

Abstract
The negative affective symptoms of opiate withdrawal powerfully motivate drug-seeking behavior and may trigger relapse to heroin abuse. To date, no medications exist that effectively relieve the negative affective symptoms of opiate withdrawal. The corticotropin-releasing factor (CRF) system has been hypothesized to mediate the motivational effects of drug dependence. The CRF signal is transmitted by two distinct receptors named CRF receptor-1 (CRF1) and CRF2. Here we report that genetic disruption of CRF1 receptor pathways in mice eliminates the negative affective states of opiate withdrawal. In particular, neither CRF1 receptor heterozygous (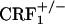 ) nor homozygous (
) nor homozygous (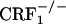 ) null mutant mice avoided environmental cues repeatedly paired with the early phase of opiate withdrawal. These results were not due to altered associative learning processes because
) null mutant mice avoided environmental cues repeatedly paired with the early phase of opiate withdrawal. These results were not due to altered associative learning processes because 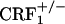 and
and 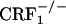 mice displayed reliable, conditioned place aversions to environmental cues paired with the κ-opioid receptor agonist U-50,488H. We also examined the impact of CRF1 receptor-deficiency upon opiate withdrawal-induced dynorphin activity in the nucleus accumbens, a brain molecular mechanism thought to underlie the negative affective states of drug withdrawal. Consistent with the behavioral indices, we found that, during the early phase of opiate withdrawal, neither
mice displayed reliable, conditioned place aversions to environmental cues paired with the κ-opioid receptor agonist U-50,488H. We also examined the impact of CRF1 receptor-deficiency upon opiate withdrawal-induced dynorphin activity in the nucleus accumbens, a brain molecular mechanism thought to underlie the negative affective states of drug withdrawal. Consistent with the behavioral indices, we found that, during the early phase of opiate withdrawal, neither 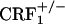 nor
nor 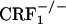 showed increased dynorphin mRNA levels in the nucleus accumbens. This study reveals a cardinal role for CRF/CRF1 receptor pathways in the negative affective states of opiate withdrawal and suggests therapeutic strategies for the treatment of opiate addiction.
showed increased dynorphin mRNA levels in the nucleus accumbens. This study reveals a cardinal role for CRF/CRF1 receptor pathways in the negative affective states of opiate withdrawal and suggests therapeutic strategies for the treatment of opiate addiction.
Keywords: dynorphin, drug dependence, heroin, dysphoria
Heroin addiction is one of the most common and serious substance-related disorders (1). In addicted individuals, heroin “highs” are inexorably followed by a severe opiate withdrawal syndrome composed of somatic signs and negative affective states, such as dysphoria and depressed mood (2). The negative affective states of opiate withdrawal dramatically motivate compulsive heroin-seeking behavior and opiate abuse (3, 4). However, available treatments do not effectively relieve the negative affective symptoms of opiate withdrawal, thus leaving opiate addicts vulnerable to relapse to heroin.
The corticotropin-releasing factor (CRF) system might mediate the motivational effects of drug dependence. Functional antagonism of CRF neurotransmission reduces stress-induced reinstatement of drug-seeking behavior and may attenuate anxiety-like and affective states associated with drug withdrawal (5). The latter findings point to the CRF system as a potential therapeutic target for treating the negative affective consequences of drug addiction. However, recent studies showing opposite functions for the two known CRF receptor pathways, CRF receptor-1 (CRF1) and CRF2, in anxiety-like and depression-like behaviors indicate a complex role for the CRF system in affect regulation (6-11). Elements in the CRF circuitry might also differentially contribute to the negative affective states of drug withdrawal. However, the specific role for the different components of the CRF system in drug withdrawal-induced negative affect is still unknown.
Mutant mice bearing targeted mutations of the CRF system are unique tools to elucidate the role of CRF in drug dependence and withdrawal. Here we used genetically engineered mice lacking functional CRF1 receptor levels to study the role for CRF/CRF1 receptor pathways in the negative affective states of opiate withdrawal (6). Moreover, in keeping with the clinical setting where signs and symptoms of opiate withdrawal “spontaneously” and gradually rise along with the drug removal from the body, behavioral and molecular studies reported here were conducted in mice undergoing spontaneous opiate withdrawal. In contrast, previous studies that have examined the role of CRF in the somatic and aversive effects of opiate withdrawal have all used opioid receptor antagonist-precipitated opiate withdrawal procedures (12-16). However, high-affinity competitive opioid receptor antagonists “precipitate” behavioral, endocrine, and molecular patterns that greatly differ from those observed upon spontaneous opiate withdrawal (17-20), thus making questionable their use to model clinical conditions.
To search for possible molecular mechanisms implicated in the negative affective states of opiate withdrawal, we also examined the impact of CRF1 receptor-deficiency upon opiate withdrawal-induced dynorphin gene expression in the nucleus accumbens, a brain region involved in addictive behaviors (21). Previous studies have shown that opiate withdrawal increases the genetic transcription of the endogenous opioid peptide dynorphin in several brain regions (22, 23). In addition, CRF-opioid interactions have been demonstrated in the locus coeruleus and the hypothalamus, brain regions relevant to the autonomic and neuroendocrine alterations associated with opiate withdrawal (24, 25). However, to date, no studies have reported on the role of CRF in the increased brain dynorphin activity associated with opiate withdrawal.
Here, we combined behavioral and molecular assays to investigate the role for CRF/CRF1 receptor pathways in the negative affective states and increased dynorphin activity in the nucleus accumbens associated with a spontaneous opiate withdrawal condition.
Materials and Methods
Subjects. Group-housed, littermate, female mice that were wild-type, CRF1-receptor heterozygous (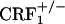 ), and homozygous (
), and homozygous (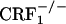 ) were used throughout (6). Mice were 4-8 months old and derived from mating
) were used throughout (6). Mice were 4-8 months old and derived from mating 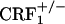 breeders. Wild-type,
breeders. Wild-type, 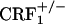 and
and 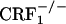 offspring of
offspring of 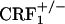 breeders were identified by PCR analysis of tail DNA. The mice were housed in a colony room maintained at 22°C on a 12-h light/dark cycle (lights on from 8 a.m. until 8 p.m.). Food and water were available ad libitum. Mice were handled on alternate days during the week preceding the tests. All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the local Animal Care and Use Committee.
breeders were identified by PCR analysis of tail DNA. The mice were housed in a colony room maintained at 22°C on a 12-h light/dark cycle (lights on from 8 a.m. until 8 p.m.). Food and water were available ad libitum. Mice were handled on alternate days during the week preceding the tests. All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the local Animal Care and Use Committee.
Opiate Withdrawal-Induced, Conditioned Place Aversion (CPA). Negative affective states associated with opiate withdrawal were examined by using the CPA paradigm, a behavioral technique commonly used to evaluate the affective-like consequences of drug withdrawal in rodents. The CPA apparatus consisted of a rectangular Plexiglas box (length, 42 cm; width, 21 cm; height, 21 cm) divided by a central partition into two chambers of equal size (21 × 21 × 21 cm). One compartment had black walls and a smooth Plexiglas floor whereas the other compartment had vertical black and white striped (2 cm) walls and a slightly rough floor. During the test sessions, an aperture (4 × 4 cm) in the central partition allowed the mice to enter both sides of the apparatus, whereas during the conditioning sessions the individual compartments were closed off from each other. Transparent Plexiglas lids allowed observation of the animal's behavior on a video monitor situated in an adjacent room and connected to a camera placed above the apparatus. The CPA experiment lasted 14 days and consisted of three phases: preconditioning test, conditioning phase, and postconditioning test. On day 1, each mouse was allowed to explore freely the entire CPA apparatus for 20 min, and time spent in each of the two compartments was measured (preconditioning test). Within each genotype, mice were then divided in two groups with similar preconditioning time values in the preferred and nonpreferred compartment of the CPA apparatus. One group was assigned to receive vehicle and the other was assigned to receive increasing doses of morphine (20-100 mg/kg). In particular, starting on day 3, every 12 h (at 8 a.m. and 8 p.m.) wild-type, 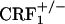 and
and 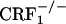 mice were treated with vehicle or morphine according to the following protocol: day 3, 20 mg/kg; day 4, 40 mg/kg; day 5, 60 mg/kg; day 6, 80 mg/kg; day 7, 100 mg/kg; and day 8, 100 mg/kg (only one injection in the morning). Conditioning sessions took place on days 5-8, while morphine-treated mice were in an opiate withdrawal state. For this purpose, 8 h after the morning injection, mice were confined for 30 min a day into their preferred compartment of the CPA apparatus, as determined on preconditioning test. Conditioning sessions were carried out 8 h after the morphine injections because in these mice expression of the somatic signs of opiate withdrawal peaks at this time point (our unpublished observations). During the conditioning sessions, we also quantified jump, wet dog shake, and diarrhea events, which are among the most important somatic signs of opiate withdrawal in mice. Postconditioning tests took place 6 days after the last conditioning session (day 14), once somatic signs of opiate withdrawal had largely dissipated in all of the three genotypes. For each mouse, a place aversion score was calculated as the postconditioning time minus the preconditioning time (expressed in seconds) spent in the conditioning compartment of the CPA apparatus.
mice were treated with vehicle or morphine according to the following protocol: day 3, 20 mg/kg; day 4, 40 mg/kg; day 5, 60 mg/kg; day 6, 80 mg/kg; day 7, 100 mg/kg; and day 8, 100 mg/kg (only one injection in the morning). Conditioning sessions took place on days 5-8, while morphine-treated mice were in an opiate withdrawal state. For this purpose, 8 h after the morning injection, mice were confined for 30 min a day into their preferred compartment of the CPA apparatus, as determined on preconditioning test. Conditioning sessions were carried out 8 h after the morphine injections because in these mice expression of the somatic signs of opiate withdrawal peaks at this time point (our unpublished observations). During the conditioning sessions, we also quantified jump, wet dog shake, and diarrhea events, which are among the most important somatic signs of opiate withdrawal in mice. Postconditioning tests took place 6 days after the last conditioning session (day 14), once somatic signs of opiate withdrawal had largely dissipated in all of the three genotypes. For each mouse, a place aversion score was calculated as the postconditioning time minus the preconditioning time (expressed in seconds) spent in the conditioning compartment of the CPA apparatus.
U-50,488H-Induced CPA. We used the same CPA apparatus and an experimental procedure very similar to that used in the opiate withdrawal experiment described above. The only differences were that the mice were not exposed to the twice daily treatment with vehicle or morphine; in addition, during the conditioning phase (days 5-8), mice were treated with vehicle or U-50,488H (5 mg/kg) just before being confined to their preferred compartment of the CPA apparatus.
Dynorphin in Situ Hybridization Histochemistry and Corticosterone Assay. A separate cohort of wild-type, 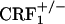 and
and 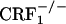 mice was treated with vehicle or morphine as described above. Eight hours after the last injection, the mice were individually confined to Plexiglas cylinders for 30 min. Their brains were removed immediately thereafter, rapidly frozen in isopentane (-40°C), and stored at -80°C. Blood samples were also collected from the trunk, and plasma samples were stored at -20°C until corticosterone assay. Plasma corticosterone levels were quantified by RIA using a specific corticosterone antibody (ICN). The intraassay and interassay coefficients of variation were ≈3.5% and 8%, respectively. Brains collected were cut in frontal sections (12 μm) with a cryostat and thaw mounted onto gelatin-covered slides to be processed for in situ hybridization. The in situ hybridization procedure was performed with oligonucleotide probes designed to recognize the dynorphin mRNA (26). Oligonucleotide probes were labeled by tailing with [35S]dATP (PerkinElmer SAS) using terminal deoxynucleotide transferase (Promega). The specific activity of the oligonucleotide probes was 56 × 107 cpm/μg. After labeling, the probes were precipitated in absolute ethanol and 5 M sodium chloride, dried, and resuspended at the concentration of 4.25 pg/μl in the hybridization buffer [50% deionized formamide/20% dextran sulfate/20% 20× SSC (1× SSC = 0.15 M sodium chloride/0.015 M sodium citrate, pH 7)/500 μg/ml denatured salmon sperm DNA/1% Denhardt/5% sarcosyl/240 μg/ml tRNA/2.4 mg/ml NaH2PO4]. The sections were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for 5 min at room temperature, rinsed twice for 30 min with 4× SSC (2% Denhardt), acetylated into 4× SSC (0.25% acetic anhydride/1.33% triethanolamine, pH 8) for 10 min at room temperature, and then dehydrated in graded alcohol. The slides were then incubated horizontally overnight at 40°C with the hybridization solution containing the labeled probe (40 μl per slide). At the end of the incubation, slides were washed in decreasing concentrations of SSC and dehydrated in ethanol. Sections were exposed at room temperature to Biomax-MR film (Kodak) over 15 days for dynorphin mRNA detection. For microautoradiographic analyses, sections were dipped into LM-1 emulsion (Amersham Biosciences, which is now GE Healthcare), diluted to a two-thirds concentration with water, exposed in the dark for 40 days at 4°C, and then developed and counterstained with Mayer's hematoxylin solution. Analysis of the core and shell portions of the nucleus accumbens was performed by counting silver grains and labeled neurons at ×50 magnification using an image analyzer system for cartography and grain countings (VisioScan-Densirag 200, Biocom, Paris). A neuron was considered labeled if its silver grain density was at least 3-fold higher than background noise. Results were expressed as silver grain density (number of silver grains in 100 μm2) by neuron density (number of labeled neurons in 0.5 mm2). The values of 100 μm2 and 0.5 mm2 were considered to closely reflect the mean size of a neuron and of the core or the shell portions of the nucleus accumbens, respectively (27). For each mouse, the nucleus accumbens of both cerebral hemispheres were quantified.
mice was treated with vehicle or morphine as described above. Eight hours after the last injection, the mice were individually confined to Plexiglas cylinders for 30 min. Their brains were removed immediately thereafter, rapidly frozen in isopentane (-40°C), and stored at -80°C. Blood samples were also collected from the trunk, and plasma samples were stored at -20°C until corticosterone assay. Plasma corticosterone levels were quantified by RIA using a specific corticosterone antibody (ICN). The intraassay and interassay coefficients of variation were ≈3.5% and 8%, respectively. Brains collected were cut in frontal sections (12 μm) with a cryostat and thaw mounted onto gelatin-covered slides to be processed for in situ hybridization. The in situ hybridization procedure was performed with oligonucleotide probes designed to recognize the dynorphin mRNA (26). Oligonucleotide probes were labeled by tailing with [35S]dATP (PerkinElmer SAS) using terminal deoxynucleotide transferase (Promega). The specific activity of the oligonucleotide probes was 56 × 107 cpm/μg. After labeling, the probes were precipitated in absolute ethanol and 5 M sodium chloride, dried, and resuspended at the concentration of 4.25 pg/μl in the hybridization buffer [50% deionized formamide/20% dextran sulfate/20% 20× SSC (1× SSC = 0.15 M sodium chloride/0.015 M sodium citrate, pH 7)/500 μg/ml denatured salmon sperm DNA/1% Denhardt/5% sarcosyl/240 μg/ml tRNA/2.4 mg/ml NaH2PO4]. The sections were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for 5 min at room temperature, rinsed twice for 30 min with 4× SSC (2% Denhardt), acetylated into 4× SSC (0.25% acetic anhydride/1.33% triethanolamine, pH 8) for 10 min at room temperature, and then dehydrated in graded alcohol. The slides were then incubated horizontally overnight at 40°C with the hybridization solution containing the labeled probe (40 μl per slide). At the end of the incubation, slides were washed in decreasing concentrations of SSC and dehydrated in ethanol. Sections were exposed at room temperature to Biomax-MR film (Kodak) over 15 days for dynorphin mRNA detection. For microautoradiographic analyses, sections were dipped into LM-1 emulsion (Amersham Biosciences, which is now GE Healthcare), diluted to a two-thirds concentration with water, exposed in the dark for 40 days at 4°C, and then developed and counterstained with Mayer's hematoxylin solution. Analysis of the core and shell portions of the nucleus accumbens was performed by counting silver grains and labeled neurons at ×50 magnification using an image analyzer system for cartography and grain countings (VisioScan-Densirag 200, Biocom, Paris). A neuron was considered labeled if its silver grain density was at least 3-fold higher than background noise. Results were expressed as silver grain density (number of silver grains in 100 μm2) by neuron density (number of labeled neurons in 0.5 mm2). The values of 100 μm2 and 0.5 mm2 were considered to closely reflect the mean size of a neuron and of the core or the shell portions of the nucleus accumbens, respectively (27). For each mouse, the nucleus accumbens of both cerebral hemispheres were quantified.
Drugs. Morphine HCl (20-100 mg/kg, i.p.; Salars, Como, Italy) and U-50,488H (5 mg/kg, s.c.; National Institute on Drug Abuse, Bethesda) were dissolved in physiological saline and injected in a volume of 10 ml/kg. Control mice were injected with the same volume of saline.
Statistical Analysis. Two-way ANOVA with genotype (wild-type, 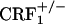 , and
, and 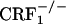 mice) and treatment (control, opiate-withdrawn, or U-50,488H-treated mice) as independent variables were used to examine place aversion scores, wet dog shakes recorded during the opiate withdrawal CPA experiment, and dynorphin mRNA levels in the nucleus accumbens core or shell. A three-way ANOVA with genotype and treatment as between-subjects factors and conditioning sessions as a within-subject repeated measure was used to analyze jumps recorded during the opiate withdrawal CPA experiment. The Student-Newman-Keuls post hoc test was used for individual group comparisons. The accepted value for significance was P < 0.05.
mice) and treatment (control, opiate-withdrawn, or U-50,488H-treated mice) as independent variables were used to examine place aversion scores, wet dog shakes recorded during the opiate withdrawal CPA experiment, and dynorphin mRNA levels in the nucleus accumbens core or shell. A three-way ANOVA with genotype and treatment as between-subjects factors and conditioning sessions as a within-subject repeated measure was used to analyze jumps recorded during the opiate withdrawal CPA experiment. The Student-Newman-Keuls post hoc test was used for individual group comparisons. The accepted value for significance was P < 0.05.
Results
Absence of Affective States Related to Opiate Withdrawal in CRF1 Receptor-Deficient Mice. During the 20-min preconditioning tests, the values (expressed in seconds) for mean times ± SEM spent in the preferred compartment of the CPA apparatus for vehicle-treated mice were 890 ± 16 (wild-type), 891 ± 20 (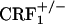 ), and 955 ± 16 (
), and 955 ± 16 (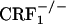 ). For morphine-treated mice, the same values were 889 ± 16 (wild-type), 899 ± 21 (
). For morphine-treated mice, the same values were 889 ± 16 (wild-type), 899 ± 21 (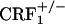 ), and 906 ± 27 (
), and 906 ± 27 (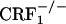 ). After conditioning, testing for opiate withdrawal-induced place aversions revealed no genotype effect (F2,69 = 2.24, P value was not significant), an opiate withdrawal effect (F1,69 = 19.21, P < 0.0001), and a genotype × opiate withdrawal interaction effect (F2,69 = 3.53, P < 0.05). Wild-type mice displayed a significant reduction in the time spent in the opiate withdrawal-paired compartment of the CPA apparatus as compared with preconditioning values (P < 0.01 versus all other groups). In contrast, opiate-withdrawn
). After conditioning, testing for opiate withdrawal-induced place aversions revealed no genotype effect (F2,69 = 2.24, P value was not significant), an opiate withdrawal effect (F1,69 = 19.21, P < 0.0001), and a genotype × opiate withdrawal interaction effect (F2,69 = 3.53, P < 0.05). Wild-type mice displayed a significant reduction in the time spent in the opiate withdrawal-paired compartment of the CPA apparatus as compared with preconditioning values (P < 0.01 versus all other groups). In contrast, opiate-withdrawn 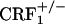 and
and 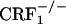 mice did not differ from control mice (Fig. 1). Thus, contrary to the wild-type mice, CRF1 receptor-deficient mice did not show any aversion for environmental cues paired with opiate withdrawal, indicating the absence of negative affective states. These results clearly show that CRF1 receptor signaling is essential to the negative affective properties of opiate withdrawal. Also, the lack of place aversions in opiate-withdrawn
mice did not differ from control mice (Fig. 1). Thus, contrary to the wild-type mice, CRF1 receptor-deficient mice did not show any aversion for environmental cues paired with opiate withdrawal, indicating the absence of negative affective states. These results clearly show that CRF1 receptor signaling is essential to the negative affective properties of opiate withdrawal. Also, the lack of place aversions in opiate-withdrawn 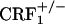 mice indicates that the negative affective states of opiate withdrawal depend on fully functional CRF1 receptor pathways.
mice indicates that the negative affective states of opiate withdrawal depend on fully functional CRF1 receptor pathways.
Fig. 1.

Lack of negative affective states in opiate-withdrawn CRF1 receptor-deficient mice. Place aversion scores of control and opiate-withdrawn wild-type, 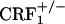 , and
, and 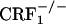 mice. For each mouse, a place-aversion score was calculated as described in Materials and Methods. Values represent mean ± SEM. n = 11-14 per group. *, P < 0.01 versus all other groups.
mice. For each mouse, a place-aversion score was calculated as described in Materials and Methods. Values represent mean ± SEM. n = 11-14 per group. *, P < 0.01 versus all other groups.
Evaluation of jumping behavior during conditioning revealed a genotype effect (F2,69 = 12.93, P < 0.0001), an opiate withdrawal effect (F1,69 = 24.74, P < 0.0001), and a genotype × opiate withdrawal × repeated measure interaction effect (F6,207 = 3.17, P < 0.01). During the third and fourth conditioning sessions, opiate-withdrawn 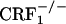 mice made more jumps than all other groups (P < 0.0001). In contrast, opiate-withdrawn wild-type and
mice made more jumps than all other groups (P < 0.0001). In contrast, opiate-withdrawn wild-type and 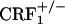 mice did not differ from control mice (Fig. 5, which is published as supporting information on the PNAS web site). Analysis of wet dog shake behavior also revealed a genotype effect (F2,69 = 9.76, P < 0.0005), an opiate withdrawal effect (F1,69 = 39.57, P < 0.0001), and a genotype × opiate withdrawal interaction effect (F2,69 = 6.07, P < 0.005). Opiate-withdrawn
mice did not differ from control mice (Fig. 5, which is published as supporting information on the PNAS web site). Analysis of wet dog shake behavior also revealed a genotype effect (F2,69 = 9.76, P < 0.0005), an opiate withdrawal effect (F1,69 = 39.57, P < 0.0001), and a genotype × opiate withdrawal interaction effect (F2,69 = 6.07, P < 0.005). Opiate-withdrawn 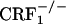 mice made more wet dog shakes than all other groups (P < 0.0005), whereas opiate-withdrawn wild-type and
mice made more wet dog shakes than all other groups (P < 0.0005), whereas opiate-withdrawn wild-type and 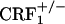 mice did not differ from control mice (Table 1, which is published as supporting information on the PNAS web site). Finally, no reliable signs of diarrhea were observed in any of the experimental groups during the four conditioning sessions.
mice did not differ from control mice (Table 1, which is published as supporting information on the PNAS web site). Finally, no reliable signs of diarrhea were observed in any of the experimental groups during the four conditioning sessions.
Lack of Dynorphin mRNA Increase in the Nucleus Accumbens of Opiate-Withdrawn CRF1 Receptor-Deficient Mice. Up-regulated dynorphin activity in the nucleus accumbens might underlie the negative affective consequences of drug withdrawal. Thus, we quantified dynorphin gene expression in the core and shell portions of the nucleus accumbens of wild-type and CRF1 receptor-deficient mice. Analysis of dynorphin mRNA levels in the shell portion of the nucleus accumbens revealed a genotype effect (F2,45 = 17.05, P < 0.0001), an opiate withdrawal effect (F1,45 = 5.11, P < 0.05), and a genotype × opiate withdrawal interaction effect (F2,45 = 5.85, P < 0.01). Opiate withdrawal elevated dynorphin mRNA content in the nucleus accumbens shell of wild-type mice (P < 0.0005 versus all other groups). In contrast, opiate withdrawal did not affect the genetic transcription of dynorphin in 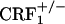 and
and 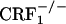 mice (Fig. 2). The dynorphin results obtained in the three different genotypes agree well with the behavioral indices of affective opiate withdrawal. That is, unlike opiate-withdrawn wild-type mice, neither
mice (Fig. 2). The dynorphin results obtained in the three different genotypes agree well with the behavioral indices of affective opiate withdrawal. That is, unlike opiate-withdrawn wild-type mice, neither 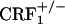 nor
nor 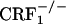 mice showed the affective-like signs and the increased dynorphin mRNA levels in the nucleus accumbens shell in response to opiate withdrawal.
mice showed the affective-like signs and the increased dynorphin mRNA levels in the nucleus accumbens shell in response to opiate withdrawal.
Fig. 2.

Opiate-withdrawn CRF1 receptor-deficient mice do not show increased dynorphin gene expression in the nucleus accumbens. (A) Dynorphin mRNA levels in the shell portion of the nucleus accumbens of control and opiate-withdrawn wild-type, 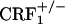 , and
, and 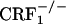 mice. Results are expressed as silver grain density (silver grains per 100 μm2) by neuron density (labeled neurons per 0.5 mm2). Values represent mean ± SEM. n = 8-10 per group. *, P < 0.0005 versus all other groups. (B) Negative images of Biomax-MR films (Upper) and high-magnification silver grains photomicrographs (Lower) of some representative brain sections illustrating dynorphin mRNA levels in the nucleus accumbens shell (S) of control and opiate-withdrawn wild-type,
mice. Results are expressed as silver grain density (silver grains per 100 μm2) by neuron density (labeled neurons per 0.5 mm2). Values represent mean ± SEM. n = 8-10 per group. *, P < 0.0005 versus all other groups. (B) Negative images of Biomax-MR films (Upper) and high-magnification silver grains photomicrographs (Lower) of some representative brain sections illustrating dynorphin mRNA levels in the nucleus accumbens shell (S) of control and opiate-withdrawn wild-type, 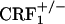 , and
, and 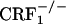 mice. (Scale bars: Upper, 0.8 mm; Lower, 10 μm.)
mice. (Scale bars: Upper, 0.8 mm; Lower, 10 μm.)
Examination of the core portion of the nucleus accumbens revealed a genotype effect (F2,45 = 5.52, P < 0.01), no opiate withdrawal effect (F1,45 = 0.00, P value was not significant) and no genotype × opiate withdrawal interaction effect (F2,45 = 0.33, P value was not significant). Overall, 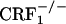 mice displayed lower dynorphin mRNA levels than wild-type and
mice displayed lower dynorphin mRNA levels than wild-type and 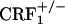 mice (P < 0.05) (Fig. 3). Thus, contrary to the shell portion, opiate withdrawal did not affect the expression of dynorphin in the nucleus accumbens core of wild-type mice. This finding is in line with a previous study showing increased neuronal activity in the shell but not in the core of the nucleus accumbens in opiate-withdrawn rats (28).
mice (P < 0.05) (Fig. 3). Thus, contrary to the shell portion, opiate withdrawal did not affect the expression of dynorphin in the nucleus accumbens core of wild-type mice. This finding is in line with a previous study showing increased neuronal activity in the shell but not in the core of the nucleus accumbens in opiate-withdrawn rats (28).
Fig. 3.

Opiate withdrawal does not affect dynorphin gene expression in the nucleus accumbens core. Shown are dynorphin mRNA levels in the core portion of the nucleus accumbens of control and opiate-withdrawn wild-type, 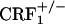 , and
, and 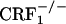 mice. Results are expressed as silver grain density (silver grains per 100 μm2) by neuron density (labeled neurons per 0.5 mm2). Values represent mean ± SEM. n = 8-10 per group. Overall,
mice. Results are expressed as silver grain density (silver grains per 100 μm2) by neuron density (labeled neurons per 0.5 mm2). Values represent mean ± SEM. n = 8-10 per group. Overall, 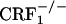 mice showed lower dynorphin mRNA levels than wild-type and
mice showed lower dynorphin mRNA levels than wild-type and 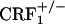 mice (P < 0.01).
mice (P < 0.01).
CRF1 Receptor-Deficient Mice Display U-50,488H-Induced Place Aversions. Contrary to the wild-type mice, 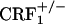 and
and 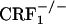 mice showed no conditioned aversions to places paired with opiate withdrawal. Thus, it can be argued that in the mutant mice absence of opiate withdrawal-induced CPA might have been due to deficits in associative learning processes required for the acquisition and expression of conditioned behaviors. To rule out this issue, wild-type,
mice showed no conditioned aversions to places paired with opiate withdrawal. Thus, it can be argued that in the mutant mice absence of opiate withdrawal-induced CPA might have been due to deficits in associative learning processes required for the acquisition and expression of conditioned behaviors. To rule out this issue, wild-type, 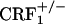 , and
, and 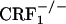 mice were tested in a CPA procedure using the U-50,488H compound, a κ-opioid receptor (KOR) agonist known to produce reliable CPA in mice (29). During the preconditioning test, the values (expressed in seconds) for mean times ± SEM spent in the preferred compartment of the CPA apparatus for vehicle-treated mice were 883 ± 25 (wild-type), 872 ± 32 (
mice were tested in a CPA procedure using the U-50,488H compound, a κ-opioid receptor (KOR) agonist known to produce reliable CPA in mice (29). During the preconditioning test, the values (expressed in seconds) for mean times ± SEM spent in the preferred compartment of the CPA apparatus for vehicle-treated mice were 883 ± 25 (wild-type), 872 ± 32 (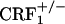 ), and 910 ± 31 (
), and 910 ± 31 (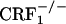 ). The same values for U-50,488H-treated mice were 889 ± 23 (wild-type), 876 ± 31 (
). The same values for U-50,488H-treated mice were 889 ± 23 (wild-type), 876 ± 31 (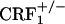 ), 905 ± 23 (
), 905 ± 23 (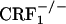 ). Analysis of place-conditioning scores revealed no genotype effect (F2,43 = 0.13, P value was not significant), a U-50,488H effect (F1,43 = 22.23, P < 0.0001), and no genotype × U-50,488H interaction effect (F2,43 = 0.21, P value was not significant). Similarly to the wild-type mice,
). Analysis of place-conditioning scores revealed no genotype effect (F2,43 = 0.13, P value was not significant), a U-50,488H effect (F1,43 = 22.23, P < 0.0001), and no genotype × U-50,488H interaction effect (F2,43 = 0.21, P value was not significant). Similarly to the wild-type mice, 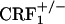 and
and 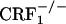 mice displayed a significant reduction in the time spent in the U-50,488H-paired compartment of the CPA apparatus as compared with preconditioning values (P < 0.0001 versus control mice) (Fig. 4). Thus, CRF1 receptor-deficient mice can acquire and display CPAs, indicating no major deficits in associative learning processes.
mice displayed a significant reduction in the time spent in the U-50,488H-paired compartment of the CPA apparatus as compared with preconditioning values (P < 0.0001 versus control mice) (Fig. 4). Thus, CRF1 receptor-deficient mice can acquire and display CPAs, indicating no major deficits in associative learning processes.
Fig. 4.

Unaltered CPA abilities in CRF1 receptor-deficient mice. Shown are place aversion scores of control and U-50,488H-treated wild-type, 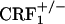 , and
, and 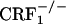 mice. For each mouse, a place aversion score was calculated as described in Materials and Methods. Values represent mean ± SEM. n = 6-11 per group. *, P < 0.0001 versus control mice.
mice. For each mouse, a place aversion score was calculated as described in Materials and Methods. Values represent mean ± SEM. n = 6-11 per group. *, P < 0.0001 versus control mice.
Discussion
This study reveals a critical role for CRF1 receptor pathways in the negative affective states of opiate withdrawal. CRF1 receptor-deficient mice withdrawn from chronic opiate exposure did not show the negative affective-like behavior or the increased expression of dynorphin in the nucleus accumbens, a brain change thought to underlie the negative affective consequences of drug addiction.
To mimic the clinical setting, we used a CPA paradigm that allowed the evaluation of the negative affective states of opiate withdrawal without using opioid receptor antagonists; that is, mice were conditioned while undergoing spontaneous opiate withdrawal. These experiments showed that, unlike wild-type mice, 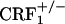 and
and 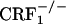 mice did not avoid environmental cues repeatedly paired with the early phase of opiate withdrawal, indicating that clearance of the opiate from the body was not accompanied by negatively charged affective states. Notably, genetic inactivation of ≈50% of CRF1 receptors in
mice did not avoid environmental cues repeatedly paired with the early phase of opiate withdrawal, indicating that clearance of the opiate from the body was not accompanied by negatively charged affective states. Notably, genetic inactivation of ≈50% of CRF1 receptors in 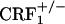 mice (30) resulted in a behavioral phenotype similar to that observed in the
mice (30) resulted in a behavioral phenotype similar to that observed in the 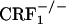 mice, indicating that fully functional CRF1 receptor systems are required for the negative affective properties of opiate withdrawal. The lack of negative affective-like behavior in opiate-withdrawn
mice, indicating that fully functional CRF1 receptor systems are required for the negative affective properties of opiate withdrawal. The lack of negative affective-like behavior in opiate-withdrawn 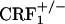 mice represents a demonstration of impaired behavioral responses in these mice.
mice represents a demonstration of impaired behavioral responses in these mice. 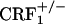 mice have in fact been shown to display anxiety-like profiles similar to wild-type mice (7). However, in the latter study,
mice have in fact been shown to display anxiety-like profiles similar to wild-type mice (7). However, in the latter study, 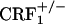 mice also showed levels of alcohol withdrawal-induced anxiety-like behaviors that were intermediate between those detected in wild-type and
mice also showed levels of alcohol withdrawal-induced anxiety-like behaviors that were intermediate between those detected in wild-type and 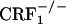 mice (7). Plasma corticosterone levels observed in
mice (7). Plasma corticosterone levels observed in 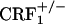 mice also rule out that lack of opiate withdrawal CPA was due to the corticosterone deficiency associated with the CRF1 receptor mutation (6). During opiate withdrawal,
mice also rule out that lack of opiate withdrawal CPA was due to the corticosterone deficiency associated with the CRF1 receptor mutation (6). During opiate withdrawal, 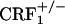 mice showed corticosterone responses similar to those detected in wild-type mice (see Fig. 6, which is published as supporting information on the PNAS web site). The latter findings are in line with previous evidence of stress-induced plasma corticosterone increases in female and male wild-type and
mice showed corticosterone responses similar to those detected in wild-type mice (see Fig. 6, which is published as supporting information on the PNAS web site). The latter findings are in line with previous evidence of stress-induced plasma corticosterone increases in female and male wild-type and 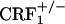 mice (6, 7). In contrast, female and male
mice (6, 7). In contrast, female and male 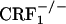 mice showed deficient corticosterone responses to stress (6, 7).
mice showed deficient corticosterone responses to stress (6, 7).
Brain CRF circuitry have been hypothesized to mediate the negative motivational effects of drug dependence. Early phases of withdrawal from chronic exposure to alcohol, cannabinoids, or cocaine are associated with increased CRF levels in the amygdala, a brain region implicated in the aversive effects of drug exposure (31-33). Accordingly, functional antagonism of CRF neurotransmission attenuated the anxiety-like and aversive effects of drug withdrawal. In particular, CRF receptor antagonist compounds have been shown to attenuate the aversive effects of naloxone in rats implanted with morphine pellets (12, 16). However, to date, no behavioral evidence exists supporting a CRF receptor subtype-selective blockade by currently available compounds. CRF1 receptor-null mutant mice do possess functional levels of CRF2 receptors, which can be activated by exogenous CRF and related peptides (34, 35). CRF1 receptor-deficient mice may thus represent a reliable tool to dissect the specific contribution of CRF1 receptor pathways to behavior. Recent studies showing opposite functions for the two known CRF receptor types in affective-like behaviors highlight a complex role for the CRF system in stress-related psychopathology (6-11). In particular, activation of CRF1 receptors may be responsible for increased anxiety-like responses, whereas stimulation of CRF2 receptors may produce anxiolytic-like and antidepressant-like effects. Thus, the investigation of CRF1 versus CRF2 receptor function in affect regulation requires appropriate and well validated experimental tools. By using 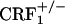 and
and 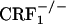 mice, we demonstrate that the CRF1 receptor is a main substrate of the negative affective states of opiate withdrawal. Accordingly,
mice, we demonstrate that the CRF1 receptor is a main substrate of the negative affective states of opiate withdrawal. Accordingly, 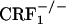 mice were previously shown to display decreased anxiety-like behaviors after withdrawal from another major substance of abuse, such as alcohol (7). Moreover, because CRF1 receptor-deficient mice possess functional levels of CRF2 receptors, our results provide indirect evidence that, during opiate withdrawal, stimulation of CRF2 receptors by increased CRF activity does not induce negative affective states.
mice were previously shown to display decreased anxiety-like behaviors after withdrawal from another major substance of abuse, such as alcohol (7). Moreover, because CRF1 receptor-deficient mice possess functional levels of CRF2 receptors, our results provide indirect evidence that, during opiate withdrawal, stimulation of CRF2 receptors by increased CRF activity does not induce negative affective states.
The lack of affective-like behaviors in CRF1 receptor-deficient mice cannot be attributed to decreased levels of somatic opiate withdrawal. During the conditioning phase of the opiate withdrawal CPA experiment, 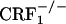 mice displayed more jumps and wet dog shakes than wild-type and
mice displayed more jumps and wet dog shakes than wild-type and 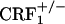 mice (see Fig. 5 and Table 1). However, wild-type but not CRF1 receptor-deficient mice showed strong aversions for the conditioning compartment of the CPA apparatus paired with opiate withdrawal. It cannot be excluded that during conditioning wild-type and
mice (see Fig. 5 and Table 1). However, wild-type but not CRF1 receptor-deficient mice showed strong aversions for the conditioning compartment of the CPA apparatus paired with opiate withdrawal. It cannot be excluded that during conditioning wild-type and 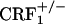 mice experienced some degree of somatic opiate withdrawal. In fact, we have found that, 8 h after treatment with the same morphine regimen used here, wild-type and
mice experienced some degree of somatic opiate withdrawal. In fact, we have found that, 8 h after treatment with the same morphine regimen used here, wild-type and 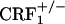 mice do show increased levels of paw tremor, chewing, and palpebral ptosis (our unpublished observations). However, the latter somatic signs of opiate withdrawal could not be measured during the CPA experiments reported here. Also, we do not yet have a direct explanation for the differences in somatic opiate withdrawal between
mice do show increased levels of paw tremor, chewing, and palpebral ptosis (our unpublished observations). However, the latter somatic signs of opiate withdrawal could not be measured during the CPA experiments reported here. Also, we do not yet have a direct explanation for the differences in somatic opiate withdrawal between 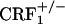 and
and 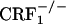 mice. However, preliminary results obtained in our laboratory indicate a major role for the hypothalamus-pituitary-adrenal (HPA) axis deficiencies associated with the absence of functional CRF1 receptors (
mice. However, preliminary results obtained in our laboratory indicate a major role for the hypothalamus-pituitary-adrenal (HPA) axis deficiencies associated with the absence of functional CRF1 receptors (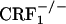 mice) in the increased expression of somatic opiate withdrawal (our unpublished observations). Interestingly, despite the large differences in HPA axis activity,
mice) in the increased expression of somatic opiate withdrawal (our unpublished observations). Interestingly, despite the large differences in HPA axis activity, 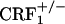 and
and 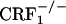 mice lacked the affective-like signs of opiate withdrawal, suggesting a minor role for the HPA axis in affective components of opiate withdrawal. Finally, the clear-cut dissociation of affective-like and somatic signs of opiate withdrawal in
mice lacked the affective-like signs of opiate withdrawal, suggesting a minor role for the HPA axis in affective components of opiate withdrawal. Finally, the clear-cut dissociation of affective-like and somatic signs of opiate withdrawal in 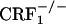 mice suggests a minor contribution of somatic malaise to the negative affective states of opiate withdrawal. This issue should be taken into consideration when treating opiate addicts with medications that mainly address the somatic components of the opiate withdrawal syndrome.
mice suggests a minor contribution of somatic malaise to the negative affective states of opiate withdrawal. This issue should be taken into consideration when treating opiate addicts with medications that mainly address the somatic components of the opiate withdrawal syndrome.
It is unlikely that the absence of opiate-withdrawal CPA in the CRF1 receptor-deficient mice was due to altered associative learning processes. The CRF system modulates learning and memory-storage processes (36). In addition, CRF1 receptors are relatively abundant in the hippocampus and amygdala, brain regions mediating stress-related cognitive functions (37, 38). To investigate cognitive abilities in CRF1 receptor-deficient mice, we tested wild-type and mutant mice in a CPA paradigm using the KOR agonist compound U-50,488H. The results of this study revealed that, similar to the wild-type mice, 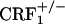 and
and 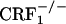 mice reliably avoided environmental cues paired with the aversive effects of U-50,488H. This finding demonstrates that CRF1 receptor deficiency does not impair the ability to acquire and display conditioned responses to aversive stimuli. Moreover, these findings provide initial evidence of functional KOR pathways, the preferential dynorphin target (39), in CRF1 receptor-deficient mice.
mice reliably avoided environmental cues paired with the aversive effects of U-50,488H. This finding demonstrates that CRF1 receptor deficiency does not impair the ability to acquire and display conditioned responses to aversive stimuli. Moreover, these findings provide initial evidence of functional KOR pathways, the preferential dynorphin target (39), in CRF1 receptor-deficient mice.
In line with the behavioral indices, opiate-withdrawn CRF1 receptor-deficient mice did not show any increase in the genetic transcription of dynorphin in the nucleus accumbens. Up-regulated dynorphin/KOR systems activity in the nucleus accumbens might subserve the negative affective consequences of drug exposure. For example, overexpression of the cyclic-AMP response-element-binding protein in the nucleus accumbens increases dynorphin mRNA levels and produces aversive responses to relatively low doses of cocaine, which are abolished by the KOR antagonist nor-binaltorphimine (40). Activation of KOR pathways also decreases dopamine release within the nucleus accumbens, which might contribute to the negative affective states of opiate withdrawal (41). However, to date, no studies have provided direct evidence in favor of a role for increased dynorphin/KOR systems activity in the negative affective components of opiate withdrawal. Here we report that opiate-withdrawn wild-type mice display negative affective-like behaviors and increased dynorphin activity in the nucleus accumbens shell. The latter results are in line with previous studies showing increased dynorphin expression and elevated endogenous dynorphin peptides levels in the nucleus accumbens of opiate-withdrawn rats and mice (22, 23, 42, 43). In contrast, CRF1 receptor-deficient mice did not show the negative affective-like behavior or the increased dynorphin activity in response to opiate withdrawal. These results provide evidence of an essential role for CRF1 receptor pathways in the increased dynorphin activity induced by chronic opiate exposure and withdrawal. These findings also point to the CRF1 receptor as a key element in the cascade of events leading to brain neurochemical changes thought to mediate the negative affective states of early phases of opiate withdrawal. The in situ hybridization studies reported here also show that, contrary to wild-type and 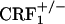 mice,
mice, 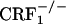 mice displayed decreased dynorphin expression in the core portion of the nucleus accumbens (see Fig. 3). Impaired hypothalamus-pituitary-adrenal axis activity might have contributed to this result.
mice displayed decreased dynorphin expression in the core portion of the nucleus accumbens (see Fig. 3). Impaired hypothalamus-pituitary-adrenal axis activity might have contributed to this result. 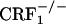 mice used in this study present a pronounced adrenal deficiency, as demonstrated by morphological analyses (atrophy of the corticosterone-producing zona fasciculata of the adrenal gland cortex) and basal and stress-related plasma corticosterone deficits (6). Accordingly, adrenalectomy has been shown to reduce dynorphin expression in the striatum and hippocampus (44, 45).
mice used in this study present a pronounced adrenal deficiency, as demonstrated by morphological analyses (atrophy of the corticosterone-producing zona fasciculata of the adrenal gland cortex) and basal and stress-related plasma corticosterone deficits (6). Accordingly, adrenalectomy has been shown to reduce dynorphin expression in the striatum and hippocampus (44, 45).
The present study reveals a role for brain CRF1 receptor pathways in opiate dependence and withdrawal. These results bear important implications for research aimed at developing treatments for opiate addiction. Human studies contrasting affective and nonaffective features of drug withdrawal suggest that, whereas physical components may have little if any motivating property, alleviation of negative affect may play a primary role in sustaining continued drug abuse (3, 4). The negative affective symptoms of drug withdrawal accurately index relapse vulnerability to drug taking and might contribute to the compulsive nature of drug-seeking behavior in addicted individuals (4). The identification of the neural substrates underlying the negative affective symptoms of opiate withdrawal is thus a primary goal of current opiate addiction research. Here we provide initial evidence showing an essential role for CRF1 receptor pathways in the negative affective states of opiate withdrawal and underlying brain molecular mechanisms. These results suggest therapeutic strategies for treating opiate addiction: dampening of CRF1 receptor signaling might alleviate the negative affective symptoms of opiate withdrawal and, thus, reduce compulsive heroin-seeking behaviors and relapse to opiate abuse.
Supplementary Material
Acknowledgments
We thank Dr. Wylie Vale (The Salk Institute, La Jolla, CA) for generously donating the CRF1 mutant mouse breeders; Mr. Massimo Rizza and Dr. Miriam Zanetti (University of Padua) for mouse breeding and genotyping; and Dr. Pier Vincenzo Piazza, Dr. Catherine Le Moine, Dr. Pierre Kitchener, and Mr. David Belin (Institut National de la Sante et de la Recherche Médicale and Centre National de la Recherche Scientifique, Bordeaux, France) for help with the in situ hybridization and corticosterone experiments. We also thank the National Institute on Drug Abuse for the generous gift of U-50,488H. A.C. was supported by a research grant from the University of Padua and European Union Grant MCIF HPMF-CT-2001-01327. F.P. was supported by a Ph.D. Fellowship grant from the University of Padua.
Author contributions: A.C. and F.P. designed research, performed research, and analyzed data; and A.C. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CRF, corticotropin-releasing factor; CPA, conditioned place aversion; KOR, κ-opioid receptor.
References
- 1.van den Brink, W. & van Ree, J. M. (2003) Eur. Neuropsychopharmacol. 13, 476-487. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien, C. P. (1996) in Goodman & Gilman's The Pharmacological Basis of Therapeutics, eds. Gilman, A., Hardman, J. & Limbird, L. (McGraw-Hill, New York), pp. 557-577.
- 3.Koob, G. F. & Le Moal, M. (2001) Neuropsychopharmacology 24, 97-129. [DOI] [PubMed] [Google Scholar]
- 4.Baker, T. B., Piper, M. E., McCarthy, D. E., Majeskie, M. R. & Fiore, M. C. (2004) Psychol. Rev. 111, 33-51. [DOI] [PubMed] [Google Scholar]
- 5.Sarnyai, Z., Shaham, Y. & Heinrichs, S. C. (2001) Pharmacol. Rev. 53, 209-243. [PubMed] [Google Scholar]
- 6.Smith, G. W., Aubry, J. M., Dellu, F., Contarino, A., Bilezikjian, L. M., Gold, L. H., Chen, R., Marchuk, Y., Hauser, C., Bentley, C. A., et al. (1998) Neuron 20, 1093-1102. [DOI] [PubMed] [Google Scholar]
- 7.Timpl, P., Spanagel, R., Sillaber, I., Kresse, A., Reul, J. M., Stalla, G. K., Blanquet, V., Steckler, T., Holsboer, F. & Wurst, W. (1998) Nat. Genet. 19, 162-166. [DOI] [PubMed] [Google Scholar]
- 8.Contarino, A., Dellu, F., Koob, G. F., Smith, G. W., Lee, K. F., Vale, W. & Gold, L. H. (1999) Brain Res. 835, 1-9. [DOI] [PubMed] [Google Scholar]
- 9.Bale, T. L., Contarino, A., Smith, G. W., Chan, R., Gold, L. H., Sawchenko, P. E., Koob, G. F., Vale, W. W. & Lee, K. F. (2000) Nat. Genet. 24, 410-414. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto, T., Radulovic, J., Radulovic, M., Lin, C. R., Schrick, C., Hooshmand, F., Hermanson, O., Rosenfeld, M. G. & Spiess, J. (2000) Nat. Genet. 24, 415-419. [DOI] [PubMed] [Google Scholar]
- 11.Bale, T. L. & Vale W. W. (2003) J. Neurosci. 23, 5295-5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinrichs, S. C., Menzaghi, F., Schulteis, G., Koob, G. F. & Stinus, L. (1995) Behav. Pharmacol. 6, 74-80. [PubMed] [Google Scholar]
- 13.Iredale, P. A., Alvaro, J. D., Lee, Y., Terwilliger, R., Chen, Y. L. & Duman, R. S. (2000) J. Neurochem. 74, 199-208. [DOI] [PubMed] [Google Scholar]
- 14.Lu, L., Liu, D., Ceng, X. & Ma, L. (2000) Eur. J. Neurosci. 12, 4398-4404. [PubMed] [Google Scholar]
- 15.McNally, G. P. & Akil, H. (2002) Neuroscience 112, 605-617. [DOI] [PubMed] [Google Scholar]
- 16.Stinus, L., Cador, M., Zorrilla, E. P. & Koob, G. F. (2005) Neuropsychopharmacology 30, 90-98. [DOI] [PubMed] [Google Scholar]
- 17.Linseman, M. A. (1977) Psychopharmacology 54, 159-164. [DOI] [PubMed] [Google Scholar]
- 18.Fukunaga, Y. & Kishioka, S. (2000) Jpn. J. Pharmacol. 82, 175-180. [DOI] [PubMed] [Google Scholar]
- 19.Cicero, T. J., Nock, B. & Meyer, E. R. (2002) Pharmacol. Biochem. Behav. 72, 691-697. [DOI] [PubMed] [Google Scholar]
- 20.Houshyar, H., Gomez, F., Manalo, S., Bhargava, A. & Dallman, M. F. (2003) Neuropsychopharmacology 28, 1960-1972. [DOI] [PubMed] [Google Scholar]
- 21.Nestler, E. J. (2001) Nat. Rev. Neurosci. 2, 119-128. [DOI] [PubMed] [Google Scholar]
- 22.Rattan, A. K., Koo, K. L., Tejwani, G. A. & Bhargava, H. N. (1992) Brain Res. 584, 207-212. [DOI] [PubMed] [Google Scholar]
- 23.Turchan, J., Lason, W., Budziszewska, B. & Przewlocka, B. (1997) Neuropeptides 31, 24-28. [DOI] [PubMed] [Google Scholar]
- 24.Almeida, O. F., Hassan, A. H. & Holsboer, F. (1993) Ciba Found. Symp. 172, 151-172. [DOI] [PubMed] [Google Scholar]
- 25.Valentino, R. J. & Van Bockstaele, E. (2001) Psychopharmacology 158, 331-342. [DOI] [PubMed] [Google Scholar]
- 26.Jaber, M., Normand, E. & Bloch, B. (1995) Brain Res. Mol. Brain Res. 32, 156-160. [DOI] [PubMed] [Google Scholar]
- 27.Le Moine, C. (2000) Methods Mol. Biol. 123, 143-156. [DOI] [PubMed] [Google Scholar]
- 28.Gracy, K. N., Dankiewicz, L. A. & Koob, G. F. (2001) Neuropsychopharmacology 24, 152-160. [DOI] [PubMed] [Google Scholar]
- 29.Funada, M., Suzuki, T., Narita, M., Misawa, M. & Nagase, H. (1993) Neuropharmacology 32, 1315-1323. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt, M., Oitzl, M. S., Muller, M. B., Ohl, F., Wurst, W., Holsboer, F., Levine, S. & De Kloet, E. R. (2003) Neuroscience 119, 589-595. [DOI] [PubMed] [Google Scholar]
- 31.Merlo Pich, E., Lorang, M., Yeganeh, M., Rodriguez de Fonseca, F., Raber, J., Koob, G. F. & Weiss, F. (1995) J. Neurosci. 15, 5439-5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez de Fonseca, F., Carrera, M. R., Navarro, M., Koob, G. F. & Weiss, F. (1997) Science 276, 2050-2054. [DOI] [PubMed] [Google Scholar]
- 33.Richter, R. M. & Weiss, F. (1999) Synapse 32, 254-261. [DOI] [PubMed] [Google Scholar]
- 34.Contarino, A., Dellu, F., Koob, G. F., Smith, G. W., Lee, K. F., Vale, W. W. & Gold, L. H. (2000) Endocrinology 141, 2698-2702. [DOI] [PubMed] [Google Scholar]
- 35.Bradbury, M. J., McBurnie, M. I., Denton, D. A., Lee, K. F. & Vale, W. W. (2000) Endocrinology 141, 2715-2724. [DOI] [PubMed] [Google Scholar]
- 36.Heinrichs, S. C. (1999) Trends Pharmacol. Sci. 20, 311-315. [DOI] [PubMed] [Google Scholar]
- 37.Chen, Y., Brunson, K. L., Muller, M. B., Cariaga, W. & Baram, T. Z. (2000) J. Comp. Neurol. 420, 305-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Pett, K., Viau, V., Bittencourt, J. C., Chan, R. K., Li, H. Y., Arias, C., Prins, G. S., Perrin, M., Vale, W. & Sawchenko, P. E. (2000) J. Comp. Neurol. 428, 191-212. [DOI] [PubMed] [Google Scholar]
- 39.Chavkin, C., James, I. F. & Goldstein, A. (1982) Science 215, 413-415. [DOI] [PubMed] [Google Scholar]
- 40.Carlezon, W. A., Jr., Thome, J., Olson, V. G., Lane-Ladd, S. B., Brodkin, E. S., Hiroi, N., Duman, R. S., Neve, R. L. & Nestler, E. J. (1998) Science 282, 2272-2275. [DOI] [PubMed] [Google Scholar]
- 41.Di Chiara, G. & Imperato, A. (1988) J. Pharmacol. Exp. Ther. 244, 1067-1080. [PubMed] [Google Scholar]
- 42.Nylander, I., Vlaskovska, M. & Terenius, L. (1995) Psychopharmacology 118, 391-400. [DOI] [PubMed] [Google Scholar]
- 43.Georges, F., Stinus, L., Bloch, B. & Le Moine, C. (1999) Eur. J. Neurosci. 11, 481-490. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe, Y., Weiland, N. G. & McEwen, B. S. (1995) Brain Res. 680, 217-225. [DOI] [PubMed] [Google Scholar]
- 45.Lucas, L. R., Pompei, P., Ono, J. & McEwen, B. S. (1998) J. Neurochem. 71, 833-843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.