

Abstract
CS1 pili are important virulence factors of enterotoxigenic Escherichia coli strains associated with human diarrheal disease. They are the prototype for a family of pili that share extensive sequence similarity among their structural and assembly proteins. Only four linked genes, cooB, cooA, cooC, and cooD, are required to produce CS1 pili in E. coli K-12. To identify amino acids important for the function of the major pilin CooA, we used alanine substitution mutagenesis targeting conserved residues in the N and C termini of the protein. To test function, we examined cooA mutants for the ability to agglutinate bovine erythrocytes. Each hemagglutination-negative (HA−) cooA mutant was examined to identify its assembly pathway defect. CooA has been shown to be degraded in the absence of CooB (K. Voegele, H. Sakellaris, and J. R. Scott, Proc. Natl. Acad. Sci. USA 94:13257-13261, 1997). We found several HA− cooA mutants that produced no detectable CooA, suggesting that recognition by CooB is mediated by residues in both the N and C termini of CooA. In addition, we found that alanine substitution for some of the conserved residues in the C-terminal motif “AGxYxG(x6)T,” which is found in all subunits of this pilus family, had no effect on pilus formation. However, alanine substitution for some of the alternating hydrophobic residues within this motif prevented CooA from interacting with CooD, which serves as both the tip adhesin and nucleation protein for pilus formation. Thus, it appears that some, but not all, of the residues in both the N and C termini of CooA play a critical role in the intermolecular interactions of the major pilin with the other structural and assembly proteins. We anticipate that the results obtained here for CS1 pili in enterotoxigenic E. coli will help develop an understanding of the pilus assembly pathway used by CS1 family members in several important human pathogens.
Enterotoxigenic Escherichia coli (ETEC) is a major cause of human diarrheal disease worldwide. Infection by ETEC occurs through the consumption of contaminated food or water. ETEC is responsible for both traveler's diarrhea and significant mortality among infants and young children in developing countries (5-7, 27). The disease process can range from mild and self-limiting to severe and life threatening, with massive fluid loss (21, 42). It is estimated that 3 to 5 million deaths annually can be attributed to diarrhea, and about half of these cases are caused by enterotoxin-producing bacteria. Among these, ETEC is responsible for the majority of cases (35). Preventing colonization of the small intestine by ETEC through the development of an effective human vaccine would have a positive impact worldwide.
Colonization of host tissues is an important first step in the establishment of infection by pathogenic organisms. Bacteria have evolved to produce different types of adherence factors to mediate attachment in the host. Pili, also known as fimbriae, are one class of bacterial adherence factor that is expressed by most gram-negative bacteria (10). Pili are filamentous structures on the surface of bacterial cells that consist predominantly of repeating subunits of a single protein. An adhesin which binds cellular receptors in the host is generally located either at the tip or along the body of the pilus structure (18).
Pili are attractive vaccine candidates because of the key role they play in the pathogenesis of many bacteria (38). Detection by the immune system would likely be aided by the significant amount of surface-exposed protein making up these structures. A vaccine aimed at blocking the interaction of pili with cellular receptors should prevent establishment and/or impede the progression of infection and lead to bacterial clearance from the host. Pilin-based vaccines for use in humans are being investigated for many bacterial pathogens (2, 24, 25, 29) and are already successfully employed in animals to block colonization by ETEC (26).
Pili can be divided into different categories based on the mechanisms used for their assembly. One type of assembly system is that of the type 4 pili found in many gram-negative bacteria, including ETEC strains associated with human diarrhea (14). Assembly of these pili requires more than 14 assembly proteins and a multicomponent assembly complex that is related to the type II secretion system (47, 48, 54). A second type of system is responsible for the assembly of coiled surface structures known as curli and proceeds by the extracellular nucleation and precipitation of subunits (16). Probably the best-studied pilus assembly system is that represented by P pili of uropathogenic E. coli (46). These pili, as well as over 30 other adhesins in gram-negative bacteria, are assembled via the classical chaperone/usher pathway (20, 50). In this system, a periplasmic chaperone (36, 37) stabilizes and traffics subunits to an outer membrane usher. The usher forms a pore through which the pilus is translocated to the bacterial cell surface (9, 51). A fourth type of pilus assembly system is represented by the CS1 (coli surface antigen 1) pili of human ETEC strains, the focus of this study. This system, termed the “alternate chaperone/usher” pathway (34, 46), is functionally similar to that of P pili, although the four proteins required for assembly share no detectable primary sequence similarity with those of the classical chaperone/usher pathway (12, 41). Based on sequence analysis, all CS1 family members appear to share a simple genetic organization (four structural and assembly genes), and all four proteins needed for their assembly have homologs to those of CS1.
Pili of the CS1 family are present in both human ETEC strains (13, 56) and other bacterial species. In addition to CS1, other ETEC-associated pili within this family include colonization factor I (CFA/I), coli surface antigen 2 (CS2), CS14, CS17, and CS19 (34). ETEC strains with CS1 family pili represent those most commonly associated with human diarrheal disease (4). CS1-related pili in other bacterial species include the cable type II pili of Burkholderia cepacia (30), which are associated with disease in patients with cystic fibrosis, and the Tcf fimbriae of Salmonella enterica serovar Typhi (11), which are associated with typhoid fever in humans.
Only four linked genes, cooB, cooA, cooC, and cooD, are required to produce functional CS1 pili in E. coli K-12 (12). All Coo proteins contain a putative signal sequence and are expected to cross the cytoplasmic membrane via the Sec pathway. CooA is the major pilin that composes the body of the pilus structure (28). In the periplasm, interaction with the periplasmic chaperone CooB stabilizes the CooA protein by preventing its degradation (53). CooB also stabilizes the outer membrane protein CooC (31) and CooD. CooD is a minor constituent of the pilus found at the tip, where it is required for adherence (32). CooD is thought to initiate pilus assembly, because (i) the major pilin, CooA, is not secreted in the absence of CooD; (ii) varying the amount of CooD protein changes the number of surface-expressed pili; and (iii) CooD is found at the pilus tip, indicating that it is the first subunit incorporated into the pilus structure (33). Because of its location in the outer membrane, CooC is thought to serve as an usher through which pilin subunits are translocated to the cell surface (31).
The current model for CS1 pilus assembly (Fig. 1) proposes that a periplasmic CooB-CooD complex initiates assembly upon binding to CooC at the outer membrane (31, 33, 53). During assembly, CooA-CooB complexes associate with CooC at the outer membrane, and CooA subunits are added to CooD at the base of the growing pilus (34). An incoming CooA-CooB complex may displace the CooB chaperone on the preceding subunit (CooD or CooA), or the chaperone may dissociate after interaction with CooC. In either case, the chaperone is not part of the final pilus structure and is most likely recycled to the periplasm (31). Extension of the pilus structure across the outer membrane to the bacterial cell surface occurs by repeated addition of CooA subunits at the usher.
FIG. 1.

Model of CS1 pilus assembly at the outer membrane protein CooC. Letters specify the periplasmic chaperone CooB (B), the nucleation protein/tip adhesin CooD (D), the outer membrane protein CooC (C), and the major pilin subunit CooA (A). Pilus assembly is initiated when a periplasmic CooB-CooD complex associates with the outer membrane protein CooC. The pilus structure grows as CooA subunits, in association with the CooB chaperone, are added to CooD. With the addition of each subunit, CooB is displaced and recycled to the periplasm.
Although they share no apparent sequence similarity, the requirement for a periplasmic chaperone and outer membrane usher for the assembly of both CS1 and P pili suggests that both may be assembled via similar pathways. During the morphogenesis of both types of pili, the interaction of periplasmic chaperones with subunits is critical for stabilization and progression of these proteins along the productive assembly pathway (36, 53). Regions in the C terminus of P pilus subunits, including a stretch of alternating hydrophobic residues, a conserved glycine (14 residues from the C terminus), and a penultimate tyrosine, are required for interaction with the chaperone (23). Hydrophobic residues in the N terminus of the subunit also stabilize interaction with the chaperone in P pili (45). An outer membrane usher is required for translocation of pilin subunits to the bacterial cell surface in both systems (9, 31, 51). However, the assembly of CS1 pili requires only four proteins (12), while that of P pili requires more than 11 structural and assembly proteins (19). CS1 pili are homopolymers of a single subunit, CooA (15, 28), with a tip adhesin, CooD (32), whereas P pili are composite structures of six different pilin subunits with a thick helical rod and a tip fibrillum (22). Despite the differences, examination of the intermolecular interactions during the assembly of CS1 pili and comparison with those of P pili will aid in understanding the interaction of the proteins required for assembly of CS1 family pili.
In this study, we focused on the major CS1 subunit, CooA, which interacts with all Coo proteins for assembly of pili. We used alanine substitution mutagenesis to identify residues in the N and C termini of CooA that are important for function. We targeted highly conserved amino acids in these regions to determine if they are essential for subunit-subunit interaction and interaction with the periplasmic chaperone, like those in the P pilus family. Herein, we report the identification of residues within the N and C termini of the major pilin, CooA, that play a critical role in the intermolecular interactions with structural and assembly proteins.
MATERIALS AND METHODS
Media.
Bacterial cultures were grown in Luria-Bertani (LB) medium with aeration or on LB agar plates at 37°C. When required, antibiotics were added to the media as follows: ampicillin (Ap), 100 μg/ml; chloramphenicol (Cm), 40 μg/ml; and kanamycin (Km), 40 μg/ml.
Bacterial strains and plasmids.
Chemically competent E. coli Top10 (Invitrogen) and XL1-Blue supercompetent cells (Stratagene) were used in routine cloning and mutagenesis applications, respectively. MC4100 (8), an E. coli K-12 strain deleted for the lac operon and repressor, was used for the expression of coo genes from the lac promoter. Plasmids used in this study are listed in Table 1.
TABLE 1.
Plasmids used in this studya
| Plasmid | Characteristic(s) | Replicon | Reference or source |
|---|---|---|---|
| pEU605 | cooBA under control of Plac, Apr | ColE1 | 28 |
| pEU152 | cooB with C-terminal Strep-tag II, Apr | ColE1 | This study |
| pUC19 | Plac, Apr | ColE1 | 57 |
| pEU8102 | cooB with C-terminal Strep-tag II and cooA under control of Plac, Apr | ColE1 | This study |
| pEU493 | cooD under control of Plac, Cmr | pSC101 | 12 |
| pTrc99A | Apr | ColE1 | 1 |
| pEU9509 | cooD with N-terminal Strep-tag II under control of Plac, Apr | ColE1 | This study |
| pHSG575 | Cmr | pSC101 | 49 |
| pEU8105 | cooD with N-terminal Strep-tag II under control of Plac, Cmr | pSC101 | This study |
| pEU478 | cooCD under control of Plac, Cmr | pSC101 | 12 |
| pFDX500 | lacIq, Kmr | p15A | 40 |
Ap, ampicillin; Cm, chloramphenicol; Km, kanamycin.
Construction of Strep-tag II recombinant proteins.
The plasmid pEU8102 (Table 1) expresses recombinant CooB with a C-terminal Strep-tag II (39, 44) and wild-type CooA, both under the control of the lac promoter. To construct pEU8102, the open reading frame (ORF) encoding Strep-tagged CooB was PCR amplified from the plasmid template pEU152 (Table 1) using Pfu Turbo DNA polymerase (Stratagene) and the primers A-152 (5′-CAGTGATAGAGAAAAGTGAAATGAATAGTTCGACAAAAATCT-3′) and B-605/152 (5′-GAAAATTAAGATACCCAAGTAATACTTATTTTTCGAACTGCGGGTGGCTC-3′). The plasmid pEU152 contains the cooB ORF cloned into the BsaI site of the C-terminal Strep-tag II expression vector pASK-IBA3 (IBA). The ORF for wild-type cooA was PCR amplified from the plasmid template pEU605 (28) using the primers C-152/605 (5′-GAGCCACCCGCAGTTCGAAAAATAAGTATTACTTGGGTATCTTAATTTTC-3′) and D-152/605 (5′-AACAGAAACAGAGCAGCATCACACCGCTGCTCTGGCTTA-3′). The 0.8-kb fragment encoding CooB-Strep-tag II was fused to the 5′ end of the 0.6-kb fragment encoding CooA by overlapping PCR (17) using Pfu Turbo DNA polymerase and the primers E-152 (5′-GCCCCTGCAGTAACGAGGGCAAAAAATGCGAAAATTATTTTTAAG-3′) and F-605 (5′-GCCCGGTCTAGACACCGCTGCTCTGGCTTAAAAGACTTCCGTTG-3′). The PstI restriction site is underlined, and the XbaI site is boldfaced in the primer sequences. The 1.4-kb PCR product was digested with PstI/XbaI and cloned into the corresponding restriction sites in pUC19 (57), thus resulting in CooB-Strep-tag II and CooA expression from the lac promoter.
To construct recombinant CooD containing an N-terminal Strep-tag II, the ompA signal sequence and the Strep-tag II from pASK-IBA6 (IBA) were PCR amplified using Pfu Turbo DNA polymerase and the primers pASK6-F1 (5′-GACACCATCGAATGGCCAGATG-3′) and strepR (5′-TTTTTCGAACTGCGGGTGGCTCCA-3′). The cooD gene (minus the signal sequence) was PCR amplified from the plasmid template pEU493 (12) using the primers strep-CooD-F (5′-TGGAGCCACCCGCAGTTCGAAAAAGTCAGTGCCGGGCGATACCCGG-3′) and CooD R1 (5′-CACATACAATGCCCAGTGTC-3′). The 0.2-kb fragment encoding the ompA signal sequence and Strep-tag II affinity tag was fused to the 5′ end of the 1.1-kb fragment encoding mature CooD by overlapping PCR using the primers pASK6 F2A (5′-GAAAAGTGAAATGAATAGTTCGAC-3′) and cooD R2A (5′-GTCATAAATTTTCGACACTGGGTG-3′). The 1.3-kb PCR product was cloned into pCR Blunt II TOPO (Invitrogen), resulting in the plasmid pEU9508 (Table 1). The 1.2-kb XbaI/HindIII fragment from pEU9508 encoding the N-terminal Strep-tag II-CooD fusion protein was then subcloned into the corresponding restriction sites in pTrc99A (1), resulting in the plasmid pEU9509 (Table 1). The presence of the Strep-tag II at the N terminus of CooD was verified by DNA sequencing. To construct pEU8105 (Table 1), the plasmid pEU9509 was digested with XmaI/HindIII, and the resulting 1.2-kb fragment encoding Strep-tag II-CooD was cloned into XmaI/ HindIII-digested pHSG575 (49).
Site-directed mutagenesis.
Mutations in cooA were constructed using the QuikChange Site-Directed Mutagenesis kit (Stratagene). The plasmids pEU605 and pEU8102 were used as templates. Complementary mutagenic primer sets were designed such that independent alanine substitutions for conserved residues were constructed during the PCR. Conserved alanine residues were replaced with serine. PCR products were treated with DpnI to digest parental template DNA. Mutagenized DNA was transformed into XL1-Blue supercompetent cells (Stratagene). The incorporation of the directed mutations in cooA was confirmed through sequence analysis of the whole gene. The cooB gene, located on the template plasmids, was sequenced to verify that no second-site mutations were introduced during amplification of the entire plasmid. In this study, amino acids in CooA are numbered from the beginning of the mature protein (minus the signal sequence).
Hemagglutination assays.
CooA residues essential for the assembly of functional CS1 pili were identified by testing the ability of cooA mutants to agglutinate bovine erythrocytes. MC4100(pEU478) (12), which expresses the outer membrane protein CooC and the nucleation/adhesin protein CooD, was transformed with either pEU605, carrying wild-type cooBA, or plasmid derivatives expressing mutant CooAs. Overnight cultures of the bacteria were resuspended in ice-cold 0.15 M NaCl to an optical density at 600 nm (OD600) of 30. On glass microscope slides, 30 μl of the bacterial suspensions was mixed with 30 μl of a 10% (vol/vol) suspension of bovine erythrocytes. Slides were incubated at 4°C for 15 to 30 min and scored for the presence of clearly visible agglutination.
Preparation of periplasmic extracts.
Periplasmic extracts were prepared by EDTA-lysozyme treatment as previously described (31), with the exception that overnight cultures were standardized to an OD600 of 120 in Buffer P (100 mM Tris-HCl, pH 8, 500 mM sucrose, 1 mM EDTA) prior to the generation of sphaeroplasts.
Expression and purification of Coo protein complexes.
Strep-tagged forms of CooB and CooD were used to examine the interaction of CooA with these proteins in hemagglutination-negative (HA−) mutants. For the analysis of CooA-CooB complex formation, MC4100(pEU8102/pFDX500), expressing CooB and CooA, or strains harboring CooA mutant derivatives of pEU8102 were grown to an OD600 of 0.5. Strains containing pEU8102 lysed in the absence of lacIq gene expression (pFDX500). Protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside for 6 h at 37°C. To examine CooA-CooD interaction, periplasmic fractions were extracted from overnight cultures of either MC4100(pEU605/pEU8105) expressing CooB, CooA, and CooD or a derivative of this strain carrying pEU605 with mutant CooAs.
Strep-tagged CooB and CooD were affinity-purified from periplasmic extracts using Strep-Tactin Spin Columns (IBA) according to the purification protocol provided by the manufacturer, with the exception of the column wash steps. The spin columns were washed five times with wash buffer containing 500 mM NaCl instead of the recommended 150 mM NaCl to eliminate nonspecific binding of proteins to the column. Protein complexes were eluted with buffer provided in the kit containing 2.5 mM desthiobiotin. Samples from the column flowthrough, wash fractions, and eluate were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot for the presence of either Strep-tagged CooB or CooD. Column fractions were also screened by Western blot for the presence of CooA as part of the purified complex.
Native gel electrophoresis, SDS-PAGE, and Western blotting.
Nondenatured proteins from periplasmic extracts were separated by native gel electrophoresis using 4 to 20% Novex Tris-glycine polyacrylamide precast gels (Invitrogen). Denatured proteins were separated by SDS-PAGE using 12% NuPage Novex Bis-Tris polyacrylamide precast gels (Invitrogen). Separated proteins were electroblotted to polyvinylidene difluoride membranes (Bio-Rad). For Western blot analysis of proteins obtained from affinity purification experiments, 5% of the input sample, 5% of the column flowthrough, 10% of each wash fraction, and 25% of the column eluate were loaded. For all other Western blots, an aliquot of each periplasmic extract, prepared from an equal amount of cells, was loaded in each lane. The presence of CooA was detected using polyclonal antiserum (1:1,000 dilution) made to whole pili (31). The presence of CooB was detected using antiserum (1:1,000 dilution) made to a maltose binding protein fusion with CooB (31). Fusion proteins containing the Strep-tag II affinity tag were detected after incubation with Strep-Tactin alkaline phosphatase conjugate (1:4,000 dilution) (IBA).
RESULTS
Experimental design to study cooA mutants.
These studies were initiated to identify functionally important residues in the major pilin subunit CooA during assembly of CS1 pili. We focused on two regions, the N and C termini of CooA, which contain amino acids highly conserved among the major pilin subunits of CS1 family members (Fig. 2). Near the N terminus of the mature protein, the major pilin subunits have conserved polar residues and a region of alternating hydrophobicity. In their C termini, CS1 family major pilins share the amino acid motif “AGxYxG(x6)T” (where x represents dissimilar amino acids) (31). Outside of this motif, major pilins share conserved hydrophobic and polar residues in the C terminus (Fig. 2). To test the importance of this cross-species conservation, we constructed alanine substitutions separately in conserved residues in the N and C termini of CooA. Next, we tested the ability of these mutants to assemble functional pili using hemagglutination assays with bovine erythrocytes. Alanine substitutions in CooA that result in a HA− phenotype may prevent the interaction of CooA with the CooB periplasmic chaperone, the CooD tip adhesin/nucleation protein, other CooA subunits, or the CooC outer membrane protein. Each HA− cooA mutant was examined to identify the step in the assembly pathway that was disrupted.
FIG. 2.

Alignment of amino acid sequences of CS1 family major pilin proteins. CLUSTAL W (52) was used to align the following: CooA, enterotoxigenic Escherichia coli CS1 pili (National Center for Biotechnology Information reference number AAA23596); CotA, enterotoxigenic Escherichia coli CS2 pili (CAA87761); CfaB, enterotoxigenic Escherichia coli CFA/I pili (P02971); CblA, Burkholderia cepacia cable type II pili (AAM56038); and TcfB, Salmonella enterica serovar Typhi Tcf pili. Identical residues are indicated by black boxes, and similar residues are indicated by gray boxes. The arrow below the alignment indicates the predicted cleavage site of the signal peptide. Asterisks below the sequence indicate conserved hydrophobic residues. Carets indicate the conserved residues comprising the C-terminal motif (AG-x-Y-x-G-x6-T). Residues that were mutated in CooA are indicated above the sequence alignment.
Does alanine substitution of conserved residues in the N and C termini of CooA affect function?
To assess the functionality of cooA mutants, we examined strains encoding all four Coo proteins (CooABCD) for the assembly of functional pili by scoring their ability to agglutinate bovine erythrocytes. We found alanine substitution mutations in both the N and C termini of CooA that affected function. Half of the mutants with substitutions in the N terminus of CooA (13 out of 26) were unable to assemble functional pili (HA−), and 9 out of 29 mutants with C-terminal substitutions were HA− (Table 2). Five out of nine residues in the C terminus (G134, Y136, V140, I142, and L144) important for the function of CooA are within the conserved C-terminal motif “AGxYxG(x6)T” shared among CS1 family major and minor pilins (Fig. 2) (31). Therefore, these residues are likely to constitute a C-terminal domain of interaction required for the assembly of the CooA major pilin into functional pili.
TABLE 2.
Result of alanine substitutions on the function of CooA as measured by hemagglutination
| N terminus
|
C terminus
|
||
|---|---|---|---|
| HA+ | HA− | HA+ | HA− |
| V1Aa | G86A | ||
| E2A | G93A | ||
| I96A | |||
| K3A | L101A | ||
| I5A | F103A | ||
| S6A | G107A | ||
| V7A | V108A | ||
| T8A | V111A | ||
| A9S | K116A | ||
| V11A | I119A | ||
| D12A | A121S | ||
| P13A | A123S | ||
| T14A | G128A | ||
| V15A | G129A | ||
| D16A | L131A | ||
| L17A | T132A | ||
| L18A | A133S | ||
| Q19A | G134A | ||
| D21A | Q135A | ||
| G22A | Y136A | ||
| A24S | Q137A | ||
| L25A | G138A | ||
| P26A | L139A | ||
| V29A | V140A | ||
| L31A | S141A | ||
| Y33A | I142A | ||
| P35A | I143A | ||
| L144A | |||
| T145A | |||
Amino acid positions are numbered in the mature protein (minus the signal sequence).
Are detectable amounts of CooA found in the periplasm of HA− cooA mutants?
Because the HA− phenotype could be due to a deficit in the amount of CooA, we examined periplasmic extracts for the presence of CooA by Western blot using CooA antiserum made to purified pili (31). We detected CooA in the periplasms of 16 HA− mutants (representative results are shown in Fig. 3). Although there was less CooA in several of these mutants than in the wild-type control, for most of these mutants the amount exceeded that made by cooA(K3A), which was HA+ (Fig. 3). Therefore, the reduced amount of CooA in some HA− mutants does not appear to be responsible for their HA− phenotype.
FIG. 3.

Western blot of CooA in the periplasm of N-terminal cooA mutants. Periplasmic extracts were prepared from strains expressing wild-type CooA (MC4100/pEU605) (WT); negative control (MC4100) (NEG); and mutant CooA proteins produced by MC4100/pEU605 derivatives (I5A-K3A) with mutations as indicated above each lane. Mutants with substitutions at I5A-D21A were HA−. The mutant with the K3A substitution was HA+. A 20-μl aliquot of each periplasmic extract at an OD600 of 30 was loaded onto the gel. Proteins were separated by SDS-PAGE using 12% NuPage Novex Bis-Tris polyacrylamide precast gels (Invitrogen). CooA was detected by immunoblot using CooA antiserum.
Little or no CooA was detected in six additional HA− mutants. Figure 3 shows the results for cooA(D21A), representative of this class. Four of the mutants with reduced amounts of CooA had N-terminal substitutions (D21A, G22A, L25A, and Y33A), and two had C-terminal substitutions (I119A and Y136A). Interaction with the CooB periplasmic chaperone is required for stabilization of CooA in the periplasm prior to assembly of CS1 pili, and lack of interaction would result in proteolytic degradation of CooA (53). Therefore, for mutants in which we were unable to detect CooA protein, periplasmic extracts were examined for the presence of CooB. We found CooB in all such mutants (data not shown), suggesting that these mutant CooA proteins may be either unable to interact with the CooB chaperone or have decreased affinity for CooB.
To test the idea that a lack of detectable CooA in the periplasm of cooA substitution mutants results from a defect in the ability to interact with CooB, it is necessary to have sufficient CooA protein to study. Therefore, we tested strains deficient in known periplasmic proteases to determine whether they produced more CooA in the absence of the CooB chaperone than did the wild-type parent. Previous experiments in our laboratory demonstrated that the DegP protease is not involved in the degradation of Coo proteins (53). We found that the amount of CooA in the absence of CooB did not differ between the wild-type strains and mutants in degQ, degS (55), prc (43), and ptr (3) (data not shown). These data indicate that CS1 pilins are degraded by some unidentified protease (53) or combination of proteases. Because these cooA mutants had too little CooA to detect, we were unable to study them further.
Does CooA interact with the CooB periplasmic chaperone in all HA− cooA mutants with detectable amounts of mutant protein?
Because interaction with the CooB chaperone has been demonstrated to be required for stabilization of CooA (53), we expected that all cooA substitution mutations that resulted in detectable amounts of CooA protein would be able to interact with CooB. To analyze CooA-CooB interaction in these mutants, we constructed cooA substitution mutations in the plasmid pEU8102, which also encodes a Strep-tagged CooB. Using periplasmic extracts from each mutant, we affinity purified CooB and assayed for the presence of CooA in the purified complex present in the column eluate (see Materials and Methods). In the wild-type control strain, MC4100/pEU8102/pFDX500, both CooA and CooB were found in the column eluate (Fig. 4), demonstrating that the presence of the Strep-tag in CooB does not prevent CooA-CooB interaction.
FIG. 4.

Western blot of column fractions after affinity purification to examine CooA-CooB interaction. Periplasmic extracts were recovered from the wild-type (WT) strain MC4100/pEU8102/pFDX500 and an isogenic strain carrying a pEU8102 derivative encoding CooA with an L31A substitution in the mature protein. To purify CooA-CooB complexes, periplasmic extracts were applied to a Strep-Tactin affinity column (see Materials and Methods). (A) An aliquot of each fraction (the input sample [I], 5%; column flowthrough [FT], 5%; wash fractions [W1 to W5], 10%; and eluate [E], 25%) was analyzed for the presence of CooA by immunoblot analysis with CooA antiserum. (B) CooB was detected by immunoblot analysis with Strep-Tactin alkaline phosphatase conjugate (IBA).
Both CooA and CooB were found in the column eluates of all but one of the HA− cooA mutants that produced detectable CooA. Therefore, CooA produced by these mutants is able to interact with the chaperone (data not shown), as predicted. However, no CooA protein was found in the CooB-containing column eluate from the cooA(L31A) mutant (Fig. 4B). Instead, for this mutant, CooA was present only in the column flowthrough and wash fractions (Fig. 4A). This indicates that although the amount of CooA protein in the periplasm of the cooA(L31A) mutant was similar to that of the wild type, CooA-L31A may not form periplasmic preassembly complexes with CooB. Alternatively, CooA-L31A may interact more weakly or only transiently with the CooB chaperone, and therefore the periplasmic CooA-CooB complex may not survive the purification process. Therefore, for this mutant, stable association with the chaperone may be unnecessary to stabilize the CooA protein and protect it from degradation. It seems possible, therefore, that the substitution of alanine for leucine at residue 31 may allow the mutant CooA protein to fold into a stable conformation that is protected from proteolysis.
Does CooA interact with the minor pilin protein CooD in HA− cooA mutants?
CooD is necessary to initiate CS1 pilus assembly (33), and interaction of CooA-CooB with CooD-CooB in the periplasm may be important for the process. For this analysis, we transformed pEU605 (28) derivatives containing cooA mutations and wild-type cooB into MC4100 (8) containing pEU8105, which is a low-copy plasmid that encodes a Strep-tagged CooD. We examined periplasmic extracts for the presence of CooA, CooB, and CooD by Western blot and found that all of the mutants, with the exception of the cooA (P26A) mutant, produced detectable amounts of CooA protein (data not shown). The CooA-P26A mutant protein may be more susceptible to degradation in the presence of CooD due to an abnormal CooA-CooD complex conformation which exposes cleavage sites in CooA. As a result of the decreased amount of CooA, the cooA(P26A) mutant could not be tested for CooA-CooD complex formation. For all other HA− mutants, we used periplasmic extracts to affinity purify CooD and then screened column eluates for the presence of CooA. As a positive control, we used periplasmic extracts from wild-type MC4100/pEU605/pEU8105 to determine whether the Strep-tag on CooD prevents its interaction with CooA. The presence of CooA with CooD in the column eluate from the wild-type control strain indicates that the tag does not interfere with CooA-CooD interaction (Fig. 5). The presence of CooD in the flowthrough indicates that the column had been saturated by the Strep-tagged CooD. For the wild-type control, the excess CooA also appeared in the column flowthrough.
FIG. 5.

Western blot of column fractions after affinity purification to examine CooA-CooD interaction. Periplasmic extracts were recovered from the wild-type (WT) strain MC4100/pEU8105/pEU605 and isogenic strains carrying pEU605 derivatives with the CooA mutations indicated. Amino acid positions are numbered from the beginning of the mature protein (minus the signal sequence). To purify CooA-CooD complexes, 500 μl of periplasmic extracts, prepared from an equal amount of cells, was applied to a Strep-Tactin affinity column (see Materials and Methods). (A) An aliquot of each fraction (the input sample [I], 5%; column flowthrough [FT], 5%; wash fractions [W1 to W5], 10%; and eluate [E], 25%) was analyzed for the presence of CooA by immunoblot analysis with CooA antiserum. (B) CooD was detected by immunoblot analysis with Strep-Tactin alkaline phosphatase conjugate (IBA).
Of the 15 HA− cooA mutants that produce detectable CooA, five mutants with alanine substitutions for C-terminal CooA residues V111, G134, V140, I142, and L144 (Fig. 5) produced CooA protein unable to interact with CooD. In all five mutants, all or most of the CooA applied to the column (Fig. 5, lane I) was present in the flowthrough (Fig. 5, lane FT). This indicates that the amount of CooA mutant protein was adequate for detection even in the periplasm of mutants that had less CooA than the wild type. The amount of Strep-tagged CooD applied to the column was similar in all cases; however, for these five mutants, only CooD (Fig. 5B, lane E) and not CooA (Fig. 5A, lane E) was found in the column eluates. In contrast, for other mutants that produced amounts of CooA as small as the five shown here, CooA protein was present in the column eluate (data not shown). Therefore, because CooA with substitutions at V111A, G134A, V140A, I142A, and L144A was not bound to the CooD in the column, it appears that these substitutions prevent formation of CooA-CooD complexes in the periplasm. Four of these substitutions are located within the highly conserved C-terminal motif “AGxYxG(x6)T,” suggesting that this region is important for interactions between the major and minor pilins, CooA and CooD.
Do HA− cooA mutants form periplasmic CooA multimers?
CooA subunits must polymerize to form the pilus structure (31). It is not known whether this occurs in the periplasm or at the outer membrane. While CooA multimers are detected in the periplasm (31), they may be either productive intermediates in the assembly pathway or nonproductive “dead end” products. If multimers are productive assembly intermediates, we would expect that the inability of some cooA substitution mutants to hemagglutinate may result from their inability to form periplasmic multimers. Finding such substitutions may aid in the identification of residues important for CooA-CooA interaction.
We analyzed the ability of CooA mutants to form CooA-CooA periplasmic multimers by native gel electrophoresis and Western blot using CooA antiserum (31). Like periplasmic extracts from strain MC4100/pEU605, which encodes wild-type CooA and CooB, periplasmic extracts from all HA− cooA mutants (Table 2) contained a ladder of different-size CooA complexes, indicative of production of periplasmic CooA oligomers. Representative results for the N-terminal alanine substitution mutants cooA(I5A), cooA(V7A), and cooA(A9S) are shown in Fig. 6. Because all of our HA− cooA mutants form periplasmic CooA multimers, we were unable to identify residues that might be required for subunit-subunit interaction of the major pilin.
FIG. 6.
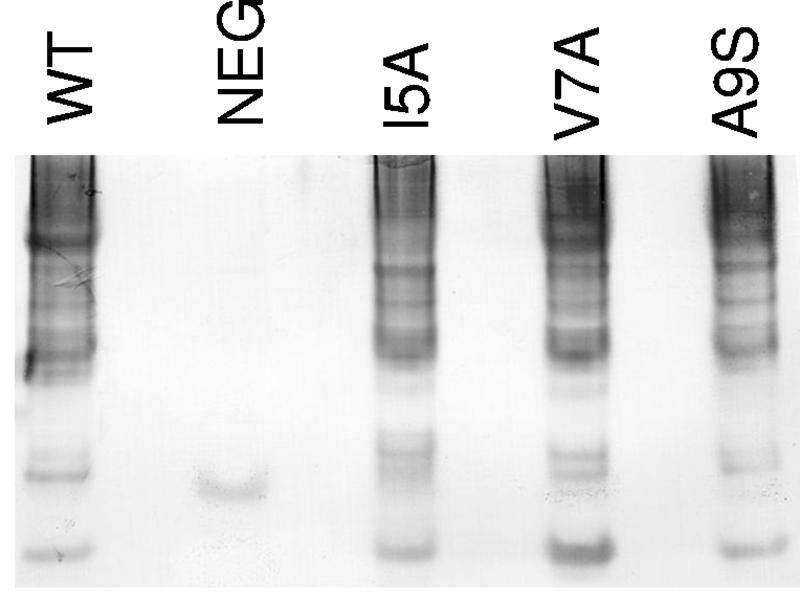
Western blot of CooA multimers in the periplasms of HA− cooA mutants. Periplasmic extracts were prepared from strains expressing the following: lane 1, wild-type (WT) CooA from MC4100/pEU605; and lane 2, negative control (NEG) from MC4100. Lanes 3 to 5 contain extracts of MC4100/pEU605 derivatives expressing CooA with the following mutations: lane 3, I5A; lane 4, V7A; and lane 5, A9S. Amino acid positions are numbered from the beginning of the mature protein (minus the signal sequence). A 12-μl aliquot of each periplasmic extract at an OD600 of 120 was loaded onto the gel. Proteins were separated by native gel electrophoresis using 4 to 20% Novex Tris-glycine polyacrylamide precast gels (Invitrogen). CooA oligomers were detected by immunoblot with CooA antiserum.
DISCUSSION
In this study, we identified conserved residues in the N and C termini of the CS1 major pilin, CooA, that are important for intermolecular interactions during assembly. We identified 13 residues in the N terminus and 9 in the C terminus that, when replaced with alanine, abolished CooA function, resulting in an HA− phenotype (Fig. 7, boxed residues). Conserved hydrophobic amino acids in these regions appear to be particularly important for the interaction of CooA with the other CS1 proteins during assembly, since they accounted for 20 out of 22 HA− substitutions.
FIG. 7.

Summary of phenotypes for N-terminal and C-terminal cooA mutants. Amino acid positions are numbered from the beginning of the mature protein (minus the signal sequence). Residues in boldface type were replaced by alanines, with the exception of alanine residues, which were replaced with serines. Boxed residues are amino acids in CooA that, when mutated, resulted in an HA− phenotype. Green-boxed residues are amino acids in CooA that, when mutated, reduced the amount of protein in the periplasm to undetectable levels. The pink-boxed residue, when mutated, eliminates interaction of CooA with the CooB chaperone but does not affect the interaction of CooA with the CooD minor pilin. Blue-boxed residues are amino acids involved in interaction of CooA with the CooD minor pilin. The orange-boxed residue, when mutated, had no effect on the interaction of CooA with the CooB chaperone but reduced the amount of CooA protein in the periplasm to undetectable levels when CooD was also expressed. Yellow-boxed residues, when replaced by alanine, had no effect on the interaction of CooA with either the CooB chaperone or the CooD minor pilin.
The function of CooA was not affected by alanine substitution for more than half of the conserved residues we examined (Fig. 7, unboxed boldfaced residues). These data indicate that either the residues are not important for CooA function or the change to alanine was a conservative substitution at that position in the protein. Although hydrophobic amino acids accounted for most of the HA− substitutions, alanine substitution for some hydrophobic residues had no effect on the function of CooA (HA+). In these cases, either the residue changed is not involved in CooA interactions with the other proteins or the change to alanine, which is a weakly hydrophobic amino acid, was a permitted substitution at that site. Surprisingly, the substitution of alanine for two acidic residues (E2 and D12), and each of two conserved lysines (K3 and K116), which are large positively charged amino acids, also resulted in an HA+ phenotype. We predict that these residues are not essential for the function of CooA and do not significantly contribute to the native conformation of the protein. In addition to these charged amino acids, no substitutions for any uncharged polar residues that we examined affected the function of CooA. Because serine and threonine are small amino acids like alanine, the substitution may have been conservative for size and thus did not cause a major change in the conformation of the protein. In addition, the tolerability of these substitutions shows that these residues are not required for functionally important hydrogen bonds. However, because glutamine is a larger polar amino acid, it is quite surprising that its replacement with alanine did not affect function. We conclude that the conserved glutamines at positions 19, 135, and 137 are not important for intermolecular interactions of CooA with other Coo proteins during assembly of CS1 pili.
We determined that all HA− cooA mutants form periplasmic CooA multimers (Fig. 6), indicating that interaction of the major pilins with each other was not affected by alanine substitutions. However, it is not known if periplasmic CooA multimers are part of the productive assembly pathway. Therefore, the presence of periplasmic multimers may not be an accurate reflection of the state of appropriate CooA-CooA interaction in these mutants.
We identified a stretch of alternating hydrophobic amino acids (I5, V7, A9, and V11) (Fig. 7) in the N terminus of CooA, each of which, when changed to alanine (serine for A9), resulted in an HA− phenotype. It was surprising that the CooA produced by these mutants formed periplasmic complexes with both the CooB chaperone and the CooD minor pilin, since these mutants were HA−. Residues that make contact to form CooA multimers in the periplasm may be different from those that mediate polymerization at the outer membrane protein, CooC. We suspect that the substitutions at I5, V7, A9, and V11 may have altered the interaction of the major subunits with each other when they are associated with CooC. In P pili, disruption of alternating hydrophobic residues in the N terminus of subunits eliminates the ability of pili to assemble, presumably through the loss of subunit-subunit interaction at the usher (45).
In P pili, it has been demonstrated that subunit residues in the N terminus and alternating hydrophobic amino acids in the C terminus are important for stabilization by the chaperone (23, 45). Although residues in the same general regions in CooA appear to be required for stabilization by the CooB chaperone (Fig. 7, green-boxed residues), alternating hydrophobic amino acids in the C terminus of CooA are not important for chaperone recognition. In addition to four N-terminal substitutions (D21A, G22A, L25A, and Y33A), we found two C-terminal substitutions (I119A and Y136A) that resulted in small to undetectable amounts of CooA in the periplasm. These data suggest that there are differences in how subunit-chaperone complexes are formed during assembly of CS1 and P pili.
Previously, it was suggested that conservation of the C-terminal motif in both the major and minor pilins of CS1 family members might be due to its importance for stabilization by the periplasmic chaperone (31). We found that substitution of alanine for the conserved tyrosine appeared to affect subunit interaction with the chaperone because CooA was not detected in the periplasm of the cooA(Y136A) mutant. However, we determined that alanine substitution for three of the absolutely conserved amino acids (underlined) within the motif [AGxYxG(x6)Tx], A133, G138, and T145, had no effect on the function of CooA, since these mutants were HA+ (Fig. 7, boldfaced residues). Instead, our work suggests that the residues within the C-terminal motif that are important for CooA function are nonconserved hydrophobic amino acids (Fig. 7, blue-boxed residues). Although the primary sequence is not identical in all CS1 family members, hydrophobic residues are always found at these positions in the major and minor pilins. Thus, our results suggest that alternating hydrophobicity rather than amino acid conservation in this region is critical for CooA-CooD association.
It was previously determined that CooD is required for nucleation of pili, based on data showing that CooD is necessary for CS1 assembly (12) and for the extracellular secretion of CooA (33). Our laboratory has also demonstrated that modulation of the level of CooD expression changes the number of pili expressed on the surface (33). Because the loss of CooA-CooD interaction in the periplasm results in an HA− phenotype, we speculate that either these CooA-CooD interactions are the same as those that occur between CooA and the CooD-CooC complex at the outer membrane or pilus formation is initiated not by CooD alone but by periplasmic CooA-CooD complexes associating with CooC.
In this study, we demonstrated that conserved residues within the N and C termini of CooA are important for pilus assembly and function, as are conserved residues in the same regions of P pili subunits. Recognition by the periplasmic chaperone CooB appears to be mediated by residues in both the N and C termini of CooA, but the residues important for recognition are not organized as alternating hydrophobic amino acids like those required during assembly of P pili (23). Instead, for CS1, conserved alternating hydrophobic residues within the C-terminal motif play a role in interaction of the major pilin CooA with the minor pilin CooD. These data, in combination with analysis of the crystal structure of CooA, will lead to a clearer understanding of the interactions of CS1 structural and assembly proteins during the morphogenesis of pili. Because of the conservation of essential residues in the major pilins, we anticipate that the results obtained here for CS1 pili in ETEC will also be relevant for CS1 family members in other serious human pathogens. Insight into the assembly of these important virulence factors should be useful for the development of vaccines and therapeutics aimed at preventing colonization.
Acknowledgments
We thank Amy Craft for technical assistance in the construction of pEU9509.
This work was supported by grant AI24870 from the National Institutes of Health. A.M.S. was supported, in part, by NIH training grant T32-AI007470.
REFERENCES
- 1.Amann, E., B. Ochs, and K. J. Abel. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301-315. [DOI] [PubMed] [Google Scholar]
- 2.Attridge, S. R., G. Wallerstrom, F. Qadri, and A. M. Svennerholm. 2004. Detection of antibodies to toxin-coregulated pili in sera from cholera patients. Infect. Immun. 72:1824-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baneyx, F., and G. Georgiou. 1991. Construction and characterization of Escherichia coli strains deficient in multiple secreted proteases: protease III degrades high-molecular-weight substrates in vivo. J. Bacteriol. 173:2696-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barry, E. M., Z. Altboum, G. Losonsky, and M. M. Levine. 2003. Immune responses elicited against multiple enterotoxigenic Escherichia coli fimbriae and mutant LT expressed in attenuated Shigella vaccine strains. Vaccine 21:333-340. [DOI] [PubMed] [Google Scholar]
- 5.Black, R. E. 1993. Epidemiology of diarrhoeal disease: implications for control by vaccines. Vaccine 11:100-106. [DOI] [PubMed] [Google Scholar]
- 6.Black, R. E. 1990. Epidemiology of travelers' diarrhea and relative importance of various pathogens. Rev. Infect. Dis. 12(Suppl. 1):S73-S79. [DOI] [PubMed] [Google Scholar]
- 7.Black, R. E., K. H. Brown, S. Becker, A. R. Alim, and I. Huq. 1982. Longitudinal studies of infectious diseases and physical growth of children in rural Bangladesh. II. Incidence of diarrhea and association with known pathogens. Am. J. Epidemiol. 115:315-324. [DOI] [PubMed] [Google Scholar]
- 8.Casadaban, M. J. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541-555. [DOI] [PubMed] [Google Scholar]
- 9.Dodson, K. W., F. Jacob-Dubuisson, R. T. Striker, and S. J. Hultgren. 1993. Outer-membrane PapC molecular usher discriminately recognizes periplasmic chaperone-pilus subunit complexes. Proc. Natl. Acad. Sci. USA 90:3670-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards, R. A., and J. L. Puente. 1998. Fimbrial expression in enteric bacteria: a critical step in intestinal pathogenesis. Trends Microbiol. 6:282-287. [DOI] [PubMed] [Google Scholar]
- 11.Folkesson, A., A. Advani, S. Sukupolvi, J. D. Pfeifer, S. Normark, and S. Lofdahl. 1999. Multiple insertions of fimbrial operons correlate with the evolution of Salmonella serovars responsible for human disease. Mol. Microbiol. 33:612-622. [DOI] [PubMed] [Google Scholar]
- 12.Froehlich, B. J., A. Karakashian, L. R. Melsen, J. C. Wakefield, and J. R. Scott. 1994. CooC and CooD are required for assembly of CS1 pili. Mol. Microbiol. 12:387-401. [DOI] [PubMed] [Google Scholar]
- 13.Gaastra, W., and A. M. Svennerholm. 1996. Colonization factors of human enterotoxigenic Escherichia coli (ETEC). Trends Microbiol. 4:444-452. [DOI] [PubMed] [Google Scholar]
- 14.Giron, J. A., M. M. Levine, and J. B. Kaper. 1994. Longus: a long pilus ultrastructure produced by human enterotoxigenic Escherichia coli. Mol. Microbiol. 12:71-82. [DOI] [PubMed] [Google Scholar]
- 15.Hall, R. H., D. R. Maneval, Jr., J. H. Collins, J. L. Theibert, and M. M. Levine. 1989. Purification and analysis of colonization factor antigen I, coli surface antigen 1, and coli surface antigen 3 fimbriae from enterotoxigenic Escherichia coli. J. Bacteriol. 171:6372-6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammar, M., Z. Bian, and S. Normark. 1996. Nucleator-dependent intercellular assembly of adhesive curli organelles in Escherichia coli. Proc. Natl. Acad. Sci. USA 93:6562-6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horton, R. M., H. D. Hunt, S. N. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61-68. [DOI] [PubMed] [Google Scholar]
- 18.Hultgren, S. J., C. H. Jones, and S. N. Normark. 1996. Bacterial adhesions and their assembly, p. 2730-2756. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, D.C.
- 19.Hultgren, S. J., S. Normark, and S. N. Abraham. 1991. Chaperone-assisted assembly and molecular architecture of adhesive pili. Annu. Rev. Microbiol. 45:383-415. [DOI] [PubMed] [Google Scholar]
- 20.Hung, D. L., and S. J. Hultgren. 1998. Pilus biogenesis via the chaperone/usher pathway: an integration of structure and function. J. Struct. Biol. 124:201-220. [DOI] [PubMed] [Google Scholar]
- 21.Kaper, J. B., J. P. Nataro, and H. L. Mobley. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123-140. [DOI] [PubMed] [Google Scholar]
- 22.Kuehn, M. J., J. Heuser, S. Normark, and S. J. Hultgren. 1992. P pili in uropathogenic E. coli are composite fibers with distinct fibrillar adhesive tips. Nature 356:252-255. [DOI] [PubMed] [Google Scholar]
- 23.Kuehn, M. J., D. J. Ogg, J. Kihlberg, L. N. Slonim, K. Flemmer, T. Bergfors, and S. J. Hultgren. 1993. Structural basis of pilus subunit recognition by the PapD chaperone. Science 262:1234-1241. [DOI] [PubMed] [Google Scholar]
- 24.Levine, M., J. G. Morris, G. Losonsky, E. Boedeker, and B. Rowe. 1986. Fimbriae (pili) adhesins as vaccines, p. 14-145. In D. L. Lark (ed.), Protein-carbohydrate interactions in biological systems. Academic Press, Inc., London, United Kingdom.
- 25.Levine, M. M., and R. Edelman. 1990. Future vaccines against enteric pathogens. Infect. Dis. Clin. N. Am. 4:105-121. [PubMed] [Google Scholar]
- 26.Moon, H. W., and T. O. Bunn. 1993. Vaccines for preventing enterotoxigenic Escherichia coli infections in farm animals. Vaccine 11:213-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Casal, J., J. S. Swartley, and J. R. Scott. 1990. Gene encoding the major subunit of CS1 pili of human enterotoxigenic Escherichia coli. Infect. Immun. 58:3594-3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts, J. A., M. B. Kaack, G. Baskin, M. R. Chapman, D. A. Hunstad, J. S. Pinkner, and S. J. Hultgren. 2004. Antibody responses and protection from pyelonephritis following vaccination with purified Escherichia coli PapDG protein. J. Urol. 171:1682-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sajjan, U. S., L. Sun, R. Goldstein, and J. F. Forstner. 1995. Cable (cbl) type II pili of cystic fibrosis-associated Burkholderia (Pseudomonas) cepacia: nucleotide sequence of the cblA major subunit pilin gene and novel morphology of the assembled appendage fibers. J. Bacteriol. 177:1030-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakellaris, H., D. P. Balding, and J. R. Scott. 1996. Assembly proteins of CS1 pili of enterotoxigenic Escherichia coli. Mol. Microbiol. 21:529-541. [DOI] [PubMed] [Google Scholar]
- 32.Sakellaris, H., G. P. Munson, and J. R. Scott. 1999. A conserved residue in the tip proteins of CS1 and CFA/I pili of enterotoxigenic Escherichia coli that is essential for adherence. Proc. Natl. Acad. Sci. USA 96:12828-12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakellaris, H., V. R. Penumalli, and J. R. Scott. 1999. The level of expression of the minor pilin subunit, CooD, determines the number of CS1 pili assembled on the cell surface of Escherichia coli. J. Bacteriol. 181:1694-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakellaris, H., and J. R. Scott. 1998. New tools in an old trade: CS1 pilus morphogenesis. Mol. Microbiol. 30:681-687. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez, J., and J. Holmgren. 2005. Virulence factors, pathogenesis and vaccine protection in cholera and ETEC diarrhea. Curr. Opin. Immunol. 17:1-11. [DOI] [PubMed] [Google Scholar]
- 36.Sauer, F. G., K. Futterer, J. S. Pinkner, K. W. Dodson, S. J. Hultgren, and G. Waksman. 1999. Structural basis of chaperone function and pilus biogenesis. Science 285:1058-1061. [DOI] [PubMed] [Google Scholar]
- 37.Sauer, F. G., S. D. Knight, G. J. Waksman, and S. J. Hultgren. 2000. PapD-like chaperones and pilus biogenesis. Semin. Cell Dev. Biol. 11:27-34. [DOI] [PubMed] [Google Scholar]
- 38.Sauer, F. G., M. A. Mulvey, J. D. Schilling, J. J. Martinez, and S. J. Hultgren. 2000. Bacterial pili: molecular mechanisms of pathogenesis. Curr. Opin. Microbiol. 3:65-72. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt, T. G., J. Koepke, R. Frank, and A. Skerra. 1996. Molecular interaction between the Strep-tag affinity peptide and its cognate target, streptavidin. J. Mol. Biol. 255:753-766. [DOI] [PubMed] [Google Scholar]
- 40.Schnetz, K., S. L. Sutrina, M. H. Saier, Jr., and B. Rak. 1990. Identification of catalytic residues in the beta-glucoside permease of Escherichia coli by site-specific mutagenesis and demonstration of interdomain cross-reactivity between the beta-glucoside and glucose systems. J. Biol. Chem. 265:13464-13471. [PubMed] [Google Scholar]
- 41.Scott, J. R., J. C. Wakefield, P. W. Russell, P. E. Orndorff, and B. J. Froehlich. 1992. CooB is required for assembly but not transport of CS1 pilin. Mol. Microbiol. 6:293-300. [DOI] [PubMed] [Google Scholar]
- 42.Sears, C. L., and J. B. Kaper. 1996. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 60:167-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silber, K. R., and R. T. Sauer. 1994. Deletion of the prc (tsp) gene provides evidence for additional tail-specific proteolytic activity in Escherichia coli K-12. Mol. Gen. Genet. 242:237-240. [DOI] [PubMed] [Google Scholar]
- 44.Skerra, A., and T. G. Schmidt. 2000. Use of the Strep-Tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol. 326:271-304. [DOI] [PubMed] [Google Scholar]
- 45.Soto, G. E., K. W. Dodson, D. Ogg, C. Liu, J. Heuser, S. Knight, J. Kihlberg, C. H. Jones, and S. J. Hultgren. 1998. Periplasmic chaperone recognition motif of subunits mediates quaternary interactions in the pilus. EMBO J. 17:6155-6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soto, G. E., and S. J. Hultgren. 1999. Bacterial adhesins: common themes and variations in architecture and assembly. J. Bacteriol. 181:1059-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone, K. D., H. Z. Zhang, L. K. Carlson, and M. S. Donnenberg. 1996. A cluster of fourteen genes from enteropathogenic Escherichia coli is sufficient for the biogenesis of a type IV pilus. Mol. Microbiol. 20:325-337. [DOI] [PubMed] [Google Scholar]
- 48.Strom, M. S., and S. Lory. 1993. Structure-function and biogenesis of the type IV pili. Annu. Rev. Microbiol. 47:565-596. [DOI] [PubMed] [Google Scholar]
- 49.Takeshita, S., M. Sato, M. Toba, W. Masahashi, and T. Hashimoto-Gotoh. 1987. High-copy-number and low-copy-number plasmid vectors for lacZ alpHA− complementation and chloramphenicol- or kanamycin-resistance selection. Gene 61:63-74. [DOI] [PubMed] [Google Scholar]
- 50.Thanassi, D. G., E. T. Saulino, and S. J. Hultgren. 1998. The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr. Opin. Microbiol. 1:223-231. [DOI] [PubMed] [Google Scholar]
- 51.Thanassi, D. G., E. T. Saulino, M. J. Lombardo, R. Roth, J. Heuser, and S. J. Hultgren. 1998. The PapC usher forms an oligomeric channel: implications for pilus biogenesis across the outer membrane. Proc. Natl. Acad. Sci. USA 95:3146-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voegele, K., H. Sakellaris, and J. R. Scott. 1997. CooB plays a chaperone-like role for the proteins involved in formation of CS1 pili of enterotoxigenic Escherichia coli. Proc. Natl. Acad. Sci. USA 94:13257-13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wall, D., and D. Kaiser. 1999. Type IV pili and cell motility. Mol. Microbiol. 32:1-10. [DOI] [PubMed] [Google Scholar]
- 55.Waller, P. R., and R. T. Sauer. 1996. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. J. Bacteriol. 178:1146-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolf, M. K. 1997. Occurrence, distribution, and associations of O and H serogroups, colonization factor antigens, and toxins of enterotoxigenic Escherichia coli. Clin. Microbiol. Rev. 10:569-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]