes. Protein expression is shown from two pTM1-derived plasmids containing A35R (A35pTM A1 and A2), a pGEM-derived plasmid containing the A35R (A35pGEM), and the luciferase control plasmid. The arrow points to the expressed A35R protein band. (D) Western blot with polyclonal anti-A35R sera. The blot shows proteins expressed in vitro from the luciferase control plasmid (pLucif), the pTM1 derived A35R expressing plasmid (pA35), the A35R deletion mutant virus-infected cells (vA35Δ), and wild-type virus-infected cells (vWT). The arrow points to the A35R protein band.
A35R partitions into the aqueous phase in TX-114 partitioning.
The hydrophobic nature of the A35R protein was assessed in TX-114 partitioning. Integral membrane proteins, glycophospholipid-linked proteins, and proteins associated with membranes through acylation all generally partition into the detergent phase, whereas soluble proteins partition into the aqueous phase (4, 31, 45). Figure 4B shows a standard radioimmunoprecipitation of infected cell lysates and immunoprecipitations from TX-114 partitioned samples. The A35R protein reproducibly partitioned into the aqueous phase and was undetectable in the detergent phase. These data suggest that A35R is a soluble cytoplasmic protein.
In vitro expression of A35R.
The VV strain WR A35R ORF was cloned into pGEM and pTM1 vectors under control of the T7 promoter and expressed with an in vitro transcription and translation system. Proteins were metabolically labeled with [35S]methionine. Autoradiography showed that vector constructs with the A35R ORF inserted after the promoter produced a protein with an apparent molecular weight (MW) of 23,000 (Fig. 4C). The quantity of protein produced was higher in the pTM1 vector due to the presence of the encephalomyocarditis leader translational enhancer in this vector. Empty vector and luciferase controls are also shown and produced either no protein or a protein of the expected MW of luciferase, respectively. The apparent MW of A35R was slightly greater than the predicted MW of 20,000. As these proteins were expressed in the absence of microsomal membranes, only the peptide backbone of the protein, without any glycosylation, was produced in this system. In vitro-expressed proteins were compared to A35R expressed in infected cells in a Western blot. Figure 4D shows that A35R protein has the same apparent MW whether expressed in an in vitro transcription/translation system or in infected cells, suggesting that the A35R protein is not glycosylated or otherwise posttranslationally modified in vivo in such a way as to significantly alter the MW.
Intracellular localization of A35R.
Clues to protein function can be inferred from the intracellular localization of a protein. To determine the distribution of A35R, uninfected, wild-type-infected, and vA35Δ-infected cells were fixed, permeabilized, and stained with anti-A35R polyclonal sera and fluorescence-labeled secondary antibody (Fig. 5, right). Hoechst stain was used to visualize nuclei and virus factories (extranuclear areas of DNA) (Fig. 5, left). A35R showed a diffuse cytoplasmic localization in wild-type-infected cells but was often concentrated in virus factories. Figure 5 shows WR-infected cells with five virus factory areas (lower left); A35R protein is shown concentrated in all five of these locations (lower right). This staining pattern indicates that A35R specifically localizes to virus factories where DNA replication, transcription, and early morphogenesis occur.
FIG. 5.

A35R intracellular localization. BS-C-1 cells were infected with wild-type WR or vA35Δ at an MOI of 2.5 for 10 h and then fixed, permeabilized, and labeled with anti A35R antisera at a 1:5,000 dilution. Secondary antibody was conjugated to the immunofluorescent dye Alexa 568, and DNA was stained with Hoechst. (Left) Hoechst staining of nuclei and cytoplasmic virus factories (arrows); (right) A35R staining in the same cells.
vA35Δ mutant virus undergoes normal morphogenesis and is infectious.
Since the vA35Δ forms normal plaques, it was expected that the virus would proceed normally through morphogenesis. To determine whether the A35Δ mutant virus formed normal amounts of the various morphogenic forms of virus, virus particles from wild-type and vA35Δ mutant infections were purified by CsCl gradient centrifugation. In the first experiment, the quantities of wrapped and unwrapped virus from supernatants were measured by absorbance of 260-nm light. vA35Δ released 98% and 103%, respectively, of the amount of wrapped and unwrapped virus released by wild-type virus-infected cells. In a second experiment, proteins were metabolically labeled with [35S]methionine, and quantities of virus were measured in duplicate by radioactive counts. vA35Δ formed from 63 to 74% of the wild-type morphogenic forms resolved on CsCl gradients (cell-associated unwrapped and wrapped particles and wrapped virions in the medium). These data were consistent with the formation of large plaques by the vA35Δ mutant and the one-step growth curve (Fig. 2), which showed that the vA35Δ produced as much intracellular and extracellular virus as wild-type-infected cells with similar kinetics.
Formation of wrapped virus particles is required for acid induced fusion of virus-infected cells (3, 9, 22, 47, 49). To further assess the effects of deletion of the A35R gene, cells were infected with wild-type and vA35Δ and then acid treated (Fig. 6). Results showed that A35R protein was not required for polykaryon formation. These data confirm that there was no significant defect in virus morphogenesis.
FIG. 6.

Acid-induced fusion of infected cells. Cells were infected with the wild type or vA35Δ for 12 h, incubated for 2 min in buffer at pH 5.5, returned to growth medium, and photographed after 3 h.
To assess the infectivity of vA35Δ EEV, cells were infected with similar numbers of WR and A35Δ EEV particles (measured by absorbance at 260 nm) and scored for plaque formation. The vA35Δ produced 87% of the infectivity of wild-type virus, indicating that there was little loss of infectivity in the A35Δ mutant virions.
Analysis of CsCl-purified virus particles by immunoprecipitation and Western blotting detected A35R protein associated with intracellular (IMV and IEV) particles; however, A35R could also be detected in nuclear pellet and cell membrane fractions, suggesting that the hydrophobic nature of the A35R protein causes nonspecific association with membranes in the cytoplasm. In support of this hypothesis, A35R protein was not detectable in similar numbers of EEV particles (estimated from absorbance at 260 nm), possibly because EEV sheds its outermost membrane (the membrane exposed to cytoplasmic A35R) upon egress from the infected cell. Since the IMV is contained within the EEV and the EEV is contained within the IEV, it is difficult to envision a model whereby a protein could be present in IMV and IEV but not EEV. Further study will be required to determine whether A35R is specifically packaged into virions or simply associates nonspecifically with cytoplasmic membranes.
vA35Δ mutant virus is attenuated in mice.
Since the absence of the A35R protein had no detectable effect on the virus life cycle in vitro and A35R orthologs are highly conserved, we hypothesized that the A35R protein functions in virulence. To test this, it was first desired to construct a rescue virus, vA35-Res, from the vA35Δ virus to show that any phenotype of the vA35Δ virus was due to loss of the A35R, rather than a mutation in another locus. Therefore, a recombinant virus was engineered from the vA35Δ in which the A35R gene was returned to its original locus. It was unlikely that the vA35Δ phenotype was due to disruption of the neighboring A34R or A36R gene expression because only the A35R-coding region was removed in the vA35Δ and the mutant virus formed large plaques. Disruption of A34R or A36R expression causes a small plaque phenotype (11, 37, 60), and the vA35Δ forms normal plaques.
To determine virulence in mice, groups of 5-week-old female BALB/c mice weighing between 14 and 17 g apiece (six mice per group) were anesthetized and intranasally challenged with 104 to 106 PFU of purified wild-type WR, vA35Δ, vA35-Res, or PBS in 20 μl. Virus titers were determined on the day of challenge to confirm virus dose. Mice were weighed to monitor health and sacrificed if they lost 30% of their original body weight. Figure 7 shows that over the course of the experiment, mice challenged with PBS or vA35Δ gained between 10 and 11% in body weight, whereas mice challenged with 104 PFU of the vA35-Res or VV strain WR lost, on average, 16% and 26% of their body weight, respectively, by day 10. Two WR-challenged mice (and no others) died between day 9 and 11. This experiment was repeated, and attenuation of the vA35Δ was also seen at higher challenge doses. At 105 PFU, one mouse challenged with vA35Δ died, compared to four with WR and six with vA35Res. When challenged with 106 PFU, all mice died, but survival was increased by 1 to 2 days in the vA35Δ. With WR, two mice died on day 6, and four died on day 7. When challenged with A35Res, all six mice died on day 6; but with vA35Δ, four mice died on day 7 and two survived until day 8. Together, these data clearly show that the A35R protein is important for virulence in mice.
FIG. 7.

vA35Δ challenge in mice. Groups of 5-week-old mice (six mice per group) were intranasally challenged with 104 PFU of WR, vA35Δ, vA35-Res, or PBS. Mice were weighed every 2 days and sacrificed if they lost 30% of their body weight (shown on righthand y axis). In a second experiment, mice were challenged with 105 and 106 PFU and weighed as before. Crosses on the 104 and 105 PFU graphs indicate the numbers of dead animals in the adjacent group. In the 106 PFU challenge dose, all animals died by day 8.
DISCUSSION
This paper presents the initial characterization of the A35R protein in VV. A35R is expressed early during infection, has an apparent MW of 23,000 in sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and is not glycosylated or secreted. It does not behave as an integral membrane protein in TX-114 analysis and concentrates intracellularly to virus factories. Deletion of the A35R gene does not significantly hinder virus morphogenesis, infectivity, or spread in vitro but has a significant impact on virulence in the mouse intranasal challenge model.
The A35R gene is predicted to encode a 176-aa protein in VV Copenhagen (21), modified vaccinia Ankara (1), and Tian Tan strains (GenBank AF095689). The VV-A35R orthologs are conserved in all poxviruses with a mammalian host range, ranging in size from 176 to 192 aa in all sequenced viruses, except for a large ortholog of 233 aa in molluscum contagiosum virus (55). However, the gene is fragmented into 60-aa and 50-aa ORFs in variola virus sequences (India, Bangladesh, and Garcia) (35, 50-52). While this fragmentation potentially preserves 63% of the coding sequence, experimental evidence will be required to determine whether these proteins are expressed or might be functional in variola virus. Its conservation in molluscum contagiosum virus, a pathogen restricted to humans, suggests that the larger molluscum contagiosum virus ortholog may have a function in infecting and surviving in the human host. It is possible that the smaller orthopox A35R protein may not function in human hosts and thus was lost from variola or that other virulence mechanisms in variola made A35R unnecessary.
The wide conservation of A35R orthologs suggests an important role for this protein in some aspect of the virus life cycle. Our characterization here indicates that the A35R is not required in vitro for replication and spread of the virus but that the A35R has a function in virulence. Since the A35R is not secreted, it must execute its function intracellularly. A wide variety of possibilities exist. A35R could block some stage of antigen processing or presentation in infected cells or interfere with regulation of apoptosis. In addition, the A35R function may be required for growth in certain cell types, e.g., macrophage, in vivo. The fact that the A35R protein has little homology to non-poxvirus proteins suggests that the A35R protein participates in a novel virulence mechanism.
Two lines of evidence suggest that the A35R may function in gene regulation; it localizes to factories where viral DNA is located and it was shown to be a constitutive transcriptional activator in a large-scale yeast two-hybrid study (36). Most A35R orthologs are acidic, ranging in pI between 4.4 and 4.6 in the genus Orthopoxvirus. The acidity suggests that these proteins may act as an acid blob in activation of transcription. There is one notable exception to the acid nature of A35R orthologs. MCV contains an A35R ortholog predicted to encode a 233-aa protein with a pI of 8.5. However, the MCV protein has only one fewer acidic residue (20) than the orthopox proteins, so depending on the protein tertiary folding, it could still potentially form an acid blob domain. In VV, it is clear that there is no major defect in gene expression in the absence of A35R, because all of the large number of proteins required for virion formation and spread (Fig. 1, 2, and 6) are apparently expressed normally. Furthermore, background protein band patterns were apparently not different between the two viruses from early to late infection times (Fig. 3 and 4). The possibility remains that A35R has a role in some gene-specific transcription activity, something that has not been demonstrated in poxviruses. The early temporal expression of the A35R is consistent with a function either in host interactions or in transcription during infection.
Acknowledgments
This work was begun at The Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, with the support and guidance of Bernard Moss. I also thank Linda Wyatt for help with one of the animal challenge experiments and Monika Fazekas for help with the one-step growth experiment.
This work was supported in part by the National Science and Engineering Research Council (RPGIN 250230) and East Carolina University.
REFERENCES
- 1.Antoine, G., F. Scheiflinger, F. Dorner, and F. G. Falkner. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365-396. [DOI] [PubMed] [Google Scholar]
- 2.Barrett, J. W., J. X. Cao, S. Hota-Mitchell, and G. McFadden. 2001. Immunomodulatory proteins of myxoma virus. Semin. Immunol. 13:73-84. [DOI] [PubMed] [Google Scholar]
- 3.Blasco, R., and B. Moss. 1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-dalton outer envelope protein. J. Virol. 65:5910-5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bordier, C. 1990. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604-1607. [PubMed] [Google Scholar]
- 5.Crandell, R. A., H. W. Casey, and W. B. Brumlow. 1969. Studies of a newly recognized poxvirus of monkeys. J. Infect. Dis. 119:80-88. [DOI] [PubMed] [Google Scholar]
- 6.Cudmore, S., I. Reckmann, and M. Way. 1997. Viral manipulations of the actin cytoskeleton. Trends Microbiol. 5:142-148. [DOI] [PubMed] [Google Scholar]
- 7.Damaso, C. R., J. J. Esposito, R. C. Condit, and N. Moussatche. 2000. An emergent poxvirus from humans and cattle in Rio de Janeiro State: Cantagalo virus may derive from Brazilian smallpox vaccine. Virology 277:439-449. [DOI] [PubMed] [Google Scholar]
- 8.Davison, A. J., and B. Moss. 1989. Structure of vaccinia virus early promoters. J. Mol. Biol. 210:749-769. [DOI] [PubMed] [Google Scholar]
- 9.Doms, R. W., R. Blumenthal, and B. Moss. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Downie, A. W. 1972. The epidemiology of tanapox and yaba virus infections. J. Med. Microbiol. 5:14. [PubMed] [Google Scholar]
- 11.Duncan, S. A., and G. L. Smith. 1992. Identification and characterization of an extracellular envelope glycoprotein affecting vaccinia virus egress. J. Virol. 66:1610-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earl, P. L., B. Moss, and R. W. Doms. 1991. Folding, interaction with GRP78-BiP, assembly, and transport of the human immunodeficiency virus type 1 envelope protein. J. Virol. 65:2047-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehlers, A., J. Osborne, S. Slack, R. L. Roper, and C. Upton. 2002. Poxvirus orthologous clusters (POCs). Bioinformatics 18:1544-1545. [DOI] [PubMed] [Google Scholar]
- 14.Engelstad, M., S. T. Howard, and G. L. Smith. 1992. A constitutively expressed vaccinia gene encodes a 42-kDa glycoprotein related to complement control factors that forms part of the extracellular virus envelope. Virology 188:801-810. [DOI] [PubMed] [Google Scholar]
- 15.Engelstad, M., and G. L. Smith. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194:627-637. [DOI] [PubMed] [Google Scholar]
- 16.Espana, C. 1971. Review of some outbreaks of viral disease in captive nonhuman primates. Lab. Anim. Sci. 21:1023-1031. [PubMed] [Google Scholar]
- 17.Espana, C., M. A. Brayton, and B. H. Ruebner. 1971. Electron microscopy of the Tana poxvirus. Exp. Mol. Pathol. 15:34-42. [DOI] [PubMed] [Google Scholar]
- 18.Esposito, J., and F. Fenner. 2001. Poxviruses, p. 2885-2921. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 19.Fenner, F. 1994. Rabbitpox virus, p. 51-57. In A. D. M. E. Osterhaus (ed.), Virus infections of rodents and lagomorphs. Elsevier Science B.V., Amsterdam, The Netherlands.
- 20.Geada, M. M., I. Galindo, M. M. Lorenzo, B. Perdiguero, and R. Blasco. 2001. Movements of vaccinia virus intracellular enveloped virions with GFP tagged to the F13L envelope protein. J. Gen. Virol. 82:2747-2760. [DOI] [PubMed] [Google Scholar]
- 21.Goebel, S. J., G. P. Johnson, M. E. Perkus, S. W. Davis, J. P. Winslow, and E. Paoletti. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247-266. [DOI] [PubMed] [Google Scholar]
- 22.Gong, S. C., C. F. Lai, and M. Esteban. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 178:81-91. [DOI] [PubMed] [Google Scholar]
- 23.Hiller, G., and K. Weber. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirt, P., G. Hiller, and R. Wittek. 1986. Localization and fine structure of a vaccinia virus gene encoding an envelope antigen. J. Virol. 58:757-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann, K., and W. Stoffel. 1993. TMbase—a database of membrane spanning proteins segments. Biol. Chem. 374:166. [Google Scholar]
- 26.Hollinshead, M., G. Rodger, H. Van Eijl, M. Law, R. Hollinshead, D. J. Vaux, and G. L. Smith. 2001. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 154:389-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutin, Y. J., R. J. Williams, P. Malfait, R. Pebody, V. N. Loparev, S. L. Ropp, M. Rodriguez, J. C. Knight, F. K. Tshioko, A. S. Khan, M. V. Szczeniowski, and J. J. Esposito. 2001. Outbreak of human monkeypox, Democratic Republic of Congo, 1996 to 1997. Emerg. Infect. Dis. 7:434-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichihashi, Y., and S. Dales. 1971. Biogenesis of poxviruses: interrelationship between hemagglutinin production and polykaryocytosis. Virology 46:533-543. [DOI] [PubMed] [Google Scholar]
- 29.Isaacs, S. N., G. J. Kotwal, and B. Moss. 1990. Reverse guanine phosphoribosyltransferase selection of recombinant vaccinia viruses. Virology 178:626-630. [DOI] [PubMed] [Google Scholar]
- 30.Isaacs, S. N., E. J. Wolffe, L. G. Payne, and B. Moss. 1992. Characterization of a vaccinia virus-encoded 42-kilodalton class I membrane glycoprotein component of the extracellular virus envelope. J. Virol. 66:7217-7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen, O. N., T. Houthaeve, A. Shevchenko, S. Cudmore, T. Ashford, M. Mann, G. Griffiths, and J. Krijnse Locker. 1996. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J. Virol. 70:7485-7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konya, J., and C. H. Thompson. 1999. Molluscum contagiosum virus: antibody responses in persons with clinical lesions and seroepidemiology in a representative Australian population. J. Infect. Dis. 179:701-704. [DOI] [PubMed] [Google Scholar]
- 33.Kupper, J. L., H. W. Casey, and D. K. Johnson. 1970. Experimental Yaba and benign epidermal monkey pox in rhesus monkeys. Lab. Anim. Care 20:979-988. [PubMed] [Google Scholar]
- 34.Mahalingam, S., P. S. Foster, M. Lobigs, J. M. Farber, and G. Karupiah. 2000. Interferon-inducible chemokines and immunity to poxvirus infections. Immunol. Rev. 177:127-133. [DOI] [PubMed] [Google Scholar]
- 35.Massung, R. F., L. I. Liu, J. Qi, J. C. Knight, T. E. Yuran, A. R. Kerlavage, J. M. Parsons, J. C. Venter, and J. J. Esposito. 1994. Analysis of the complete genome of smallpox variola major virus strain Bangladesh-1975. Virology 201:215-240. [DOI] [PubMed] [Google Scholar]
- 36.McCraith, S., T. Holtzman, B. Moss, and S. Fields. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc. Natl. Acad. Sci. USA 97:4879-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntosh, A. A., and G. L. Smith. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNulty, W. P., W. C. Lobitz, F. Hu, C. A. Maruppo, and A. S. Hall. 1968. A pox disease in monkeys trasmitted to man. Arch. Dermatol. 97:286-293. [PubMed] [Google Scholar]
- 39.Moss, B. 2001. Poxviruses, p. 2849-2884. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 40.Moss, B., and J. L. Shisler. 2001. Immunology 101 at poxvirus U: immune evasion genes. Semin. Immunol. 13:59-66. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen, H., S. Brunak, and G. von Heijne. 1999. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 12:3-9. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen, H., J. Engelbrecht, S. Brunak, and G. von Heijne. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10:1-6. [DOI] [PubMed] [Google Scholar]
- 43.Parkinson, J. E., and G. L. Smith. 1994. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology 204:376-390. [DOI] [PubMed] [Google Scholar]
- 44.Payne, L. G., and E. Norrby. 1976. Presence of haemagglutinin in the envelope of extracellular vaccinia virus particles. J. Gen. Virol. 32:63-72. [DOI] [PubMed] [Google Scholar]
- 45.Roper, R. L., and B. Moss. 1999. Envelope formation is blocked by mutation of a sequence related to the HKD phospholipid metabolism motif in the vaccinia virus F13L protein. J. Virol. 73:1108-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roper, R. L., L. G. Payne, and B. Moss. 1996. Extracellular vaccinia virus envelope glycoprotein encoded by the A33R gene. J. Virol. 70:3753-3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roper, R. L., E. J. Wolffe, A. Weisberg, and B. Moss. 1998. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72:4192-4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmelz, M., B. Sodeik, M. Ericsson, E. J. Wolffe, H. Shida, G. Hiller, and G. Griffiths. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol. 68:130-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seki, M., M. Oie, Y. Ichihashi, and H. Shida. 1990. Hemadsorption and fusion inhibition activities of hemagglutinin analyzed by vaccinia virus mutants. Virology 175:372-384. [DOI] [PubMed] [Google Scholar]
- 50.Shchelkunov, S. N., A. V. Totmenin, I. V. Babkin, P. F. Safronov, V. V. Gutorov, S. G. Pozdnaikov, V. M. Blinov, S. M. Resenchuk, and L. S. Sandakhchiev. 1996. Study of the structure-activity organization of the smallpox viral genome. V. Sequencing and analysis of the nucleotide sequence of the left terminus of the India-1967 strain genome. Mol. Biol. (Moscow) 30:595-612. (In Russian.) [PubMed] [Google Scholar]
- 51.Shchelkunov, S. N., A. V. Totmenin, V. N. Loparev, P. F. Safronov, V. V. Gutorov, V. E. Chizhikov, J. C. Knight, J. M. Parsons, R. F. Massung, and J. J. Esposito. 2000. Alastrim smallpox variola minor virus genome DNA sequences. Virology 266:361-386. [DOI] [PubMed] [Google Scholar]
- 52.Shchelkunov, S. N., A. V. Totmenin, and L. S. Sandakhchiev. 1996. Analysis of the nucleotide sequence of 23.8 kbp from the left terminus of the genome of variola major virus strain India-1967. Virus Res. 40:169-183. [DOI] [PubMed] [Google Scholar]
- 53.Shida, H. 1986. Nucleotide sequence of the vaccinia virus hemagglutinin gene. Virology 150:451-462. [DOI] [PubMed] [Google Scholar]
- 54.Shisler, J. L., and B. Moss. 2001. Immunology 102 at poxvirus U: avoiding apoptosis. Semin. Immunol. 13:67-72. [DOI] [PubMed] [Google Scholar]
- 55.Upton, C., S. Slack, A. L. Hunter, A. Ehlers, and R. L. Roper. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J. Virol. 77:7590-7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Eijl, H., M. Hollinshead, G. Rodger, W. H. Zhang, and G. L. Smith. 2002. The vaccinia virus F12L protein is associated with intracellular enveloped virus particles and is required for their egress to the cell surface. J. Gen. Virol. 83:195-207. [DOI] [PubMed] [Google Scholar]
- 57.Ward, B. M., and B. Moss. 2001. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 75:11651-11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolffe, E. J., S. N. Isaacs, and B. Moss. 1993. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 67:4732-4741. (Erratum) 67:5709-5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolffe, E. J., E. Katz, A. Weisberg, and B. Moss. 1997. The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. J. Virol. 71:3904-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolffe, E. J., A. S. Weisberg, and B. Moss. 1998. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 244:20-26. [DOI] [PubMed] [Google Scholar]
- 61.Zhang, W. H., D. Wilcock, and G. L. Smith. 2000. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 74:11654-11662. [DOI] [PMC free article] [PubMed] [Google Scholar]