

Abstract
Experimental evidence suggests that the folding and aggregation of the amyloid β-protein (Aβ) into oligomers is a key pathogenetic event in Alzheimer's disease. Inhibiting the pathologic folding and oligomerization of Aβ could be effective in the prevention and treatment of Alzheimer's disease. Here, using all-atom molecular dynamics simulations in explicit solvent, we probe the initial stages of folding of a decapeptide segment of Aβ,Aβ21–30, shown experimentally to nucleate the folding process. In addition, we examine the folding of a homologous decapeptide containing an amino acid substitution linked to hereditary cerebral hemorrhage with amyloidosis–Dutch type, [Gln-22]Aβ21–30. We find that: (i) when the decapeptide is in water, hydrophobic interactions and transient salt bridges between Lys-28 and either Glu-22 or Asp-23 are important in the formation of a loop in the Val-24–Lys-28 region of the wild-type decapeptide; (ii) in the presence of salt ions, salt bridges play a more prominent role in the stabilization of the loop; (iii) in water with a reduced density, the decapeptide forms a helix, indicating the sensitivity of folding to different aqueous environments; and (iv) the “Dutch” peptide in water, in contrast to the wild-type peptide, fails to form a long-lived Val-24–Lys-28 loop, suggesting that loop stability is a critical factor in determining whether Aβ folds into pathologic structures.
Keywords: molecular dynamics, Alzheimer's disease, hydrophobic interactions, salt bridges
The amyloid cascade hypothesis, first proposed in the early 1990s, posits that the deposition of amyloid fibrils is the seminal event in the pathogenesis of Alzheimer's disease (1, 2). However, recent biophysical, biological, and clinical data indicate that the formation of oligomeric assemblies much smaller than fibrils may be a key pathologic event (3–10). This paradigm shift suggests an attractive therapeutic approach: prevent or disrupt the assembly of amyloid β-protein (Aβ) monomer into toxic oligomers. To do so requires an understanding of the molecular dynamics (MD) involved in the transition from nontoxic monomeric to toxic oligomeric states.
Elucidation of the initial events in Aβ monomer folding and assembly is complicated by the solvent dependence of the process (11). Monomeric Aβ is largely helical in a membrane or membrane-mimicking environment such as ionic detergents (12–15). In contrast, Aβ monomers in aqueous solution show negligible α-helix or β-sheet content (11, 16, 17). Studies of Aβ10–35-amide at micromolar concentrations in water show a pH-dependent folding transition in which the conformation is not helical but contains several turns and at least two short strands (18). Solution-state NMR combined with diffusion-ordered spectroscopy indicate that variation of the anionic strength in the buffer shifts the equilibrium between monomeric and oligomeric states, possibly allowing for the stabilization of intermediate structures (19). MD studies of Aβ16–22 monomer in aqueous urea solution show that increasing the concentration of urea promotes a transition from a compact random coil to β-strand structures (20).
Aβ folding and oligomerization are also influenced significantly by amino acid structure at specific sites. The dipeptide Ile-41–Ala-42 at the C terminus of Aβ1–42 is responsible for the different biophysical behaviors of Aβ1–42 and Aβ1–40 (21, 22). Oxidation of Met-35 disrupts Aβ1–42 oligomer formation but does not affect Aβ1–40 oligomer formation, consistent with the possibility that there are structural differences between oligomers of the two alloforms involving Met-35 and neighboring residues (23). Simulations of the oligomerization of Aβ1–40 and Aβ1–42 (24, 25), using discrete MD with implicit solvent (26–29), are consistent with in vitro data, yield additional structural predictions, and offer a plausible mechanistic explanation for the Met-35 oxidation experiments (25). MD studies show that the presence of glycines induce a transition from an α-helix to a β-strand conformation in Aβ1–40 monomers in aqueous solution (30). Simulation studies of the Dutch [Gln-22]Aβ10–35 peptide in water indicate that interactions between the peptide and the solvent are stronger in the wild-type (WT) than in the mutant peptide (31).
Recent limited proteolysis experiments on Aβ1–40 and Aβ1–42, under conditions favoring oligomerization, identify in both peptides a protease-resistant segment, Ala-21–Ala-30. The homologous decapeptide Aβ21–30 shows identical protease resistance (32). Structure calculations based on distance constraints from proton solution-state NMR of Aβ21–30 reveal a turn structure at Val-24–Lys-28. Lazo et al. (32) postulate that this structure nucleates the intramolecular folding of the Aβ monomer and that partial unfolding of the Ala-21–Ala-30 region may be necessary for the subsequent fibrillization of Aβ. The observations on full-length Aβ and the Aβ21–30 decapeptide are consistent with previous work showing that peptide fragments containing the folding nuclei of globular proteins are, by themselves, structured (33). Furthermore, the structures found in the isolated folding nuclei are similar to those found in the full-length proteins (33). MD simulations of the folding nucleus, therefore, provide insights into the earliest events in the folding of the full-length protein. Borreguero et al. (34) recently used discrete MD with implicit solvent and a united-atom protein model to simulate folding of the putative Aβ folding nucleus Aβ21–30. The united-atom peptide model considers explicitly all protein atoms except hydrogen. Important findings in Borreguero et al. (34) are: (i) the existence of a loop that is stabilized by hydrophobic interactions in the Val-24–Lys-28 region; (ii) a high degree of flexibility in the termini; and (iii) electrostatic interactions between the charged groups of Glu-22, Asp-23, and Lys-28 that modulate the stability of the folded structure.
Here, we test whether the stability of the Val-24–Lys-28 loop described by Borreguero et al. (34) persists in simulations that consider an explicit solvent (all atoms are included in the simulation). In addition, we determine the effects of solvent alterations on the folding dynamics and investigate the changes in the dynamics caused by amino acid substitutions. To this end, we present results of long-time, all-atom MD simulations of Aβ21–30 in explicit water. We also study the dynamics of the monomer containing the Dutch [Gln-22]Aβ21–30 mutation. This mutation leads to hereditary cerebral hemorrhage with amyloidosis–Dutch type, characterized by extensive deposition of Aβ1–40 in arterioles and small cerebral vessels but only limited senile plaque formation. In vitro studies indicate that Dutch Aβ has a greater tendency to form protofibrils and fibrils than does the WT Aβ (3, 35). To investigate dynamical differences in the folding caused by changes in the composition and density of the solvent, we study the system with solvated ions in normal density water and in water with a reduced density. Our results are consistent with previous results (34) and, in addition, reveal a helix conformation of the decapeptide when the encompassing water has a lower density than normal water.
Simulations with solvated ionic atoms in water illustrate how a different solvent composition can influence the role of salt bridges (SBs) between the charged amino acids by strengthening the stability of the Val-24–Lys-28 loop. In addition, simulations of Aβ21–30 containing the Dutch [Gln-22]Aβ21–30 mutation suggest that the experimentally observed increased fibril formation of this peptide may be a consequence of the increased number of aggregation-prone unpacked conformations.
Methods
MD Simulations. We performed long-time MD simulations of Aβ21–30 monomer in water at normal and reduced density, normal density water with dissociated salt ions, and the Dutch [Gln-22]Aβ21–30 monomer in normal density water. We explicitly considered all atoms with potential energies given by the charmm-27 force-field (36) with the namd package (37). We used the tip3p (38) model for the water molecules. We used the nvt ensemble and confined the system in a box with periodic boundary conditions. We carried out the MD simulations at a constant temperature of T = 283 K, corresponding to the in vitro experiments (32).
Aβ21–30 has the primary structure Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala, where Glu-22 and Asp-23 are negatively charged and Lys-28 is positively charged, whereas the Dutch mutation (39) substitutes Glu-22 by Gln-22 (polar). Following ref. 32, 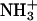 and
and 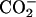 groups were placed at the N and C termini, respectively. We generated five trajectories with the following initial conditions: [RC], WT random coil conformation in normal density water; [P1] and [P2], WT loop conformations from Lazo et al. (32) in water with reduced density (corresponding to families I and II from ref. 32, respectively); [DU], Dutch peptide in a random coil conformation in normal density water; and [RCS], WT random coil conformation in salted water.
groups were placed at the N and C termini, respectively. We generated five trajectories with the following initial conditions: [RC], WT random coil conformation in normal density water; [P1] and [P2], WT loop conformations from Lazo et al. (32) in water with reduced density (corresponding to families I and II from ref. 32, respectively); [DU], Dutch peptide in a random coil conformation in normal density water; and [RCS], WT random coil conformation in salted water.
We chose these five trajectories because they provide information about the dynamics of the WT Aβ21–30 in normal water and water with a reduced density, starting from experimentally postulated conformations ([P1] and [P2]) and a control [RC]. These were then compared with the Dutch [DU] and the WT peptide in a salted solvent [RCS].
We solvated each monomer by inserting it in the center of a previously equilibrated cube of water molecules of side 43 Å. This insertion deleted all water molecules overlapping or in close proximity (<2.4 Å) to any of the monomer atoms, resulting in a system with ≈2,542 water molecules (see Supporting Text, which is published as supporting information on the PNAS web site, for details). To maintain system neutrality (for WT only), we inserted a single Na+ ion far from the monomer. To obtain the “salted” system, we inserted 25 dissociated molecules of NaCl, resulting in a system with 2,399 water molecules. We carried out all of the insertions and preparations of the systems by using the vmd package (40). We used the Particle Mesh Ewald method (41) to calculate the electrostatic interactions and a cut-off distance of 12 Å for the direct electrostatic and van der Waals interactions. We used a time step of 1 fs for all simulations and saved configurations (monomer and water) every 2 ps.
We first minimized the energy of the system for 20,000 steps by applying the conjugate gradient algorithm that relaxed all atoms except the Cα atoms. Next, we released the Cα atoms and minimized again for another 20,000 steps. Then, we gradually heated the system in the nvt ensemble from 0 to 283 K by harmonically constraining all Cα atoms for 100 ps. After this step, and still constraining the Cα atoms, we then performed another 200 ps in the npt ensemble at 283 K, followed by a brief 50-ps npt simulation with no constraints to generate the starting configuration for the production runs.
Because of the way that we deleted overlapping water molecules in the solvation step above, these npt equilibration steps allowed for water molecules to fill in holes left from the insertion of the decapeptide, thus shrinking the size of the system by ≈1 Å in all directions (see Supporting Text for details). For trajectories [P1] and [P2], we skipped the npt equilibration, not allowing the system to shrink, thus creating a system in which the effective density of the water was reduced by ≈7.5%. In Table 3, which is published as supporting information on the PNAS web site, we list all of the thermodynamic quantities for each of the trajectories. Total simulation times for the production runs were 102.6 ns for [RC], 65 ns for [P1], 83.6 ns for [P2], 80.0 ns for [DU], and 145.0 ns for [RCS]. Although these simulation times are too small to study the totality of the folding process of the Aβ21–30, they are long enough to exhibit characteristic elements of the dynamics of the folding for this decapeptide.
Structural Determinants. One of the most important quantities characterizing the structure of Aβ21–30 is the distance between atoms. The notation R(x,y) signifies the distance R between two specified atoms from amino acid x and amino acid y. The quantities we used to characterize the structure of Aβ21–30 as a function of simulation time are: (i) the distance between the two Cα atoms of Ala-21 and Ala-30 [denoted by R(1,10)] and Val-24 and Lys-28 [denoted by R(4,8)]; the distance between Lys-28(Nζ) and Glu-22(Cδ) [denoted by R(2,8)]; the distance between Lys-28(Nζ) and Asp-23(Cγ) [denoted by R(3,8)]; (ii) the radius of gyration Rg of the monomer given by
 |
where 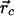 is the center of mass; (iii) the combined solvent accessible surface area (42) of the Val-24 and Lys-28 side chains (denoted by S); (iv) the hydrogen bond (HB) contact maps between amino acids; (v) the secondary structure propensity for each amino acid; (vi) the normalized scalar product D between the vector joining Cα and Cβ of Val-24 and the vector joining Cα and Cε of Lys-28; and (vii) the number and type of HBs between atoms in the decapeptide (40). We calculated secondary structure propensity by using the program stride (43) available through the vmd package. A HB exists if the distance between donor and acceptor is ≤3 Å, and the angle θ between the donor–hydrogen–acceptor atoms is 160° ≥ θ ≤ 200°. If, in addition, two hydrogen-bonded atoms are of opposite charge, they form a SB (44).
is the center of mass; (iii) the combined solvent accessible surface area (42) of the Val-24 and Lys-28 side chains (denoted by S); (iv) the hydrogen bond (HB) contact maps between amino acids; (v) the secondary structure propensity for each amino acid; (vi) the normalized scalar product D between the vector joining Cα and Cβ of Val-24 and the vector joining Cα and Cε of Lys-28; and (vii) the number and type of HBs between atoms in the decapeptide (40). We calculated secondary structure propensity by using the program stride (43) available through the vmd package. A HB exists if the distance between donor and acceptor is ≤3 Å, and the angle θ between the donor–hydrogen–acceptor atoms is 160° ≥ θ ≤ 200°. If, in addition, two hydrogen-bonded atoms are of opposite charge, they form a SB (44).
The quantities defined above provide a broad qualitative and quantitative description of the structure of Aβ21–30. In particular, the distance R(4,8) monitors possible formation of a loop involving the Val-24–Lys-28 region and close proximity of the hydrophobic parts of the Val-24 and Lys-28 side chains. The secondary structure propensity per amino acid determines general structure. A decrease of the combined solvent accessible surface area of two side chains indicates that the side chains are packed, reducing their contact with the solvent. A reduced normalized scalar product D indicates that the side chains of Val-24 and Lys-28 are constrained in their movements, implying that they are bound to either the backbone or other side chains. A small radius of gyration Rg along with a reduced R(1,10) indicate packed conformations. Small values of R(2,8) or R(3,8) indicate the possible formation of SBs between Glu-22 and Lys-28 or Asp-23 and Lys-28, respectively. Finally, the contact maps along with the number and type of HBs determine the nature and strength of interactions between different amino acids.
Results
Distances. In Fig. 1 we show results for R(2,8), R(3,8), R(4,8), and Rg as a function of time. As expected, these quantities fluctuate, but they also show “events” that last extended periods of time in which their values are substantially reduced and their fluctuations are uncharacteristically small. R(1,10) did not exhibit events in any of the trajectories, indicating that the termini are flexible (results not shown). The graph of S as a function of time has a close functional resemblance to R(4,8) (results not shown). This functional resemblance arises because whenever Val-24 and Lys-28 are in close proximity [R(4,8) < 6.5 Å], S is reduced from ≈575 Å2 (separated and accessible to solvent) to 475 Å2.
Fig. 1.

Distances between Cα atoms of Val-24 and Lys-28 [R(4,8)], charged atoms of Glu-22–Lys-28 [R(2,8), black], Asp-23–Lys-28 [R(3,8), gray], and radius of gyration Rg as a function of time for each trajectory. All of the distances are measured in Å.
We define events in Fig. 1 by: (i) R*(4,8), in which R(4,8) < 6.5 Å (Val-24–Lys-28 loop); (ii) R*(2,8) or R*(3,8), in which R(2,8) or R(3,8) < 4.2 Å (mostly SBs); (iii) S*, in which S < 525 Å2 (Val-24 and Lys-28 side-chain packing); and (iv) R*g, in which Rg < 6.5 Å (peptide packing). We classify i and iii as hydrophobic events and ii as electrostatic events with the formation of SBs.
Events within a trajectory occur at different times and time intervals (Fig. 1). By adding up the duration in time of these events, we obtain the total event time per trajectory (Table 1). Values in Table 1 are reported in ns rather than as percentages because the total simulation times are too short to obtain statistically significant percentage results. From these data, we see that S* and R*(4,8) (hydrophobic events) total times in each trajectory are comparable. We also see that hydrophobic events last for much longer periods of time than SB [R*(2,8) and R*(3,8)] events.
Table 1. Total accumulated times of events in each of the five trajectories.
| Trajectory
|
|||||
|---|---|---|---|---|---|
| Time/event | [RC] | [P1] | [P2] | [DU] | [RCS] |
| Total time, ns | 102.6 | 65.0 | 83.6 | 80.0 | 145.0 |
| Events, ns | |||||
| S* | 37.1 | 45.9 | 27.0 | 12.6 | 34.0 |
| R* (4,8) | 35.2 | 46.6 | 21.4 | 9.6 | 30.8 |
| R* (2,8) | 7.6 | 8.1 | 5.9 | 0.4 | 19.0 |
| R* (3,8) | 5.9 | 14.7 | 1.7 | 1.7 | 18.0 |
We next determined the amount of temporal overlap between pairs of events (Table 2), which is helpful for establishing correlations between events. We define the percentage of overlap for event X between two events X and Y as the time that X and Y overlap divided by the total duration of event X, multiplied by 100. In Table 2, we divide each overlap into two subcolumns, one for each of the events. In the first overlap column [S*  R*(4,8)], we observe that in all trajectories the S* and R*(4,8) events are correlated (high percentage of overlap), indicating that the proximity of Val-24 and Lys-28 is linked to an energetically favorable hydrophobic interaction. In the next column, the values for the overlap between the R*(4,8) and the SB events [R*(2,8) and R*(3,8) taken together] demonstrate that, except for trajectories [P2] and [DU], the SB events mostly occur conditional on an existing R*(4,8) event. Trajectory [P2] does not allow for this overlap because of its different secondary structure and HB contacts (see Supporting Text). In trajectory [DU] the lack of a negative charge in Gln-22 that effectively eliminates all R*(2,8) events might be key to the lack of R*(4,8) and SB overlaps. Thus, the prominent overlaps of SB with R*(4,8) suggest that interactions between amino acids in positions 22, 23, and 28 in the WT may be intermediary (or necessary) for extended R*(4,8) events to exist.
R*(4,8)], we observe that in all trajectories the S* and R*(4,8) events are correlated (high percentage of overlap), indicating that the proximity of Val-24 and Lys-28 is linked to an energetically favorable hydrophobic interaction. In the next column, the values for the overlap between the R*(4,8) and the SB events [R*(2,8) and R*(3,8) taken together] demonstrate that, except for trajectories [P2] and [DU], the SB events mostly occur conditional on an existing R*(4,8) event. Trajectory [P2] does not allow for this overlap because of its different secondary structure and HB contacts (see Supporting Text). In trajectory [DU] the lack of a negative charge in Gln-22 that effectively eliminates all R*(2,8) events might be key to the lack of R*(4,8) and SB overlaps. Thus, the prominent overlaps of SB with R*(4,8) suggest that interactions between amino acids in positions 22, 23, and 28 in the WT may be intermediary (or necessary) for extended R*(4,8) events to exist.
Table 2. Percentage of overlap between pairs of events per trajectory.
| Overlaps
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
S* ∩ R* (4,8)
|
R* (4,8) ∩ SB
|
R* (4,8) ∩ Rg*
|
R* (2,8) ∩ R* (3,8)
|
|||||
| Trajectory | S* | R* (4,8) | R* (4,8) | SB | R* (4,8) | Rg* | R* (2,8) | R* (3,8) |
| [RC] | 91.9 | 96.7 | 34.9 | 90.8 | 72.3 | 79.7 | 0.0 | 0.0 |
| [P1] | 97.1 | 95.6 | 40.6 | 84.9 | 95.9 | 85.8 | 5.3 | 2.9 |
| [P2] | 76.0 | 95.8 | 1.2 | 3.4 | 73.9 | 45.7 | 0.0 | 0.0 |
| [DU] | 72.6 | 95.7 | 4.3 | 19.7 | 68.1 | 41.3 | 5.7 | 1.4 |
| [RCS] | 85.7 | 94.6 | 54.1 | 70.4 | 81.1 | 60.2 | 70.1 | 73.9 |
Next, examining R*(4,8)  R*g we observe that for most trajectories there is a high degree of correlation between the R*(4,8) and R*g events, indicating that the decapeptide has a smaller effective size whenever Val-24 and Lys-28 are in close proximity. Finally, in the last column [R*(2,8)
R*g we observe that for most trajectories there is a high degree of correlation between the R*(4,8) and R*g events, indicating that the decapeptide has a smaller effective size whenever Val-24 and Lys-28 are in close proximity. Finally, in the last column [R*(2,8)  R*(3,8)] we see that the formation of the R*(2,8) and R*(3,8) SBs is mutually exclusive (except in trajectory [RCS]). This result indicates that whereas Glu-22 (Asp-23) interacts with Lys-28, Asp-23 (Glu-22) interacts with other amino acids or water. In contrast, in trajectory [RCS] the large overlap between the two SB events suggests that the ions in the solvent facilitate simultaneous formation of both SBs possibly by disrupting properties of the water molecules surrounding the decapeptide.
R*(3,8)] we see that the formation of the R*(2,8) and R*(3,8) SBs is mutually exclusive (except in trajectory [RCS]). This result indicates that whereas Glu-22 (Asp-23) interacts with Lys-28, Asp-23 (Glu-22) interacts with other amino acids or water. In contrast, in trajectory [RCS] the large overlap between the two SB events suggests that the ions in the solvent facilitate simultaneous formation of both SBs possibly by disrupting properties of the water molecules surrounding the decapeptide.
Secondary Structure and Packing of Val-24 and Lys-28. In Fig. 2 we show the time evolution of the secondary structure propensity of each amino acid for all of the trajectories. As with other structural determinants, there is a high degree of correlation between these secondary structures and other events. Typically, R*(4,8) events are correlated with increased turn propensities that span at least the Gly-25–Lys-28 region (Fig. 4, which is published as supporting information on the PNAS web site). In Fig. 2 [P1], the π-helix correlates with lowered values of all of the R(2,8), R(3,8), and R(4,8) distances and with a very compact conformation with the lowest Rg (from Fig. 1). We observe a similar behavior in trajectory [P2] during the helix formation in the later part of the trajectory (75–80 ns). The helices in [P1] and [P2] both are formed under a preexisting R*(4,8) event [compare R(4,8) from Fig. 1 with Fig. 2]. We note that the prominent helices in [P1] and [P2] both occur in the reduced density water environments, whereas in the trajectories with normal density water there is no observable helix. In trajectory [DU], in Fig. 2 the row corresponding to Gly-25 (label “25” on the y axis) is less populated (turn) than the others, suggesting two turn regions separated at Gly-25 (see Fig. 4 [DU] for trajectory averages).
Fig. 2.

Secondary structure of each amino acid as a function of time for each of the trajectories. The amino acid numbering is sequential from 21 (Ala-21) to 30 (Ala-30). The colors in the graphs correspond to: turn (green), bridge (tan), α-helix (pink), π-helix (red), and coil (white, none of the above).
We calculate the normalized scalar product D between the vectors formed by the side chains of Val-24 and Lys-28. By comparing results on D (Fig. 5, which is published as supporting information on the PNAS web site) with Fig. 1, we observe a persistence of the relative orientation of the side chains of Val-24 and Lys-28 during R*(4,8) events. These rather stable values for D indicate that during R*(4,8) events the side chains of Val-24 and Lys-28 are constrained in their relative orientation by being bound to atoms in the monomer, increasing packing and possibly providing stability to the hydrophobic interaction. Trajectory [DU], however, does not show the same persistence in D values, suggesting that the lack of R*(4,8) and R*(2,8) events permits an unconstrained motion of the Val-24 and Lys-28 side chains.
HBs. An examination of the number of HBs within the decapeptide as a function of time (Fig. 6 Upper, which is published as supporting information on the PNAS web site) reveals that only a handful of short-lived HBs are active during events. In SB events, there is one HB involving one of the Glu-22(Oε) [or Asp-23(Oδ)] oxygens and one of the Lys-28(Hζ) hydrogens. An exception is trajectory [RCS], in which the two SBs are present at the same time. The hydrophobic events [R*(4,8) and S*], on the other hand, are not characterized by unique HBs, but consist instead of different HBs. In Fig. 3, we show contact maps where the strength of contacts is proportional to the number of HBs between two amino acids. In most trajectories, Lys-28 is the most active amino acid in HB formation. In trajectory [RCS], the strongest contacts are the SBs between Glu-22, Asp-23, and Lys-28.
Fig. 3.

HB contact maps for each of the trajectories. The level of darkness of each contact is proportional to the number of HBs between two amino acids (from Table 4). The grayscale values for each contact map are relative to the maximum and minimum number of HBs from each trajectory. The strongest contacts per trajectory are: in [RC] between Gly-25 and Lys-28; in [P1] between Asp-23 and Lys-28; in [P2] between Val-24 and Gly-29; in [DU] between Gly-25 and Lys-28; and in [RCS] between Glu-22 and Lys-28 and between Asp-23 and Lys-28.
By analyzing the frequency of formation of individual HBs during hydrophobic events (Table 4, which is published as supporting information on the PNAS web site), we determine that the highest percentage of HBs is formed between atoms belonging to the backbone. The most prevalent backbone HBs are Lys-28(O)–Gly-25(H) and Asp-23(O)–Lys-28(H), and in trajectory [RC] the longest hydrophobic event that is stabilized by Lys-28(O)–Gly-25(H) also shows the tightest (smallest) loop formation. The NH +3 group of Lys-28 also participate in SB formation (Table 4), which is consistent with experimental results indicating that Lys-28 in WT Aβ21–30 is protected from hydrogen exchange (32). These results suggest that although transient backbone HBs may help stabilize an already existing loop, loop formation and long-term stability depend on the hydrophobic interactions of Val-24 and Lys-28 rather than on specific HBs. On the other hand, loop formation in trajectory [RCS] is not only driven by hydrophobic interactions but, to a large extent, by SB interactions, suggested by the large number of SBs in comparison with backbone HBs (Table 4).
Discussion
In this work, we present a series of all-atom MD simulations of the decapeptide folding nucleus of Aβ monomer (Aβ21–30) in explicit water and in water with the addition of salt ions. We also present all-atom MD simulations of the Dutch [Gln-22]Aβ21–30 peptide. Our results indicate that for all five trajectories studied (four for WT and one for Dutch) hydrophobic events, characterized by packing of the isopropyl group of Val-24 and the butyl group of Lys-28, predominate over electrostatic events (SB between Glu-22 and Lys-28 or between Asp-23 and Lys-28).
The hydrophobic events are highly correlated with a smaller value for the radius of gyration Rg, and they last longer than SB events (Tables 1 and 2). In WT Aβ21–30 in normal density water, results on the distance between termini and lifetimes of backbone HBs indicate the formation of a semirigid loop in the Val-24–Lys-28 region with highly flexible termini, in close agreement with discrete MD studies (34), replica exchange MD simulations (A. Baumketner, S. Bernstein, T. Wyttenbach, N.D.L., D.B.T., M. Bowers, and J.-E. Shea, unpublished work), and consistent with NMR experiments of Aβ21–30 (32).
In WT Aβ21–30 in reduced density water, the Val-24–Lys-28 region also forms a loop that during periods of time may belong to a larger helical structure (mostly π- and α-helices). The formation of a helix in our [P1] and [P2] trajectories is not surprising in light of prior studies of Aβ and other proteins that form helices when the density of water is effectively reduced by other solvated components (12–15, 45). The formation of helices in reduced density water is consistent with the possibility that hydrophobic assembly in proteins is facilitated by removal of water molecules (“vaporization”) from regions between hydrophobic amino acids (46). Future work in this area will have relevance for understanding how Aβ folding is affected by different intracellular and extracellular milieus, including endoplasmic and plasma cell membranes, endosomal/lysosomal compartments, cytoplasm, plasma, and cerebrospinal fluid.
Although in our trajectories with pure normal water the Glu-22–Lys-28 and Asp-23–Lys-28 SBs are transient in time, they could be precursors to the SBs proposed in recent MD simulations (47) and modeling based on NMR data (48), where it is suggested that the turn or bend in Aβ fibrils is stabilized by a SB involving Asp-23–Lys-28. In Lazo et al. (32), however, the R(2,8) and R(3,8) distances (>9 Å) seem to rule out the existence of the SBs seen here. This finding can be reconciled by considering that NMR experiments measure averages of interatomic distances and the measurements are done in the microsecond to millisecond time regime. Our simulations are done in the nanosecond time regime.
In WT Aβ21–30 in normal water with solvated salt ions [RCS], we observe the longest “waiting time” for a hydrophobic event that can be attributed to the well known salting-in effect in which a cloud of ions increases solubility of a protein by lowering its electrostatic free energy (49). In this same trajectory, the coexistence of the Glu-22–Lys-28 and Asp-23–Lys-28 SBs during the R*(4,8) event might be a consequence of a disruption of the HB network of the water by the solvated ions. In this scenario, ions locally reorient water molecules, changing the HB interactions between water and Glu-22 and Asp-23, enhancing the simultaneous formation of these SBs.
In the [DU] trajectory, the lack of hydrophobic and SB events, and the unconstrained motion of the Val-24 and Lys-28 side chains, indicate a more flexible structure than the WT in agreement with ref. 50. Taken as a whole, results on the [DU] trajectory suggest that the Dutch mutation alters the folding pathway of the peptide. According to a postulate of Lazo et al. (32), partial unfolding of the Ala-21–Ala-30 region of Aβ should occur before fibril formation. If this is so, then our observation of the inability of the [DU] trajectory to form a stable Val-24–Lys-28 loop might explain the higher propensity of the Dutch peptide to form protofibrils and fibrils.
Conclusions
Our results show that hydrophobic interactions play a crucial role in Aβ21–30 folding dynamics, assisted by the formation of SBs between the charged amino acids. By performing secondary structure and HB analysis, we find that there is no regular secondary structure or permanent hydrogen bonding, suggesting that folding involves formation of a loop stabilized by the packing of the side chains of Val-24 and Lys-28. We also show that by reducing the density of water we may induce formation of a π-helix.
Interestingly, we find that in normal density water, if the solvent contains desolvated ions, the SBs play a prominent role in the stabilization of the Val-24–Lys-28 loop. Finally, we find that for the Dutch [Gln-22]Aβ21–30 decapeptide in water, elimination of charge at position 22 disrupts the natural tendency of the monomer to form a long-lived Val-24–Lys-28 loop. This substitution likely alters the Aβ folding pathway, leading to the formation of alternative turn structures, including those stabilized solely by an Asp-23–Lys-28 SB.
Supplementary Material
Acknowledgments
Financial support was provided by National Institutes of Health Grants AG23661, NS38328, AG18921, and NS44147 and Stephen Bechtel, Jr.
Author contributions: L.C., B.U., and H.E.S. designed research; L.C., B.U., N.D.L., D.B.T., and H.E.S. performed research; L.C. analyzed data; and L.C., B.U., J.M.B., N.D.L., D.B.T., and H.E.S. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: Aβ, amyloid β-protein; HB, hydrogen bond; MD, molecular dynamics; SB, salt bridge.
References
- 1.Pike, C. J., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. (1991) Brain Res. 563, 311–314. [DOI] [PubMed] [Google Scholar]
- 2.Hardy, J. & Allsop, D. (1991) Trends Pharmacol. Sci. 12, 383–388. [DOI] [PubMed] [Google Scholar]
- 3.Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M. & Teplow, D. B. (1997) J. Biol. Chem. 272, 22364–22372. [DOI] [PubMed] [Google Scholar]
- 4.Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T. (1997) Chem. Biol. 4, 119–125. [DOI] [PubMed] [Google Scholar]
- 5.Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., Morgan, T. E., Rozovsky, I., Trommer, B., Viola, K. L., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein, W. L., Krafft, G. A. & Finch, C. E. (2001) Trends Neurosci. 24, 219–224. [DOI] [PubMed] [Google Scholar]
- 7.Yong, W., Lomakin, A., Kirkitadze, M. D., Teplow, D. B., Chen, S. H. & Benedek, G. B. (2002) Proc. Natl. Acad. Sci. USA 99, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J. & Selkoe, D. J. (2002) Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- 9.Kirkitadze, M. D., Bitan, G. & Teplow, D. B. (2002) J. Neurosci. Res. 69, 567–577. [DOI] [PubMed] [Google Scholar]
- 10.Klein, W. L., Stine, W. B., Jr. & Teplow, D. B. (2004) Neurobiol. Aging 25, 569–580. [DOI] [PubMed] [Google Scholar]
- 11.Barrow, C. J., Yasuda, A., Kenny, P. T. M. & Zagorski, M. (1992) J. Mol. Biol. 225, 1075–1093. [DOI] [PubMed] [Google Scholar]
- 12.Sticht, H., Bayer, P., Willbold, D., Dames, S., Hilbich, C., Beyreuther, K., Frank, R. W. & & Rosch, P. (1995) Eur. J. Biochem. 233, 293–298. [DOI] [PubMed] [Google Scholar]
- 13.Coles, M., Bicknell, W., Watson, A. A., Fairlie, D. P. & Craik, D. J. (1998) Biochemistry 37, 11064–11077. [DOI] [PubMed] [Google Scholar]
- 14.Shao, H. Y., Jao, S. C., Ma, K. & Zagorski, M. G. (1999) J. Mol. Biol. 285, 755–773. [DOI] [PubMed] [Google Scholar]
- 15.Crescenzi, O., Tomaselli, S., Guerrini, R., Salvadori, S., D'Ursi, A. M., Temussi, P. A. & Picone, D. (2002) Eur. J. Biochem. 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- 16.Zhang, S., Iwata, K., Lachenmann, M. J., Peng, J. W., Li, S., Stimson, E. R., Lu, Y., Felix, A. M., Maggio, J. E. & Lee, J. P. (2000) J. Struct. Biol. 130, 130–141. [DOI] [PubMed] [Google Scholar]
- 17.Gursky, O. & Aleshkov, S. (2000) Biochim. Biophys. Acta/Protein Struct. Mol. Enzymol. 1476, 93–102. [DOI] [PubMed] [Google Scholar]
- 18.Lee, J. P., Stimson, E. R., Ghilardi, J. R., Mantyh, P. W., Lu, Y. A., Felix, A. M., Llanos, W., Behbin, A., Cummings, M., Vancriekinge, M., et al. (1995) Biochemistry 34, 5191–5200. [DOI] [PubMed] [Google Scholar]
- 19.Narayanan, S. & Reif, B. (2005) Biochemistry 44, 1444–1452. [DOI] [PubMed] [Google Scholar]
- 20.Klimov, D. K., Straub, J. E. & Thirumalai, D. (2004) Proc. Natl. Acad. Sci. USA 101, 14760–14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bitan, G., Kirkitadze, M. D., Lomakin, A., Vollers, S. S., Benedek, G. B. & Teplow, D. B. (2003) Proc. Natl. Acad. Sci. USA 100, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bitan, G., Vollers, S. S. & Teplow, D. B. (2003) J. Biol. Chem. 278, 34882–34889. [DOI] [PubMed] [Google Scholar]
- 23.Bitan, G., Tarus, B., Vollers, S. S., Lashuel, H. A., Condron, M. M., Straub, J. E. & Teplow, D. B. (2003) J. Am. Chem. Soc. 125, 15359–15365. [DOI] [PubMed] [Google Scholar]
- 24.Urbanc, B., Cruz, L., Ding, F., Sammond, D., Khare, S., Buldyrev, S. V., Stanley, H. E. & Dokholyan, N. V. (2004) Biophys. J. 87, 2310–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urbanc, B., Cruz, L., Yun, S., Ding, F., Buldyrev, S. V., Bitan, G., Teplow, D. B. & Stanley, H. E. (2004) Proc. Natl. Acad. Sci. USA 101, 17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith, S. W., Hall, C. K. & Freeman, B. D. (1997) J. Comput. Phys. 134, 16–30. [Google Scholar]
- 27.Dokholyan, N. V., Buldyrev, S. V., Stanley, H. E. & Shakhnovich, E. I. (1998) Folding Des. 3, 577–587. [DOI] [PubMed] [Google Scholar]
- 28.Dokholyan, N. V., Buldyrev, S. V., Stanley, H. E. & Shakhnovich, E. I. (2000) J. Mol. Biol. 296, 1183–1188. [DOI] [PubMed] [Google Scholar]
- 29.Smith, A. V. & Hall, C. K. (2001) Proteins Struct. Funct. 44, 344–360. [DOI] [PubMed] [Google Scholar]
- 30.Xu, Y., Shen, J., Luo, X., Zhu, W., Chen, K., Ma, J. & Jiang, H. (2005) Proc. Natl. Acad. Sci. USA 102, 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Massi, F. & Straub, J. E. (2003) J. Comput. Chem. 24, 143–153. [DOI] [PubMed] [Google Scholar]
- 32.Lazo, N. D., Grant, M. A., Condron, M. C., Rigby, A. C. & Teplow, D. B. (2005) Protein Sci. 14, 1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fersht, A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 34.Borreguero, J. M., Urbanc, B., Lazo, N. D., Buldyrev, S. V., Teplow, D. B. & Stanley, H. E. (2005) Proc. Natl. Acad. Sci. USA 102, 6015–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miravalle, L., Tokuda, T., Chiarle, R., Giaccone, G., Bugiani, O., Tagliavini, F., Frangione, B. & Ghiso, J. (2000) J. Biol. Chem. 275, 27110–27116. [DOI] [PubMed] [Google Scholar]
- 36.MacKerell, A. D., Jr., Bashford, D., Bellott, M., Dunbrack, R. L., Jr., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 37.Kalé, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Phys. 151, 283–312. [Google Scholar]
- 38.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1982) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 39.Levy, E., Carman, M. D., Fernandez Madrid, I. J., Power, M. D., Lieberburg, I., Van Duinen, S. G., Bots, G. T. A. M., Luyendijk, W. & Frangione, B. (1990) Science 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- 40.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 41.Darden, T. A., York, D. M. & Pedersen, L. G. (1993) J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- 42.Sanner, M. F., Olson, A. J. & Spehner, J. (1996) Biopolymers 38, 305–320. [DOI] [PubMed] [Google Scholar]
- 43.Frishman, D. & Argos, P. (1995) Proteins Struct. Funct. 23, 566–579. [DOI] [PubMed] [Google Scholar]
- 44.Marqusee, S. & Baldwin, R. L. (1987) Proc. Natl. Acad. Sci. USA 84, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fezoui, Y. & Teplow, D. B. (2002) J. Biol. Chem. 277, 36948–36954. [DOI] [PubMed] [Google Scholar]
- 46.Chandler, D. (2002) Nature 417, 491. [DOI] [PubMed] [Google Scholar]
- 47.Ma, B. Y. & Nussinov, R. (2002) Proc. Natl. Acad. Sci. USA 99, 14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petkova, A. T., Ishii, Y., Balbach, J. J., Antzutkin, O. N., Leapman, R. D., Delaglio, F. & Tycko, R. (2002) Proc. Natl. Acad. Sci. USA 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creighton, T. (1993) Proteins: Structures and Molecular Properties (Freeman, New York), 2nd Ed.
- 50.Massi, F. & Straub, J. E. (2001) Biophys. J. 81, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.