

Abstract
Dansyl-to-heme distance distributions [P(r)] during folding have been determined in five variants of Saccharomyces cerevisiae iso-1 ferricytochrome c (labeled at mutant Cys residues 4, 39, 50, 66, and 99) by analysis of fluorescence energy-transfer kinetics. Moment analysis of the P(r) distributions clearly indicates that cytochrome c refolding is not a simple two-state process. After 1 ms of folding, the polypeptide ensemble is not uniformly collapsed and there are site variations in the relative populations of collapsed structures. P(r) distributions reveal structural features of the multiple intermediate species and evolution of the polypeptide ensemble.
Keywords: fluorescence energy transfer
Extensive theoretical work suggests that proteins can fold via multiple parallel pathways (1–3). Owing to the potential complexity of this transformation, multiple experimental probes are required to develop a complete description of the process. We have shown that fluorescence energy-transfer (FET) kinetics can be used to monitor folding of dynamic polypeptide structures to native states (4, 5). These measurements yield discrete probability distributions [P(r)] of the distance between a fluorescent donor (D) and an energy acceptor (A). Stopped-flow triggered folding with FET kinetics detection provides nanosecond time scale snapshots of D–A distance distributions as a denatured polypeptide evolves to its native structure.
In a prior study of Saccharomyces cerevisiae iso-1 cytochrome c (cyt c) refolding, we determined distributions of distances between a dansyl (Dns) fluorophore (D) attached to the C-terminal Cys-102 residue and the heme (A) (4). The data revealed that a bimodal P(r) distribution develops immediately after the stopped-flow mixing dead time (≈1 ms), consistent with roughly equal populations of collapsed and extended polypeptide structures. The two populations disappear in parallel to yield native protein, suggesting that they exchange on a time scale faster than folding.
A valid concern about studies of labeled proteins is that the dye might perturb the structure and thus alter the folding mechanism. Indeed, cyt c is destabilized by Dns derivatization of Cys-102, a residue that lies in a region of hydrophobic packing. Furthermore, we cannot be certain that the results of studies on a single labeling site are representative of the entire cyt c folding landscape.
Herein, we report the folding dynamics of five new variants of yeast cyt c. Dns fluorophores were linked to mutant Cys residues (4, 39, 50, 66, and 99) at sites corresponding to different structural elements within the folded protein and all have a high degree of solvent exposure (Fig. 1a). The FET kinetics data on the five labeling sites have allowed us to develop a thorough structural description of the cyt c polypeptide ensemble as it evolves to its native state.
Fig. 1.

Cyt c folding kinetics. (a) Structure of S. cerevisiae iso-1 cyt c showing the labeling positions. (Inset) Structure of the Dns label. (b) Refolding kinetics of wild-type and five Dns-labeled variants monitored by absorbance changes at 695 nm. (c) Refolding kinetics monitored by changes in fluorescence intensity of the Dns group (conventional ensemble-average approach). The refolding conditions were 0.2 M GuHCl, pH 7, and 20°C.
Materials and Methods
Dns-Labeled Derivatives. Protein expression, Dns-labeling, purification, and characterization of Dns-derivatives are described in ref. 5.
Stopped-Flow Experiments. Protein refolding was triggered by using a Bio-Logic SFM-4S stopped-flow mixer. Unfolded Dns-labeled cyt c (2 M GuHCl/100 mM NaPi, pH 7) was diluted 10-fold with a refolding buffer (100 mM NaPi, pH 7). The final protein concentrations were 10–15 μM. For refolding experiments in which heme misligation by His residues was inhibited, excess imidazole ([im] = 0.15 M) was added to both the denatured protein solution and the diluting buffer. Refolding kinetics in the presence of imidazole were performed at 2°C to bring the reaction into the stopped-flow time regime. At 2 M GuHCl all of the variants were fully denatured, as suggested by their circular dichroism (CD) and heme absorption spectra, as well as the midpoints of their unfolding transitions (1.3 ± 0.2 M GuHCl). Absorbance changes at 695 nm were probed by using a Hitachi laser diode (HL6738MG) and detected with a photodiode. The signal was amplified with a 100-kHz bandwidth linear amplifier and recorded with a digital oscilloscope (CompuScope 1602, Gage Applied Science). A 10-mm zigzag cuvette (TC-100-15, Bio-Logic) was used. The final protein concentrations in the absorbance experiments were 40–60 μM. The experimental setup for fluorescence measurements has been previously described (6). The sample in a 0.8 mm cuvette (FC-08, Bio-Logic) was excited with an arc lamp (330 nm ≤ λex ≤ 370 nm) and the luminescence at 510 nm was selected with a monochromator. The final protein concentrations in these experiments were 10–15 μM.
FET Measurements During Refolding and Data Analysis. The experimental configuration and data analysis methods are described in detail in refs. 4, 5, and 7. The fluorescence decay rate constant of the unquenched Dns group, k0 = 9.8 × 107 s–1, is well suited for accessing the static regime (k0 is faster than the rates of conformational interconversions) and generating snapshots of D–A distance distributions as the protein folds to the native structure. Hence, we neglect intrachain diffusion in our analyses of FET kinetics. Fluorescence anisotropy data indicate that the Dns label is extremely mobile both in folded and unfolded variants (5), so the isotropic value of κ2 = 2/3 was used in all distance calculations. The Förster critical length, R0, for the Dns–Fe(III)-heme pair is 39 Å under both native and denatured conditions; it was assumed to remain constant during folding. Analysis of the FET kinetics data for folded variants yielded D–A distances that are consistent with crystallographic data (5). At distances longer than 1.5 R0, energy transfer quenching of D is not competitive with excited-state decay, and D–A distances cannot be obtained reliably. Therefore, different structures in the protein ensembles with r ≥ 59 Å cannot be resolved.
FET kinetics were collected on both short (5 ns) and long (20 or 50 ns) time scales. Between 20 and 30 single-shot streak camera traces were averaged. The resulting short- and long-time-scale data were spliced together, and the combined traces were compressed logarithmically before fitting. In the absence of specific information about the nature of the transient species during refolding, we have used a maximum-entropy regularization method to analyze the data (8). This approach yields upper limits for the widths of D–A distance distributions [P(r)] consistent with our experimental data (7). In addition to a maximum-entropy analysis, FET kinetics for the unfolded and folded protein have been fitted to a Gaussian distribution of D–A distances (Eq. 1) (9) to yield continuous P(r) distributions appropriate for computer simulations.
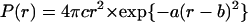 |
[1] |
Moment Analysis. The moments (Mn), variance (V), and standard deviations (S) of the P(r) distributions were calculated according to Eqs. 2 and 3.
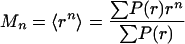 |
[2] |
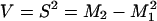 |
[3] |
The time evolution of Mn and V in a two-state folding reaction was simulated by using experimental folding rate constants (from conventional stopped-flow experiments) and continuous distributions (taken from fits of FET kinetics to Eq. 1) describing the initial and final states of variants 4, 39, 50, 66, and 99. In additional simulations, the continuous distributions describing the protein ensemble at specific times during the folding process were also converted to discrete distributions (by using the experimental distance vector) before calculation of moments. The latter procedure was intended to correct for systematic errors resulting from the use of discrete distributions on an unequally spaced distance vector.
Results and Discussion
Dns-Cyt c Folding Kinetics. Labeling of cyt c variants with a Dns group introduced no detectable perturbations of the protein secondary structure or the environment of the heme group (Fig. 4, which is published as supporting information on the PNAS web site). Moreover, the combined effects of Cys mutation and Dns labeling did not appreciably alter the stability of the variants (5). To assess the effects of these modifications on folding kinetics, we used the absorbance change at 695 nm to probe cyt c refolding (Fig. 1b). The 695-nm absorption band in native Fe(III)-cyt c, attributed to S(Met-80) to Fe(III) ligand-to-metal charge transfer, is a key indicator of native ligation and, by implication, native structure. The refolding kinetics of wild type and the five Dns-labeled cyt c variants (stopped-flow triggered; [GuHCl]initial = 2 M; [GuHCl]final = 0.2 M; pH 7, 20°C) were all quite similar (Fig. 1b). The kinetics were dominated by a single exponential phase with a rate constant 0.5 (1) s–1. Under these conditions, the final step in cyt c folding involves replacement of a misligated His residue on the heme by the native axial ligand, Met-80 (10).
Measurements of integrated Dns fluorescence reveal distinct variations in refolding behavior of the five labeled proteins (Fig. 1c), both in the size of the “burst” amplitude (i.e., the signal change during the dead time of the stopped-flow mixer) and in the overall kinetics. In general, as the number of residues between the heme and the Dns label decreases, burst amplitudes increase and folding times decrease. The exception is variant 4, in which there is no measurable change in Dns fluorescence between folded and unfolded forms. The kinetics probed by using integrated Dns fluorescence are generally biphasic: ≈60% of the signal amplitude can be described by a rate constant 2–3 s–1; the remaining amplitude corresponds to a rate constant similar to that found with 695-nm absorbance measurements [0.5 (1) s–1]. Imidazole at high concentrations (0.15 M) can displace the His residue, substantially accelerating cyt c refolding. In this case, lowering the solution temperature to 2°C brings the refolding kinetics into the stopped-flow time regime. The folded structure of the imidazole adduct is identical with that of the native protein, except for increased conformational flexibility in the vicinity of Met-80 (11).
Burst-Phase Ensemble. The burst amplitudes in our integrated fluorescence measurements indicate that some compaction of the polypeptide occurs during the stopped-flow dead time. There are varying interpretations of the nature and magnitude of this early collapse in cyt c. Roder and coworkers have observed a substantial activation barrier associated with the submillisecond decrease in Trp-59 fluorescence; they interpreted this result in terms of a specific collapse to an intermediate with native-like contacts (12). On the other hand, Englander has shown that shorter cyt c fragments that do not have a discrete folded structure exhibit comparable Trp-59 burst-phase signals; the rates and barriers for this collapse are consistent with nonspecific contraction of the polypeptide upon dilution of the denaturing solution (13–15).
Our FET kinetics measurements of the five Dns-cyt c variants provide additional insight into the cyt c burst-phase collapse. The distance distributions extracted from FET kinetics measured 1 ms after the mixing dead time (at 2°C, with excess imidazole to inhibit misligation) indicate that the mean D–A distances in four of the five labeled proteins (39, 50, 66, and 99) decrease to ≈75% of the unfolded protein values (Fig. 2). Similar behavior is observed when the heme is misligated by a His residue from the polypeptide chain (Fig. 3).
Fig. 2.

Distributions of D–A distances, P(r), and moments of the P(r) distributions for refolding of five Dns-labeled variants of cyt c at 0.2 M GuHCl (pH 7, 2°C) in the presence of 0.15 M imidazole. FET kinetics have been measured at 1 ms, 10 ms, 100 ms, and 16 s after initiation of refolding. The P(r) distributions were extracted from maximum-entropy analysis of these data. (a) P(r) distributions for the unfolded state at 2 M GuHCl/0.15 M imidazole (pH 7) (U); the transient folding species formed 1 ms after the initiation of refolding (B*, “burst phase”); and for the refolded protein at 16 s after the mix (F). (b) First moments (M1). (c) Second moments (M2). (d) Variance (V). Solid curves show simulated time dependence of M1, M2, and V for the two-state folding process using τ = 10 ms; the curves were constructed on the basis of experimental P(r) distributions: red, continuous (Eq. 1); blue, discrete.
Fig. 3.

Distributions of D–A distances, P(r), and moments of the P(r) distributions for refolding of five Dns-labeled variants of cyt c at 0.2 M GuHCl (pH 7, 20°C). FET kinetics have been measured at 1 ms, 10 ms, 100 ms, 760 ms, 5 s, and 16 s after initiation of refolding. The P(r) distributions were extracted from maximum-entropy analysis of these data. (a) P(r) distributions for the unfolded state at 2 M GuHCl (pH 7) (U), the transient folding species formed 1 ms after the initiation of refolding (B*, “burst phase”), and the refolded protein at 16 s after the mix (F). (b) First moments (M1). (c) Second moments (M2). (d) Variance (V). Solid curves show simulated time dependence of M1, M2, and V for the two-state folding process using τ = 0.6 s; the curves were constructed on the basis of experimental P(r) distributions: red, continuous (Eq. 1); blue, discrete.
As was the case with the integrated measurements, the change in FET kinetics during folding of 4 is too small to interpret reliably (Figs. 4–6, which are published as supporting information on the PNAS web site). The Dns–heme distance distributions in denatured and folded proteins, as well as those extracted during folding, are virtually indistinguishable. When denatured, the four other labeled proteins have Flory characteristic ratios (Cn = 〈r2 〉/nl2, where n is the number of residues between D and A, and l is usually taken to be 3.8 Å, the length of a peptide segment) between 3.8 (39) and 2.1 (99). The Dns–heme distribution in 4, however, leads to a characteristic ratio of 5.8, implying a stiffer polypeptide chain than in the other four Dns variants. These observations raise the possibility that the N-terminal polypeptide region does not unfold substantially in the presence of 2 M GuHCl.
The initial compaction that results from denaturant dilution is not uniform. Instead, the burst-phase distributions for three of the variants (50, 66, and 99) are clearly bimodal, revealing both compact and extended cyt c structures (Figs. 2a and 3a). The compact members of the burst-phase ensemble (r ≤ 26 Å) comprise 30–50% of the total population. The extended structures are not as elongated as in the fully denatured protein, possibly owing to nonspecific compaction associated with the transition to a poorer polymer solvent. In the case of variant 39, there is a substantial population of very compact structures (r ≤ 21 Å) in the burst-phase ensemble. The absence of a bimodal distribution for this variant is likely due to the small D–A distance range accessible in this labeled protein. In general, the extended fraction of the burst-phase ensemble tends to increase as the Dns-label is moved farther from the heme in the polypeptide chain.
The bimodality of the cyt c D–A distance distributions in the burst phase is inconsistent with an ensemble of random-coil polypeptides that have simply collapsed in response to a change in solvent. The substantial populations of very short heme–Dns distances (<26 Å) 1 ms after the folding trigger are compelling evidence for the presence of globular structures. However, unlike in the cyt c A-state (molten globule) (7, 16), these compact structures represent only a fraction (albeit considerable) of the polypeptide ensemble.
The globular fold of cyt c makes it difficult to distinguish nonspecifically collapsed structures from those in which native contacts have formed. Some of the compact structures in Figs. 2a and 3a have dimensions similar to those in the native protein, whereas others are more loosely packed. The absence of a discrete folded structure in apo-cyt c indicates that contacts between the heme and polypeptide are essential for stabilizing the native structure. When cyt c refolding is conducted in the absence of imidazole, misligation by His-26, His-33, and His-39 will disrupt many of the native interactions between the polypeptide and the heme. Hence, owing to this topological disruption arising from His misligation, it seems unlikely that the collapsed structures observed after the cyt c burst phase arise from a preponderance of native contacts. The burst-phase distance distributions determined when misligation is inhibited by imidazole (Fig. 2a) are quite similar to those found in the misligated protein (Fig. 3a). Because similar forces drive the collapse under both conditions, it is possible that the compact structures observed at early folding times in the absence of misligation may contain a substantial population of nonnative contacts as well. The distance distributions do not rule out the possibility that a small population of native structures has formed after 1 ms (17). On the basis of our FET kinetics measurements, it appears that these “fast-track” folders likely represent no more than 10% of the total protein population.
Site Variations. The relative populations of compact and extended structures in the burst-phase ensemble are site-dependent. Moment analysis of the distance distributions provides a quantitative assessment of the degree of compaction. The standard deviations (S) of P(r) for denatured and burst phase structures populated in the presence of imidazole exhibit interesting trends. For labeling sites 39 and 50, the burst-phase values of S decrease to ≈76% of their values in the denatured state. In contrast, the burst-phase S value in variant 66 is 91% of the denatured value, and for variant 99, S in the burst-phase ensemble is greater than in the denatured protein (130%). As expected, the trends in S values mirror those observed in the burst-phase signal amplitudes. The large populations of compact structures in variants 39 and 50 are fully consistent with an EPR spin labeling study of cyt c refolding (18), which found earlier immobilization of spin labels at these positions compared with other sites in the protein. The results for labeling sites 50 and 66 also are consistent with the large burst-phase signal amplitude found when refolding is probed by using Trp-59 fluorescence.
The interfacial contacts between N- and C-helices are among the key stabilizing interactions in folded cyt c. Hydrogen exchange pulsed labeling studies and recent theoretical simulations have suggested the early formation of folding intermediates with fully developed N- and C-helices and native-like interhelix interactions (19, 20). Furthermore, it has been proposed that such compact intermediates accumulate even in the presence of His misligation (20). Our burst-phase distance distribution for variant 99, however, suggests that only ≈25% of the protein ensemble has a native Dns–heme distance. In the remaining fraction of the ensemble, the helical secondary structure of the C terminus may be formed, but its native tertiary interaction with the N-terminal helix is disrupted. Extended structures in variant 99 remain populated throughout the course of folding, indicating that the N- and C-terminal helices have not adopted native structures. It is important to note that the hydrogen-exchange pulsed labeling studies were performed with the equine protein, which is substantially more stable than S. cerevisiae cyt c.
Evolution of the Polypeptide Ensemble. Snapshots of the D–A distance distributions at various time points during refolding track the evolution of the polypeptide ensemble (Figs. 2, 3, 5, and 6). Extended structures observed in the burst-phase ensembles are present throughout the course of folding, with and without misligation; they disappear in parallel with the populations of nonnative compact structures to yield folded protein. This behavior is consistent with rapid equilibration between nonnative compact and extended structures, and a slower transformation to folded protein. The persistence of extended structures at different labeling sites is consistent with our prior observations with a Dns102-labeled protein (4). The compact-extended equilibrium appears to be a general feature of the cyt c refolding mechanism. Reminiscent of the mechanism of folding chaperones (21), reextension of nonnative collapsed species and repetitive collapse guide the polypeptide structures away from topological traps.
We have characterized the folding of the protein ensemble through an analysis of the time dependence of the moments and variances of the P(r) distributions (Figs. 2 and 3). The first and second moments of the P(r) distributions (Eq. 2) characterize the ensemble mean and mean-squared D–A distances (〈rn 〉= Mn); the variance (V) reflects the breadth of the distribution. The behavior of the moments both in the presence and absence of misligation is roughly similar; we will focus our discussion on the latter case. In the presence of 0.15 M imidazole at 2°C, the first and second moments of P(r) decrease substantially in the burst phase (t < 10–3 s), then continue to decrease monotonically as the protein refolds (10–3 to 101 s). We have simulated the time evolution of the moments and variances of P(r) assuming a two-state folding mechanism (Fig. 3), using the unfolded distributions and P(r) recorded at 16 s as the initial and final states. The most significant discrepancy between the simulations and the experimental data is the large burst-phase signal amplitude. As discussed above, this signal change cannot be attributed wholly to nonspecific collapse in response to the change in solvent conditions. A second difference is the more gradual reduction in Mn; the experimental values tend to decrease continuously throughout the 10–3 to 101-s time interval. A single-exponential, two-state process is expected to show a more abrupt transition as unfolded protein converts to the native state. Deviations from a two-state model are also apparent when the burst-phase P(r) distributions were used to represent the initial state of cyt c. Clearly, the simple two-state process is not an adequate model for the refolding behavior of cyt c.
The time evolution of the variance of P(r) provides some interesting insights. For the protein labeled at 39, the variance reaches its final value within 100 ms. In contrast, the breadths of the Dns–heme distributions in proteins with labels at positions 50 and 66 decrease continuously throughout the 10–3 to 101-s time interval. For variant 99, the variance increases in the burst phase, then decreases monotonically over the next 10 s. The behavior of 99 is closest to that simulated for two-state folding. The variance is expected to increase to a maximum with a rate constant equal to twice the folding rate constant. After reaching this maximum value, the variance should then decrease with the folding rate constant to its equilibrium value. The initial increase in variance is due to the early population of two distinct and widely separated distributions. The two-state simulations predicted that both 66 and 99 would show the biphasic time evolution of the variance. The increase in the variance for 99 is not as great as predicted by the simulation, presumably owing to the burst-phase drop in distances sampled by the extended population. Similar reasoning can explain the absence of biphasic character in the time evolution of V for 66.
Conclusions
The mechanism of cyt c folding is a subject of intense discussion, with the number, origin, and structural features of kinetic intermediates being among the main topics (12, 13, 22, 23). Our FET snapshots of the distributions of structures within the refolding ensemble shed light on the folding mechanism. In particular, the time dependences of M1, M2, and V predicted on the basis of a two-state folding mechanism show significant discrepancies with the experimental moments. Overall, the magnitude of the burst-phase collapse is greater than expected, and the subsequent time evolution of the moments and variance is more gradual than predicted. Intermediate species must be involved in the refolding process, and our snapshots of the polypeptide ensemble provide a glimpse of these structures. The lack of global collapse and distinct site variations in the population of compact structures point to multiple transient species and the likelihood of multiple refolding pathways.
The variations in behavior at different labeling sites highlight the importance of employing multiple probes in the study of protein folding. Heme–Trp-59 energy-transfer data have strongly influenced ideas about cyt c folding. Studies employing other residue-specific probes have shown, however, that there is substantial diversity in folding behavior in different regions of the protein (18, 20, 24–26). The time evolution of D–A distributions from five different labeling sites provides important insights into cyt c folding, contributing to a growing body of data that will ultimately lead to a consensus about the folding mechanism of this protein.
Supplementary Material
Acknowledgments
We acknowledge Yuling Sheng for her help with protein expression and purification during the final stages of this work. We also thank Jennifer C. Lee and Judy E. Kim for help with FET kinetics measurements and numerous discussions. This work was supported by National Institutes of Health Grant GM068461 (to J.R.W.) and an Ellison Medical Foundation Senior Scholar Award in Aging (to H.B.G.).
Author contributions: E.V.P., H.B.G., and J.R.W. designed research; E.V.P. performed research; E.V.P. and J.R.W. analyzed data; and E.V.P., H.B.G., and J.R.W. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: cyt c, cytochrome c; FET, fluorescence energy-transfer; Dns, dansyl.
References
- 1.Onuchic, J. N., Luthey-Schulten, Z. & Wolynes, P. G. (1997) Annu. Rev. Phys. Chem. 48, 545–600. [DOI] [PubMed] [Google Scholar]
- 2.Dill, K. A. & Chan, H. S. (1997) Nat. Struct. Biol. 4, 10–19. [DOI] [PubMed] [Google Scholar]
- 3.Dobson, C. M., Sali, A. & Karplus, M. (1998) Angew. Chem. Int. Ed. 37, 868–893. [DOI] [PubMed] [Google Scholar]
- 4.Lyubovitsky, J. G., Gray, H. B. & Winkler, J. R. (2002) J. Am. Chem. Soc. 124, 5481–5485. [DOI] [PubMed] [Google Scholar]
- 5.Pletneva, E. V., Gray, H. B. & Winkler, J. R. (2005) J. Mol. Biol. 345, 855–867. [DOI] [PubMed] [Google Scholar]
- 6.Lee, J. C., Engman, K. C., Tezcan, F. A., Gray, H. B. & Winkler, J. R. (2002) Proc. Natl. Acad. Sci. USA 99, 14778–14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pletneva, E. V., Gray, H. B. & Winkler, J. R. (2005) J. Am. Chem. Soc. 127, 15370–15371. [DOI] [PubMed] [Google Scholar]
- 8.Istratov, A. D. & Vyvenko, O. F. (1999) Rev. Sci. Instrum. 70, 1233–1257. [Google Scholar]
- 9.Beals, J. M., Haas, E., Krausz, S. & Scheraga, H. A. (1991) Biochemistry 30, 7680–7692. [DOI] [PubMed] [Google Scholar]
- 10.Elöve, G., Bhuyan, A. K. & Roder, H. (1994) Biochemistry 33, 6925–6935. [DOI] [PubMed] [Google Scholar]
- 11.Banci, L., Bertini, I., Liu, G., Lu, J., Reddig, T., Tang, W., Wu, Y., Yao, Y. & Zhu, D. (2001) J. Biol. Inorg. Chem. 6, 628–637. [DOI] [PubMed] [Google Scholar]
- 12.Shastry, M. C. R. & Roder, H. (1998) Nat. Struct. Biol. 5, 385–392. [DOI] [PubMed] [Google Scholar]
- 13.Sosnick, T. R., Shtilerman, M. D., Mayne, L. & Englander, S. W. (1997) Proc. Natl. Acad. Sci. USA 94, 8545–8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagen, S. J. (2003) Proteins Struct. Funct. Genet. 50, 1–4.12471594 [Google Scholar]
- 15.Krantz, B. A., Mayne, L., Rumbley, J., Englander, S. W. & Sosnick, T. R. (2002) J. Mol. Biol. 324, 359–371. [DOI] [PubMed] [Google Scholar]
- 16.Lyubovitsky, J. G., Gray, H. B. & Winkler, J. R. (2002) J. Am. Chem. Soc. 124, 14840–14841. [DOI] [PubMed] [Google Scholar]
- 17.Mirny, L. A., Abkevich, V. & Shakhnovich, E. I. (1996) Fold. Des. 1, 103–116. [DOI] [PubMed] [Google Scholar]
- 18.DeWeerd, K., Grigoryants, V., Sun, Y., Fetrow, J. S. & Scholes, C. P. (2001) Biochemistry 40, 15846–15855. [DOI] [PubMed] [Google Scholar]
- 19.Weinkam, P., Zong, C. & Wolynes, P. (2005) Proc. Natl. Acad. Sci. USA 102, 12401–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishna, M. M. G., Lin, Y., Mayne, L. & Englander, S. W. (2003) J. Mol. Biol. 334, 501–513. [DOI] [PubMed] [Google Scholar]
- 21.Shtilerman, M., Lorimer, G. H. & Englander, S. W. (1999) Science 284, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shastry, M. C. R., Sauder, J. M. & Roder, H. (1998) Acc. Chem. Res. 31, 717–725. [Google Scholar]
- 23.Winkler, J. R. (2004) Curr. Opin. Chem. Biol. 8, 169–174. [DOI] [PubMed] [Google Scholar]
- 24.Grigoryants, V. M., DeWeerd, K. A. & Scholes, C. (2004) J. Phys. Chem. B 108, 9463–9468. [Google Scholar]
- 25.Sagle, L. B., Zimmermann, J., Dawson, P. E. & Romesberg, F. E. (2004) J. Am. Chem. Soc. 126, 3384–3385. [DOI] [PubMed] [Google Scholar]
- 26.Hoang, L., Bédard, S., Krishna, M. M. G., Lin, Y. & Englander, S. W. (2002) Proc. Natl. Acad. Sci. USA 99, 12173–12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.