

Abstract
Background
The laboratory rat (Rattus norvegicus) is an important model for studying many aspects of human health and disease. Detailed knowledge on genetic variation between strains is important from a biomedical, particularly pharmacogenetic point of view and useful for marker selection for genetic cloning and association studies.
Results
We show that Single Nucleotide Polymorphisms (SNPs) in commonly used rat strains are surprisingly well represented in wild rat isolates. Shotgun sequencing of 814 Kbp in one wild rat resulted in the identification of 485 SNPs as compared with the Brown Norway genome sequence. Genotyping 36 commonly used inbred rat strains showed that 84% of these alleles are also polymorphic in a representative set of laboratory rat strains.
Conclusion
We postulate that shotgun sequencing in a wild rat sample and subsequent genotyping in multiple laboratory or domesticated strains rather than direct shotgun sequencing of multiple strains, could be the most efficient SNP discovery approach. For the rat, laboratory strains still harbor a large portion of the haplotypes present in wild isolates, suggesting a relatively recent common origin and supporting the idea that rat inbred strains, in contrast to mouse inbred strains, originate from a single species, R. norvegicus.
Background
Genetic variation exists between individuals (or strains) of all organisms and it makes up the genetic basis for phenotypic differences between individuals. In addition, genetic variation functions as a valuable resource for mapping phenotypic traits in model organisms. Single Nucleotide Polymorphisms (SNPs) are the most abundant form of genetic variation and therefore dominate high-resolution genetic mapping strategies. Moreover, numerous well-performing high-throughout SNP detection technologies have been developed, like oligonucleotide array-based technology, mass-spectrometry-based technology (MALDI-TOF), and sequence-based technology (pyrosequencing, DHPLC) [1], which makes automated SNP detection favored above the more labor-intensive detection of microsatellite markers [2].
Since the availability of its genome, the laboratory rat is gaining influence as a genetic model organism [3]. In addition, over 200 well-characterized inbred strains that are models for a wide variety of human diseases are available [4,5]. However, the availability of genetic tools, like a dense genome-wide SNP marker set, is still subordinate compared to other commonly used model organisms. This is illustrated by the number of entries in dbSNP, the central SNP repository of NCBI [6]: the amount of human (>10,000,000), chicken (>3,000,000), and mouse (>500,000) entries surpass the amount of rat entries (>43,000) spectacularly. In search for rat SNPs, experimental [7,8] and computational [9] approaches have been employed, but these efforts primarily resulted in SNPs associated with coding regions. For genetic mapping purposes, a much denser marker set, preferentially equally distributed over the genome, is required.
Laboratory rat strains are thought to be established from a limited number of founder animals originating from a domesticated wild population [10,11]. The value of inbred strains emanates from the close genetic uniformity that facilitates phenotyping and genotyping. In principle, inbred strains are selectively bred for certain traits from a genetically diverse pool, comprising diverse genetic information about the trait. However, since many of the current rat strains were derived from common ancestral stocks and simply inbred to increase genetic uniformity, inbred strains clearly share alleles [12]. Although such simplified models are essential for biomedical research, modulating effects on the clinical manifestation of a trait resulting from genetic heterogeneity in a population can only be studied to a limited extent in F1 hybrids. The use of a carefully chosen selection of inbred strains may address this issue, but the choice depends on knowledge on the relationship between the strains and hence the degree of genetic variation. Alternatively, wild-derived strains may be good alternatives to introduce sufficient genetic variation in laboratory experiments [13,14].
Based on a preliminary observation that alleles from laboratory rat strains are frequently detected in wild-derived samples, we developed a wild rat-based SNP discovery approach. The method consists of shotgun sequencing of a wild rat-derived genomic library followed by comparison with the published rat genome (strain Brown Norway). Genotyping commonly used rat strains for newly identified SNPs revealed that 84% of SNP-alleles (and 87% of all genetic variation) occurring between BN and a single wild individual is also represented in one or more laboratory strains. A user-friendly webtool allows exploration of the genetic variation between any arbitrary combinations of two strains that were used in this study, making all information directly available for experimental use.
Results
Wild rat-based SNP discovery
It is generally believed that commonly used rat strains originate from a wild-derived founder population of limited size [10]. To examine whether polymorphisms found in laboratory strains are still represented in individuals of the wild population, we typed two wild-derived samples for confirmed SNPs of the CASCAD database [9]. Interestingly, about 53% of alleles (n = 147), which were confirmed to exist in laboratory strains, were also represented in wild 1, wild 2 or both (not shown). Hence, a preselection of highly likely candidate SNPs could potentially be made by genotyping wild individuals and comparing the sequences to the rat genome sequence (Brown Norway).
Accordingly, we performed random shotgun sequencing on a genomic library of a wild rat (wild 1). We generated shotgun traces (814 Kbp) by bidirectional sequencing of about 1,600 colonies (Table 1). 85.5% of the reads (2545/2975; Table 1) could be mapped to a unique location in the Brown Norway rat genome using BLAT [15], resulting in the automated identification of nearly 5,000 ambiguous nucleotide positions (potential polymorphisms). Manual inspection of the sequencing reads reduced this set of potential polymorphisms to a set of 746 real SNPs and 122 indels. The average SNP rate between BN (BN/SsNMcw; genome sequencing project) and this single wild rat is estimated to be about 1 per 900 bp and, hence, discovery of a novel SNP can be expected every second shotgun read. A subset of the discovered SNPs was verified and genotyped in 36 commonly used strains (including BN). To this end, we designed primers for 451 SNP-containing amplicons (about 300 bp) of which 416 (92.2%) were successfully read by unidirectional sequencing of the PCR products, resulting in roughly 119 Kbp high quality sequence per strain or individual (Table 1).
Table 1.
Statistics on shotgun sequencing of the wild rat-derived genomic library
| picked colonies | 1632 |
| readable sequence reads/sequenced bases | 2975/814,440 |
| uniquely mapped (BLAT) reads/bases | 2545/768,683 |
| ambiguous positions | 4902 |
| strong candidates after manual inspection | 868 (746 SNPs + 122 indels) |
| successfully read/amplicons designed* | 416/451 (~1.65 candidate SNP/amplicon) (92.2%) |
| amplified bases per strain or wild individual | 118,971 |
* Amplicons are designed for the 746 SNP candidates.
Wild rat-derived SNP characteristics
The verification of 746 candidate SNPs by amplicon-based resequencing in 36 inbred rat strains and three wild-derived samples (wild 1, 2, and 3) revealed 960 polymorphisms, consisting of 90 indels, seven 2-bp substitutions, one 3-bp substitution, one 5-bp substitution, and 861 SNPs, of which only one was tri-allelic. The amplicons are randomly distributed over the genome (Fig. 1). We observed heterozygous positions in the outbred strains, but unexpectedly some were also found in the inbred strains (for detailed information: [see Additional file 1] or [6]). For our analysis, we considered these loci to be polymorphic as compared to the BN genome sequence.
Figure 1.

Distribution of amplicons (451 loci) designed for verification and subsequent genotyping of candidate shotgun-based SNPs in 36 commonly used inbred strains.
From the 746 shotgun-based candidate SNPs, 685 were located in the 416 PCR amplicons that worked, and 485 (71%) were reconfirmed by resequencing (shotgun-based; Table 2). Strikingly, for 408 (84%) of the confirmed SNPs, the wild rat allele is also present in one or more commonly used strains, with only 36 (7.4%) being specific to BN (Table 2). Of the remaining 77 (16%) SNPs, wild rat alleles are not present in any of the 36 selected strains and could be considered wild rat-specific. These results illustrate that shotgun sequencing one wild individual efficiently identifies shared polymorphisms among commonly used rat strains.
Table 2.
SNP discovery results
| shotgun- based | genotyping-based (only wild 1*) | genotyping-based (wild 1, 2, 3*) | total (only wild 1*) | total (wild 1, 2, 3*) | |
| BN specific | 36 (7.4 %) | 9 (2.5 %) | 7 (1.9 %) | 45 (5.3 %) | 43 (5.0 %) |
| wild specific | 77 (15.9 %) | 12 (3.4 %) | 30 (8.0 %) | 89 (10.6 %) | 107 (12.4 %) |
| in 35 strains, not in wild | 0 | 204 (57.0 %) | 156 (41.5 %) | 204 (24.2 %) | 156 (18.1 %) |
| in 35 strains, shared with wild | 372 (76.7 %) | 133 (37.1 %) | 183 (48.7 %) | 505 (59.9 %) | 555 (64.5 %) |
| total | 485 (100 %) | 358 (100 %) | 376 (100 %) | 843 (100 %) | 861 (100 %) |
* By genotyping two other wild individuals (wild 2 and 3), additional polymorphisms were identified, which could not have been found by shotgun sequencing only wild 1.
While genotyping by resequencing, 358 novel SNPs were discovered that were not identified in the shotgun sequencing experiment (genotyping-based; Table 2). About 39% (139) of this set can be accounted for by differences in the sequence coverage between the shotgun reads and the resequencing genotyping reads (Table 2), whereas the remaining part of this set is strongly biased towards SNPs that are not polymorphic between BN and wild rat 1 and thus could not have been discovered in the shotgun experiment. Interestingly, about 37% of the newly discovered SNPs are polymorphic between the shotgun sequenced wild rat and any of the inbred strains (Table 2). When considering all SNPs that are polymorphic in the set of 36 commonly used laboratory strains, of the majority (66%) the wild rat allele is found back in one of the strains (total; Table 2) and this percentage increases only slightly (70%) when two other wild individuals (wild 2 and 3) are included in the analysis. This indicates that wild rat-based SNP discovery is already highly efficient using a single wild sample.
Based on the genotyping results, the SNP rate between BN and the shotgun sequenced wild rat (wild 1) is 1 SNP per 190 bp (626 SNPs/119 Kbp). The SNP rate within the 36 rat strains, including BN, is 1 in 158 (Table 2; 45+204+505 SNPs/119 Kbp) and the SNP rate in the entire experiment, including the wild rat (wild 1), BN, and the other strains is 1 in 141 bp (Table 2; 843 SNPs/119 Kbp). To compare wild rat inter-individual variation with the inter-strain variation for commonly used inbred strains, we calculated the number of SNPs that are polymorphic when comparing arbitrary combinations of 3 strains. Genotyping of 861 SNP positions in the three wild rats resulted in 438 polymorphic positions, whereas the most polymorphic combination of inbred strains in this experiment (BN, BH, and SHR) yielded 427 SNPs. This indicates that three random, but potentially related, Dutch wild rats are about equally polymorphic as three carefully selected inbred strains. Inclusion of wild isolates from other locations worldwide may increase the efficiency of the SNP discovery approach.
Intraspecific phylogenetic network
Relationships among different rat strains have been determined previously by phylogenetic tree reconstruction based on microsatellite markers [16,17]. However, intraspecific relationships for laboratory strains are often very challenging to determine, due to small genetic distances and complex gene flow. The resulting multitude of plausible trees is best expressed by a network, which displays alternative potential evolutionary paths in the form of cycles [18]. We used Network software (v4.111 Reduced-Joining, [19]) to construct a spatial network, based on 861 SNP markers in 36 rat strains and three wild rat individuals (Fig. 2). The three wild individuals are grouped together, possibly due to the geographic and possibly genetic relation between the samples, but in accordance with the last paragraph of the previous section, they appear relatively unrelated as compared to the set of inbred strains.
Figure 2.
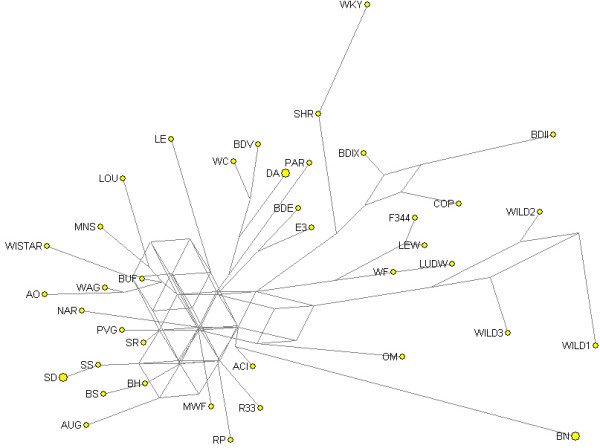
Strain relationships in a network structure. End nodes (yellow dots) represent strains. Some end nodes are double-size, meaning that they are supported by two samples. Interconnecting nodes where lines come together, represent a possible precursor.
The majority of the SNPs (485 of 861) was selected for being polymorphic between wild 1 and BN. As a result, different BN substrains (BN/Ztm, BN/Crl), depicted as a double-sized end node because of high similarity, and different wild rat individuals (wild 1, wild 2, and wild 3) are grouped together as the outliers. Several strains that are known to be closely related (source RGD-strains: [20]) are also grouped together, like DA and COP or SS and SR. Interestingly, WKY is also an outlier, indicating that besides BN, this strain can be utilized as an alternative mapping strain. WKY is already commonly used as a normotensive control strain in genetic mapping of blood pressure quantitative trait loci [21]. WKY is known to be closely related to SHR and these strains are indeed grouped together (Fig. 2). Additionally, BDII and BDIX are related and BDE is an RI strain from E3. These strain combinations are also grouped together. Wistar is contributing to a large subset of these strains, like WKY, WC, BDII, MWF, LEW, and WF, which contributes to the complexity of the network structure.
Data availability
The use of genetic markers for mapping traits in rat strains has been exploited for long time already. Current marker sets in rats are mostly limited to microsatellites [22,23], which are not abundantly available and are commonly detected in a more laborious way than SNPs. In this study, we have determined a total of about 35,000 genotypes (about 960 loci in 36 inbred strains), out of which the vast majority are SNPs. This data is accessible via a versatile webtool [24]. Pairs of strains of interest can be selected and explored on presence of verified genetic variation. Besides a graphical representation of the location of the SNPs on a genome map, primer sequences that were successfully used in our experiments are also provided. In a pairwise comparison matrix (Table 3), we plotted the absolute number of polymorphic positions for each of the (sub-)strains or individuals used. Interestingly, for some strains different alleles are observed in substrains (e.g. BN/Crl differs from BN/Ztm at 4 positions), in line with previous observations [8].
Table 3.
Absolute number of polymorphic positions between strains in a pairwise comparison.
| ACI | AO | AUG | BDE | BDII | BDIX | BDV | BH | BN | BN2 | BS | BUF | COP | DA | DA2 | E3 | F344 | LEW | LE | LOU | LUDW | MWF | MNS | NAR | OM | PAR | PVG | R33 | RP | SD | SD2 | SHR | SR | SS | WAG | WC | WF | WIST | WKY | wild3 | wild2 | wild1 | |
| ACI | x | |||||||||||||||||||||||||||||||||||||||||
| AO | 131 | x | ||||||||||||||||||||||||||||||||||||||||
| AUG | 111 | 167 | x | |||||||||||||||||||||||||||||||||||||||
| BDE | 145 | 178 | 158 | x | ||||||||||||||||||||||||||||||||||||||
| BDII | 130 | 166 | 143 | 148 | x | |||||||||||||||||||||||||||||||||||||
| BDIX | 129 | 196 | 191 | 159 | 115 | x | ||||||||||||||||||||||||||||||||||||
| BDV | 109 | 131 | 145 | 137 | 78 | 142 | x | |||||||||||||||||||||||||||||||||||
| BH | 145 | 189 | 192 | 212 | 156 | 177 | 167 | x | ||||||||||||||||||||||||||||||||||
| BN | 225 | 263 | 279 | 266 | 206 | 258 | 244 | 274 | x | |||||||||||||||||||||||||||||||||
| BN2 | 227 | 270 | 285 | 268 | 222 | 271 | 251 | 274 | 4 | x | ||||||||||||||||||||||||||||||||
| BS | 159 | 167 | 163 | 178 | 166 | 185 | 143 | 180 | 246 | 251 | x | |||||||||||||||||||||||||||||||
| BUF | 148 | 158 | 181 | 171 | 168 | 190 | 151 | 166 | 250 | 263 | 158 | x | ||||||||||||||||||||||||||||||
| COP | 84 | 194 | 187 | 166 | 155 | 147 | 155 | 191 | 262 | 275 | 203 | 192 | x | |||||||||||||||||||||||||||||
| DA | 62 | 107 | 116 | 134 | 115 | 130 | 117 | 130 | 210 | 216 | 150 | 126 | 116 | x | ||||||||||||||||||||||||||||
| DA2 | 76 | 141 | 144 | 157 | 149 | 167 | 127 | 176 | 261 | 269 | 193 | 167 | 151 | 2 | x | |||||||||||||||||||||||||||
| E3 | 136 | 189 | 170 | 86 | 154 | 180 | 144 | 204 | 244 | 253 | 172 | 182 | 171 | 148 | 180 | x | ||||||||||||||||||||||||||
| F344 | 132 | 165 | 178 | 178 | 152 | 150 | 138 | 144 | 170 | 176 | 141 | 156 | 166 | 121 | 165 | 168 | x | |||||||||||||||||||||||||
| LEW | 156 | 178 | 213 | 197 | 160 | 163 | 141 | 169 | 213 | 222 | 166 | 160 | 191 | 133 | 181 | 186 | 16 | x | ||||||||||||||||||||||||
| LE | 131 | 144 | 136 | 155 | 148 | 152 | 122 | 142 | 221 | 224 | 157 | 127 | 147 | 110 | 140 | 167 | 142 | 149 | x | |||||||||||||||||||||||
| LOU | 145 | 146 | 165 | 191 | 153 | 203 | 120 | 178 | 242 | 250 | 138 | 171 | 212 | 111 | 142 | 192 | 126 | 150 | 149 | x | ||||||||||||||||||||||
| LUDW | 153 | 175 | 186 | 198 | 161 | 169 | 153 | 195 | 252 | 263 | 183 | 177 | 189 | 123 | 163 | 215 | 115 | 125 | 133 | 161 | x | |||||||||||||||||||||
| MWF | 148 | 147 | 172 | 166 | 136 | 166 | 111 | 167 | 209 | 222 | 135 | 148 | 185 | 133 | 164 | 158 | 115 | 134 | 148 | 136 | 163 | x | ||||||||||||||||||||
| MNS | 151 | 167 | 178 | 173 | 158 | 186 | 122 | 169 | 239 | 250 | 155 | 194 | 210 | 128 | 176 | 166 | 123 | 132 | 156 | 141 | 159 | 137 | x | |||||||||||||||||||
| NAR | 147 | 169 | 184 | 212 | 168 | 177 | 145 | 177 | 233 | 249 | 146 | 155 | 197 | 134 | 193 | 188 | 137 | 166 | 122 | 170 | 153 | 164 | 151 | x | ||||||||||||||||||
| OM | 127 | 161 | 153 | 170 | 120 | 158 | 143 | 150 | 216 | 222 | 154 | 138 | 176 | 125 | 147 | 183 | 125 | 156 | 127 | 156 | 139 | 143 | 144 | 147 | x | |||||||||||||||||
| PAR | 140 | 182 | 168 | 166 | 149 | 158 | 133 | 175 | 225 | 227 | 155 | 138 | 167 | 136 | 159 | 169 | 128 | 136 | 127 | 151 | 159 | 133 | 160 | 150 | 162 | x | ||||||||||||||||
| PVG | 95 | 164 | 152 | 153 | 153 | 181 | 129 | 184 | 252 | 263 | 170 | 151 | 164 | 110 | 150 | 148 | 142 | 175 | 145 | 143 | 196 | 146 | 161 | 172 | 160 | 147 | x | |||||||||||||||
| R33 | 155 | 198 | 183 | 213 | 169 | 198 | 186 | 177 | 261 | 257 | 173 | 210 | 204 | 142 | 184 | 223 | 183 | 209 | 164 | 185 | 196 | 191 | 187 | 182 | 159 | 173 | 189 | x | ||||||||||||||
| RP | 146 | 159 | 171 | 171 | 132 | 164 | 109 | 186 | 216 | 230 | 113 | 161 | 176 | 138 | 175 | 153 | 119 | 141 | 134 | 141 | 166 | 108 | 157 | 136 | 147 | 132 | 139 | 166 | x | |||||||||||||
| SD | 121 | 154 | 156 | 177 | 153 | 160 | 129 | 118 | 233 | 247 | 149 | 134 | 174 | 100 | 148 | 184 | 131 | 138 | 135 | 125 | 145 | 144 | 141 | 149 | 103 | 133 | 130 | 147 | 138 | x | ||||||||||||
| SD2 | 95 | 116 | 134 | 150 | 117 | 130 | 122 | 85 | 219 | 220 | 117 | 96 | 145 | 104 | 115 | 168 | 109 | 115 | 92 | 99 | 110 | 118 | 121 | 107 | 90 | 121 | 109 | 126 | 109 | 16 | x | |||||||||||
| SHR | 159 | 212 | 166 | 186 | 179 | 175 | 168 | 204 | 264 | 275 | 188 | 178 | 180 | 135 | 189 | 205 | 156 | 176 | 173 | 202 | 188 | 180 | 200 | 184 | 182 | 177 | 192 | 207 | 187 | 185 | 139 | x | ||||||||||
| SR | 129 | 171 | 172 | 170 | 160 | 161 | 147 | 138 | 235 | 244 | 163 | 131 | 170 | 120 | 172 | 174 | 146 | 169 | 156 | 157 | 175 | 150 | 160 | 164 | 136 | 117 | 134 | 166 | 160 | 60 | 64 | 184 | x | |||||||||
| SS | 114 | 145 | 167 | 183 | 142 | 150 | 139 | 111 | 249 | 253 | 149 | 152 | 175 | 117 | 161 | 186 | 136 | 161 | 121 | 134 | 156 | 146 | 153 | 128 | 130 | 143 | 145 | 145 | 144 | 69 | 46 | 191 | 83 | x | ||||||||
| WAG | 120 | 105 | 156 | 147 | 143 | 158 | 115 | 160 | 197 | 200 | 96 | 125 | 164 | 94 | 136 | 151 | 110 | 132 | 127 | 96 | 145 | 120 | 121 | 129 | 140 | 126 | 128 | 154 | 108 | 118 | 84 | 181 | 119 | 110 | x | |||||||
| WC | 140 | 164 | 158 | 155 | 129 | 180 | 92 | 188 | 214 | 232 | 151 | 177 | 177 | 125 | 158 | 157 | 131 | 160 | 156 | 145 | 171 | 87 | 157 | 179 | 152 | 158 | 138 | 213 | 126 | 153 | 122 | 195 | 170 | 157 | 126 | x | ||||||
| WF | 155 | 183 | 195 | 211 | 162 | 164 | 152 | 183 | 266 | 274 | 176 | 173 | 193 | 129 | 175 | 224 | 120 | 123 | 133 | 156 | 50 | 180 | 158 | 148 | 141 | 155 | 196 | 189 | 157 | 141 | 97 | 201 | 179 | 135 | 149 | 186 | x | |||||
| WIST | 101 | 116 | 116 | 113 | 98 | 101 | 112 | 93 | 160 | 162 | 107 | 91 | 119 | 83 | 114 | 133 | 82 | 86 | 82 | 110 | 95 | 89 | 94 | 85 | 105 | 101 | 96 | 115 | 100 | 83 | 51 | 112 | 71 | 67 | 76 | 114 | 91 | x | ||||
| WKY | 169 | 208 | 198 | 213 | 164 | 188 | 168 | 208 | 264 | 276 | 208 | 181 | 205 | 128 | 170 | 228 | 180 | 210 | 165 | 229 | 206 | 200 | 220 | 195 | 189 | 197 | 208 | 205 | 194 | 196 | 148 | 115 | 196 | 189 | 183 | 216 | 203 | 101 | x | |||
| wild3 | 137 | 181 | 153 | 177 | 162 | 164 | 134 | 149 | 175 | 194 | 156 | 173 | 153 | 111 | 172 | 169 | 162 | 182 | 156 | 184 | 173 | 171 | 160 | 149 | 134 | 140 | 161 | 154 | 165 | 161 | 120 | 152 | 160 | 152 | 140 | 158 | 178 | 108 | 163 | x | ||
| wild2 | 197 | 233 | 207 | 223 | 182 | 213 | 194 | 187 | 256 | 268 | 203 | 221 | 211 | 181 | 227 | 218 | 204 | 243 | 180 | 234 | 220 | 213 | 210 | 203 | 177 | 198 | 219 | 202 | 214 | 210 | 163 | 190 | 208 | 205 | 208 | 213 | 210 | 141 | 210 | 52 | x | |
| wild1 | 334 | 414 | 368 | 406 | 329 | 405 | 339 | 372 | 520 | 551 | 369 | 400 | 395 | 315 | 392 | 397 | 386 | 446 | 338 | 415 | 410 | 413 | 391 | 403 | 331 | 352 | 377 | 371 | 387 | 373 | 321 | 334 | 375 | 388 | 372 | 404 | 410 | 280 | 357 | 134 | 157 | x |
The matrix is built from genotyping data of 960 polymorphisms in 36 strains and three wild individuals. Two inbred strains are represented by two substrains (BN and DA) and outbred SD is represented by two individuals from different stocks. Sets of polymorphisms, including a graphical representation, can be retrieved from [24].
Simulation experiment wild rat-based SNP discovery
To get insight in the benefits of using wild rats in SNP discovery studies, we simulated larger scale experiments based on the results obtained in the experiments described above. Shotgun sequencing of 814 Kbp resulted in the identification of 485 SNPs. For 408 of those, the wild rat allele was also represented in laboratory rat strains and hence of interest for research purposes. The maximum amount of SNPs that can be discovered by fully sequencing this single rat is calculated by multiplying the SNP frequency (408/814,440) with the rat genome size (2,48 Gbp), which is 1,252,911 SNPs. Since none of our shotgun reads were overlapping, we can calculate the relation between shotgun sequencing reads of the wild rat and the amount of SNPs that will be found by scaling up this methodology, assuming random distribution of 400 bp shotgun reads over the genome (Fig. 3a). One million shotgun reads of a single wild rat would already result in the discovery of 200,000 novel SNPs that are polymorphic in commonly used rat strains. This simulation indicates that a relatively small sequencing effort could potentially result in a vast expansion of the amount of genetic variation for the rat.
Figure 3.
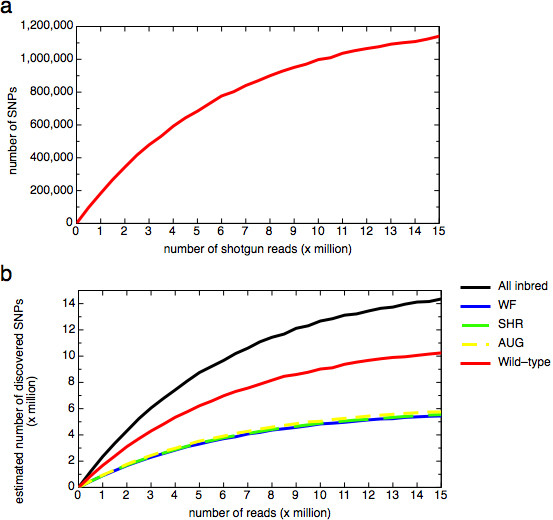
a) Simulation of wild rat-based SNP discovery experiment. Simulation is based on the discovery of 485 SNPs between wild 1 and BN in 814 Kbp of shotgun sequence. For 408 of those, the wild rat alleles is found back in one or more inbred strains. The relation between generation of randomly distributed 400 bp shotgun reads and estimated number of newly discovered SNPs is plotted. b) Simulation of SNP discovery experiment, using carefully selected (most polymorphic compared to BN) rat strains (SHR, AUG, and WF) or all rat strains, in comparison with wild rat-based SNP discovery. Simulation is based on 539, 304, 292, 287, and 754 SNPs for wild 1, AUG, SHR, WF, and all strains respectively, in 119 Kbp of genotyped sequence.
Because shotgun sequencing was only done in the wild rat 1, we cannot make a direct comparison between wild rat-based SNP discovery and SNP discovery based on rat strains separately. However, a similar simulation experiment can be performed by treating the genotyping resequencing as shotgun reads. For wild 1, this would result in the identification of 577 SNPs as compared to the BN genome sequence. For 539 of those, the wild rat allele is found back in one of the inbred strains. For the combination of three strains most polymorphic as compared to BN in this experiment, the latter number would be 304, 292, and 287 for AUG, SHR, and WF, respectively. Simulations based on these numbers show that it requires nearly two times as much shotgun sequencing in different inbred strains separately to discover the same amount of SNPs that can be found using the wild rat shotgun sequencing approach. It should be mentioned that parallel shotgun sequencing of all 36 inbred strains until saturation has the potential to yield 1.6 times as many SNPs as compared to the wild-derived approach (Fig. 3b). An advantage of using inbred strains for SNP discovery is that the genotype of the strain is immediately known. Nevertheless, reconfirmation of the SNP or genotyping of other strains of interest may be necessary anyway, minimizing the relevance of this advantage.
Discussion
An increase in the amount of documented genetic variation for the rat will be essential to allow for high-resolution genetic mapping of the many inherited traits that have now been described for a wide variety of rat inbred strains. In addition, insight into genetic variation between rat strains provides valuable information on genetic relationships between strains, which can be instrumental to dissect the genetic basis of phenotypic differences. The wild rat-based shotgun sequencing method described here provides an efficient approach to generate such a dense map of genetic variation. To be able to benefit from haplotype-based mapping approaches [25-28] a high marker density is needed to first reliably define haplotype blocks in strains of interest [29]. For the mouse, it has recently been announced that 15 inbred strains will be fully resequenced to achieve this goal [30]. With extreme dense genotype maps, it may even become possible to clone traits by haplotype-based in silico mapping [25], but to achieve this, it is estimated that complete sequences of over 50 strains are needed [29]. Although densities needed for these approaches are not reached, we do show here that wild rat-based SNP discovery is potentially much more effective than shotgun sequencing different inbred strains. We propose that the most effective SNP discovery strategy for the rat would be one based on shotgun sequencing of a single wild-derived sample and subsequent low-cost high-throughput genotyping of the resulting candidates in the laboratory strains of interest. Many other model organisms are currently undergoing full coverage sequencing and SNP discovery in these organisms will become increasingly important, especially for those organisms that are selectively bred for specific traits, such as cow and pig. Pilot experiments using for example wild-derived swine samples could be performed to test whether it is eligible to efficiently transfer the wild isolate-based SNP discovery strategy to other organisms.
Our results do provide insight in the genetic descent of the laboratory rat. It is generally accepted that current rat strains underwent two major genetic bottlenecks. First, they originate from a small founder population of domesticated wild rats and second, they were selectively inbred to obtain homogeneity [11]. The three Dutch wild rats used in this study are potentially relatively closely related as compared to wild rats from different parts of the world, but the genetic variation between them is mostly larger than or sporadically equal to any combination of three inbred strains, indeed suggesting the existence a common genetic bottleneck for laboratory strains. In addition, the laboratory rat does not show an extensive polymorphism rate in the MHC (major histocompatibilty complex) as compared to other species [31], like human, cattle etc. Cramer et al. has analyzed the MHC of wild rats and compared the data with those from inbred strains [32]. In line with our observation, there were not many new haplotypes.
We observed that wild rat genetic variation is to a large extent represented in the inbred strains, which is in sharp contrast to genetic variation in wild-derived mouse strains that is mostly unique [33]. Contrary to classical mouse inbred strains, where multiple subspecies contribute to the genetic make-up [13,34] and recent mouse strains, derived from different Mus species [35], laboratory rat strains are most likely descending from a single rat species, Rattus norvegicus [10].
An independent study using 42 microsatellites in German and Japanese wild-derived samples showed that the genetic profiles were quite divergent, partially owing to different geographic locations [36]. Our study involved only Dutch wild rats, suggesting that the inclusion of wild rats from different parts of the world could result in even more efficient SNP discovery, although it also remains to be demonstrated what proportion of the additional discovered alleles is present in the inbred strains and if a geographic bias for this exists.
When multiple SNPs are present per locus/amplicon, independent haplotypes can be discerned. The genetic variation identified here is mostly organized in a limited amount of haplotypes per locus (Table 4). Theoretically, an amplicon containing two or three SNPs can be represented by four and eight haplotypes, respectively, but in our dataset the vast majority of amplicons harboring multiple SNPs is represented by only two or three haplotypes (Table 4). Again, these observations suggest the existence of a common and small founding population with very limited haplotype diversity and/or a very narrow genetic bottleneck before inbred strain selection. The observed small genetic basis in a wide selection of laboratory rat strains does not mimic genetic variation in the human population and as a result, studies and pharmacological tests in rat models neglect potential modulatory effects caused by genetic variation. Although the use of F1 crosses and mosaic populations [37] could address this issue, our data suggests that wild-derived rats may be very useful to this end, since a large amount of all genetic variation present in a large selection of inbred strains, is already represented in a limited number of individuals. Therefore, it would be very interesting to investigate genetic variation in recently domesticated inbred [38] and outbred rats such as wild-type Groningen rats (WTG) [39]. Alternatively, careful selection of inbred strains based on genotyping data and subsequent random breeding may also expose the wild side of laboratory rats.
Table 4.
Haplotype analysis in 36 strains for all SNP-containing amplicons
| number of haplotypes | ||||||||||||
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| 2 SNPs | 46 | 57 | 8 | |||||||||
| 3 SNPs | 11 | 26 | 8 | 3 | 0 | 0 | 0 | |||||
| 4 SNPs | 4 | 11 | 5 | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| 5 SNPs | 1 | 3 | 3 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 6 SNPs | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 7 SNPs | 1 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 8 SNPs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 9 SNPs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| 10 SNPs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 11 SNPs | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| total | 64 | 97 | 27 | 9 | 5 | 1 | 0 | 0 | 0 | 0 | 1 | 204* |
*) Total number of amplicons that contains at least two SNPs. Amplicons containing no SNPs or only indels were excluded from this analysis. Amplicons containing 1 SNP are also excluded, since two-state SNPs always give rise to two haplotypes.
Conclusion
We describe a SNP discovery platform for the rat that is based on two steps. First, candidate SNPs are discovered by shotgun sequencing a wild rat, followed by genotyping laboratory strains of interest. We show that 84% of alleles in wild rats as compared to the sequenced Brown Norway rat genome are also represented in a set of 36 laboratory strains. Hence, the approach described here would be an efficient strategy for the discovery of novel informative SNPs in the laboratory rat. Inclusion of other wild samples, preferably from different locations in the world could result in an even more effective SNP discovery platform, as the three wild rats in our study, caught in relative close vicinity to each other, were already more polymorphic than the most polymorphic combination of carefully selected inbred strains. Based on the more than 34,000 genotyping datapoints obtained in this study, we postulate two things. First, laboratory rats originate from a single rat species, and inbred stains are relatively closely related with a limited number of haplotypes, reflecting known genetic bottlenecks in strain establishment. Second, wild rats have the potential to represent the degrees of genetic variation as present in the human population much more efficiently than a random selection of inbred strains. This makes them or wild-derived strains potentially well-suited for studying modulatory effects of genetic background variation on specific phenotypes, such as behavior or responses to drug treatment.
Methods
Genomic DNA isolation, shotgun library construction
Wild rat 1 (Rattus norvegicus) was caught in the canals of Utrecht and kindly provided by the Pest Control Service of the City of Utrecht (Utrecht, The Netherlands). Wild rat 2 was trapped in Gassel, a village located approximately 100 km south-east of Utrecht and was kindly provided by Tien Derks (Gassel, The Netherlands). Wild rat 3 was caught in a basement in Amsterdam, located 50 km north of Utrecht and kindly provided Romke Koch (Amsterdam, The Netherlands). Rat strains BN/Crl and Crl:Wistar (outbred) were obtained from Charles River The Netherlands. Liver samples of commonly used rat strains ACI/Ztm, BDE/Ztm, BDII/Ztm, BDIX/Ztm, BDV/Ztm, BH/Ztm, BN/Ztm, BS/Ztm, DA/Ztm, E3/Ztm, F344/Ztm, LE/Ztm, LEW/Ztm, LOU/CZtm, MNS/Ztm, MWF/Ztm, NAR/Ztm, OM/Ztm, PAR/Ztm, R33/Ztm, WC/Ztm, WF/Ztm, WKY/Ztm were provided by D.W. (Hannover Medical School, Germany) and liver samples of strains AO/OlaHsd, AUG/OlaHsd, BUF/SimRijHsd, COP/Hsd, DA/OlaHsd, LUDW/OlaHsd, PVG/OlaHsd, RP/AEurRijHsd, SHR/NHsd, SR/JrHsd, SS/JrHsd, WAG/RijHsd and 2 individuals of Hsd:SD (outbred) were kindly provided by Harlan (Horst, The Netherlands). Samples were lysed overnight in 20 ml lysis buffer, containing 100 mM Tris (pH 8.5), 200 mM of NaCl, 0.2% of SDS, 5 mM of EDTA, and 100 μg/ml of freshly added Proteinase K at 55°C under continuous rotation. Tissue debris was spinned down for 20 min at 10,000 × g and supernatant was transferred to a fresh tube. DNA was purified by phenol-chloroform extraction and precipitated by adding an equal volume of isopropanol, mixing and centrifugation for 20 min, 10,000 × g at 4°C. The supernatant was removed by gently inverting the tube and the pellets were washed with 70% ethanol and dissolved in 1000 μl water. The concentration was measured by Optical Densitometry at 260 nm.
Wild rat-derived genomic library construction and shotgun sequencing
Sheared wild rat-derived genomic DNA of approximately 1–2 Kbp in size was cloned into the SmaI-site of pUC19. Fractions of the glycerol stock of the transformed library (E. coli DH10B) were plated on LB-plates containing 50 μg/ml ampicilin, 200 μg/ml IPTG, and 0.01% X-gal for standard blue/white screening on inserts. White colonies were picked in 20 μl water. Lysis occurred at 95°C for 10 min. 5 μl of 5× diluted lysate was used for the PCR reaction. For PCR, universal M13 primers were used, namely M13F: TGTAAAACGACGGCCAGT, M13R: AGGAAACAGCTATGACCAT. PCR, sequencing and cycling conditions were similar as for strain genotyping, described below. Sequencing was performed using universal M13 primers.
PCR conditions for strain genotyping
PCR was carried out using a touchdown thermocycling program (92°C for 60 sec; 12 cycles of 92°C for 20 sec, 65°C for 20 sec with a decrement of 0.6°C per cycle, 72°C for 30 sec; followed by 20 cycles of 92°C for 20 sec, 58°C for 20 sec and 72°C for 30 sec; 72°C for 180 sec; GeneAmp9700, Applied Biosystems) and contained 30–50 ng genomic DNA, 0.2 μM of each forward primer and 0.2 μM of each reverse primer, 400 μM of each dNTP, 25 mM Tricine, 7.0% Glycerol (w/v), 1.6% DMSO (w/v), 2 mM MgCl2, 85 mM Ammonium acetate pH 8.7 and 0.2 U Taq Polymerase in a total volume of 10 μl.
Sequencing reactions, purification, and analysis
PCR products were diluted with 25 μl water and 1 μl was directly used as template for the sequencing reactions. Sequencing reactions, containing 0.25 μl BigDYE (v3.1; Applied Biosystems, Foster City, CA, USA), 3.75 μl 2.5× dilution buffer (Applied Biosystems) and 0.4 μM universal M13 primer in a total volume of 10 μl, were performed using cycling conditions recommended by the manufacturer (40 cycles of 92°C for 10 sec, 50°C for 5 sec and 60°C for 120 sec). Of sequencing products, 5 μl was purified by ethanol precipitation in the presence of 40 mM sodium-acetate and analyzed on 96-capillary 3730XL DNA analyzers (Applied Biosystems), using the standard RapidSeq protocol. Sequences were analyzed for presence of heterozygous mutations using PolyPhred [40], followed by manual inspection of the polymorphic positions.
Automation
All PCR and sequencing reactions were set up on a Tecan Genesis RSP200 liquid handling workstation, with a robotic and an 8-channel pipetting arm, an integrated 96-channel pipetting head (TEMO96, Tecan), and four integrated dual-384 well PCR blocks (Applied Biosystems).
Mapping of shotgun reads and SNP discovery
Shotgun reads were assigned to positions in the RGSC 3.1 rat genome assembly using blat search [15]. Shotgun reads that complied with our mapping criteria, namely those having at least 80 identical bp for the best hit and no more than 60 identical bp for second blat hit were retained for further analysis. Blast nucleotide sequence alignments between shotgun read and corresponding genomic segment were used for discovery of single base variations (including single base indels). A site was treated as polymorphic only in the case when it has identical 5'- and 3'-flanks of at least 5 bp. A custom designed web-application was employed for manual chromatogram inspection and confirmation of a correct shotgun base-call for every polymorphic SNP locus. Primer design for resequencing was performed using a local web-interface [41] to the PRIMER3 program [42].
Simulation model for wild rat-based SNP discovery
To estimate the number of SNPs to be discovered by the wild rat resequencing approach we performed computer simulations using the observed sample-specific polymorphism frequencies and the rat genome size of 2.48 Gbp as an input. We used a Monte-Carlo method for the placement of N 400-bp shotgun reads to the genome and calculated the total size of genome covered by N shotgun reads. To obtain a conservative estimate by assuming low heterozygosity in wild-derived strain the estimate of number of SNPs is given by product of covered genome size and polymorphism rate.
Authors' contributions
BMGS contributed to the production of the results, supervised the ongoing of the study, and drafted the manuscript. VG contributed to the computational support of the results, and contributed to the writing of the manuscript. DZ contributed to the production of sequencing reads and initial analysis of the results. DW contributed to the preparation of samples for the study and revised the manuscript. HJH participated in the interpretation of the results and revision of the manuscript. EC outlined and supervised the study, and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Genotyping details; Detailed genotyping information, including allele information for the inbred and wild rat strains
Acknowledgments
Acknowledgements
We thank Harlan (Horst – Netherlands), the Pest Control Service of the City of Utrecht (Utrecht, The Netherlands), Romke Koch (Amsterdam, The Netherlands), and Tien Derks (Gassel, The Netherlands) for kindly providing rat tissue samples. This work was supported by the Dutch Ministry of Economic Affairs through the Innovation Oriented Research Program on Genomics.
Contributor Information
Bart MG Smits, Email: smits@niob.knaw.nl.
Victor Guryev, Email: guryev@niob.knaw.nl.
Dimphy Zeegers, Email: dimphy@niob.knaw.nl.
Dirk Wedekind, Email: wedekind.dirk@mh-hannover.de.
Hans J Hedrich, Email: hedrich.hans@mh-hannover.de.
Edwin Cuppen, Email: ecuppen@niob.knaw.nl.
References
- Kristensen VN, Kelefiotis D, Kristensen T, Borresen-Dale AL. High-throughput methods for detection of genetic variation. Biotechniques. 2001;30:318–322. doi: 10.2144/01302tt01. [DOI] [PubMed] [Google Scholar]
- Sellick GS, Longman C, Tolmie J, Newbury-Ecob R, Geenhalgh L, Hughes S, Whiteford M, Garrett C, Houlston RS. Genomewide linkage searches for Mendelian disease loci can be efficiently conducted using high-density SNP genotyping arrays. Nucleic Acids Res. 2004;32:e164. doi: 10.1093/nar/gnh163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Greenhouse DD, Festing MFW, Hasan S, Cohen AL. Catalogue of Inbred Strains of Rats. In: Hedrich HJ, editor. Genetic Monitoring of Inbred Strains of Rats. Stuttgard , Gustav Fisher; 1990. pp. 410–480. [Google Scholar]
- Hedrich HJ. The Laboratory Rat. Academic Press; 2005. Taxonomy and stocks and strains; pp. 71–91, in press. [Google Scholar]
- NCBI dbSNP http://www.ncbi.nlm.nih.gov/SNP/
- Zimdahl H, Nyakatura G, Brandt P, Schulz H, Hummel O, Fartmann B, Brett D, Droege M, Monti J, Lee YA, Sun Y, Zhao S, Winter EE, Ponting CP, Chen Y, Kasprzyk A, Birney E, Ganten D, Hubner N. A SNP map of the rat genome generated from cDNA sequences. Science. 2004;303:807. doi: 10.1126/science.1092427. [DOI] [PubMed] [Google Scholar]
- Smits BM, van Zutphen BF, Plasterk RH, Cuppen E. Genetic variation in coding regions between and within commonly used inbred rat strains. Genome Res. 2004;14:1285–1290. doi: 10.1101/gr.2155004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, Berezikov E, Malik R, Plasterk RH, Cuppen E. Single nucleotide polymorphisms associated with rat expressed sequences. Genome Res. 2004;14:1438–1443. doi: 10.1101/gr.2154304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JR. Historical Foundations. In: Baker HJ, Lindsey JR, Weisbroth SH, editor. The Laboratory Rat. Vol. 1. New York , Academic Press; 1979. pp. 1–36. [Google Scholar]
- Hedrich HJ. History, Strains and Models. In: Krinke GJ, editor. The laboratory Rat. London , Academic Press; 2000. [Google Scholar]
- Koch LG, Britton SL. Strains. In: Whishaw IQ, Kolb B, editor. The Behavior of the Laboratory Rat. Oxford, UK , Oxford University Press; 2005. [Google Scholar]
- Ideraabdullah FY, de la Casa-Esperon E, Bell TA, Detwiler DA, Magnuson T, Sapienza C, de Villena FP. Genetic and haplotype diversity among wild-derived mouse inbred strains. Genome Res. 2004;14:1880–1887. doi: 10.1101/gr.2519704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Fullerton J, Miller S, Keays DA, Brady S, Bhomra A, Jefferson A, Volpi E, Copley RR, Flint J, Mott R. Unexpected complexity in the haplotypes of commonly used inbred strains of laboratory mice. Proc Natl Acad Sci U S A. 2004;101:9734–9739. doi: 10.1073/pnas.0401189101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzian F. Phylogenetics of the laboratory rat Rattus norvegicus. Genome Res. 1997;7:262–267. doi: 10.1101/gr.7.3.262. [DOI] [PubMed] [Google Scholar]
- Thomas MA, Chen CF, Jensen-Seaman MI, Tonellato PJ, Twigger SN. Phylogenetics of rat inbred strains. Mamm Genome. 2003;14:61–64. doi: 10.1007/s00335-002-2204-5. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Homepage Fluxus Engineering http://fluxus-engineering.com
- Rat Genome Database (RGD) strain information http://rgd.mcw.edu/strains
- Rapp JP. Genetic analysis of inherited hypertension in the rat. Physiol Rev. 2000;80:135–172. doi: 10.1152/physrev.2000.80.1.135. [DOI] [PubMed] [Google Scholar]
- Kwitek AE, Gullings-Handley J, Yu J, Carlos DC, Orlebeke K, Nie J, Eckert J, Lemke A, Andrae JW, Bromberg S, al. High-density rat radiation hybrid maps containing over 24,000 SSLPs, genes, and ESTs provide a direct link to the rat genome sequence. Genome Res. 2004;14:750–757. doi: 10.1101/gr.1968704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen RG, Kwitek-Black AE, Glenn C, Gullings-Handley J, Van Etten W, Atkinson OS, Appel D, Twigger S, Muir M, Mull T, al. A high-density integrated genetic linkage and radiation hybrid map of the laboratory rat. Genome Res. 1999;9:AP1–8. [PubMed] [Google Scholar]
- CASCAD SNPview http://cascad.niob.knaw.nl/snpview
- Grupe A, Germer S, Usuka J, Aud D, Belknap JK, Klein RF, Ahluwalia MK, Higuchi R, Peltz G. In silico mapping of complex disease-related traits in mice. Science. 2001;292:1915–1918. doi: 10.1126/science.1058889. [DOI] [PubMed] [Google Scholar]
- Liao G, Wang J, Guo J, Allard J, Cheng J, Ng A, Shafer S, Puech A, McPherson JD, Foernzler D, Peltz G, Usuka J. In silico genetics: identification of a functional element regulating H2-Ealpha gene expression. Science. 2004;306:690–695. doi: 10.1126/science.1100636. [DOI] [PubMed] [Google Scholar]
- Pletcher MT, McClurg P, Batalov S, Su AI, Barnes SW, Lagler E, Korstanje R, Wang X, Nusskern D, Bogue MA, Mural RJ, Paigen B, Wiltshire T. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2004;2:e393. doi: 10.1371/journal.pbio.0020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Korstanje R, Higgins D, Paigen B. Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res. 2004;14:1767–1772. doi: 10.1101/gr.2668204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Pearson H. Mouse sequencing plan aims to boost models. Nature. 2004;432:5. doi: 10.1038/432005a. [DOI] [PubMed] [Google Scholar]
- Gunther E, Walter L. The major histocompatibility complex of the rat (Rattus norvegicus) Immunogenetics. 2001;53:520–542. doi: 10.1007/s002510100361. [DOI] [PubMed] [Google Scholar]
- Cramer DV, Chakravarti A, Arenas O, Humprieres J, Mowery PA. Genetic diversity within and between natural populations of Rattus norvegicus. J Hered. 1988;79:319–324. doi: 10.1093/oxfordjournals.jhered.a110522. [DOI] [PubMed] [Google Scholar]
- Campino S, Behrschmidt C, Bagot S, Guenet JL, Cazenave PA, Holmberg D, Penha-Goncalves C. Unique genetic variation revealed by a microsatellite polymorphism survey in ten wild-derived inbred strains. Genomics. 2002;79:618–620. doi: 10.1006/geno.2002.6570. [DOI] [PubMed] [Google Scholar]
- Abe K, Noguchi H, Tagawa K, Yuzuriha M, Toyoda A, Kojima T, Ezawa K, Saitou N, Hattori M, Sakaki Y, Moriwaki K, Shiroishi T. Contribution of Asian mouse subspecies Mus musculus molossinus to genomic constitution of strain C57BL/6J, as defined by BAC-end sequence-SNP analysis. Genome Res. 2004;14:2439–2447. doi: 10.1101/gr.2899304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenet JL, Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/S0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- Voigt B, Kitada K, Kloting I, Serikawa T. Genetic comparison between laboratory rats and Japanese and German wild rats. Mamm Genome. 2000;11:789–790. doi: 10.1007/s003350010137. [DOI] [PubMed] [Google Scholar]
- Cholnoky E, Fischer J, Varga M, Gyorffy G. Aspects of genetically defined populations in toxicity testing. II. Genotypic differences in sensitivity to a toxic dextran preparation. Z Versuchstierkd. 1974;16:43–48. [PubMed] [Google Scholar]
- Ohno K, Niwa Y, Kato S, Kondo K, Oda S, Inouye M, Yamamura H. Establishment of new inbred strains derived from Japanese wild rats (Rattus norvegicus) Jikken Dobutsu. 1994;43:251–255. doi: 10.1538/expanim1978.43.2_251. [DOI] [PubMed] [Google Scholar]
- de Boer SF, Lesourd M, Mocaer E, Koolhaas JM. Selective antiaggressive effects of alnespirone in resident-intruder test are mediated via 5-hydroxytryptamine1A receptors: A comparative pharmacological study with 8-hydroxy-2-dipropylaminotetralin, ipsapirone, buspirone, eltoprazine, and WAY-100635. J Pharmacol Exp Ther. 1999;288:1125–1133. [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Local PRIMER3 interface http://primers.niob.knaw.nl
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genotyping details; Detailed genotyping information, including allele information for the inbred and wild rat strains
Data Availability Statement
The use of genetic markers for mapping traits in rat strains has been exploited for long time already. Current marker sets in rats are mostly limited to microsatellites [22,23], which are not abundantly available and are commonly detected in a more laborious way than SNPs. In this study, we have determined a total of about 35,000 genotypes (about 960 loci in 36 inbred strains), out of which the vast majority are SNPs. This data is accessible via a versatile webtool [24]. Pairs of strains of interest can be selected and explored on presence of verified genetic variation. Besides a graphical representation of the location of the SNPs on a genome map, primer sequences that were successfully used in our experiments are also provided. In a pairwise comparison matrix (Table 3), we plotted the absolute number of polymorphic positions for each of the (sub-)strains or individuals used. Interestingly, for some strains different alleles are observed in substrains (e.g. BN/Crl differs from BN/Ztm at 4 positions), in line with previous observations [8].
Table 3.
Absolute number of polymorphic positions between strains in a pairwise comparison.
| ACI | AO | AUG | BDE | BDII | BDIX | BDV | BH | BN | BN2 | BS | BUF | COP | DA | DA2 | E3 | F344 | LEW | LE | LOU | LUDW | MWF | MNS | NAR | OM | PAR | PVG | R33 | RP | SD | SD2 | SHR | SR | SS | WAG | WC | WF | WIST | WKY | wild3 | wild2 | wild1 | |
| ACI | x | |||||||||||||||||||||||||||||||||||||||||
| AO | 131 | x | ||||||||||||||||||||||||||||||||||||||||
| AUG | 111 | 167 | x | |||||||||||||||||||||||||||||||||||||||
| BDE | 145 | 178 | 158 | x | ||||||||||||||||||||||||||||||||||||||
| BDII | 130 | 166 | 143 | 148 | x | |||||||||||||||||||||||||||||||||||||
| BDIX | 129 | 196 | 191 | 159 | 115 | x | ||||||||||||||||||||||||||||||||||||
| BDV | 109 | 131 | 145 | 137 | 78 | 142 | x | |||||||||||||||||||||||||||||||||||
| BH | 145 | 189 | 192 | 212 | 156 | 177 | 167 | x | ||||||||||||||||||||||||||||||||||
| BN | 225 | 263 | 279 | 266 | 206 | 258 | 244 | 274 | x | |||||||||||||||||||||||||||||||||
| BN2 | 227 | 270 | 285 | 268 | 222 | 271 | 251 | 274 | 4 | x | ||||||||||||||||||||||||||||||||
| BS | 159 | 167 | 163 | 178 | 166 | 185 | 143 | 180 | 246 | 251 | x | |||||||||||||||||||||||||||||||
| BUF | 148 | 158 | 181 | 171 | 168 | 190 | 151 | 166 | 250 | 263 | 158 | x | ||||||||||||||||||||||||||||||
| COP | 84 | 194 | 187 | 166 | 155 | 147 | 155 | 191 | 262 | 275 | 203 | 192 | x | |||||||||||||||||||||||||||||
| DA | 62 | 107 | 116 | 134 | 115 | 130 | 117 | 130 | 210 | 216 | 150 | 126 | 116 | x | ||||||||||||||||||||||||||||
| DA2 | 76 | 141 | 144 | 157 | 149 | 167 | 127 | 176 | 261 | 269 | 193 | 167 | 151 | 2 | x | |||||||||||||||||||||||||||
| E3 | 136 | 189 | 170 | 86 | 154 | 180 | 144 | 204 | 244 | 253 | 172 | 182 | 171 | 148 | 180 | x | ||||||||||||||||||||||||||
| F344 | 132 | 165 | 178 | 178 | 152 | 150 | 138 | 144 | 170 | 176 | 141 | 156 | 166 | 121 | 165 | 168 | x | |||||||||||||||||||||||||
| LEW | 156 | 178 | 213 | 197 | 160 | 163 | 141 | 169 | 213 | 222 | 166 | 160 | 191 | 133 | 181 | 186 | 16 | x | ||||||||||||||||||||||||
| LE | 131 | 144 | 136 | 155 | 148 | 152 | 122 | 142 | 221 | 224 | 157 | 127 | 147 | 110 | 140 | 167 | 142 | 149 | x | |||||||||||||||||||||||
| LOU | 145 | 146 | 165 | 191 | 153 | 203 | 120 | 178 | 242 | 250 | 138 | 171 | 212 | 111 | 142 | 192 | 126 | 150 | 149 | x | ||||||||||||||||||||||
| LUDW | 153 | 175 | 186 | 198 | 161 | 169 | 153 | 195 | 252 | 263 | 183 | 177 | 189 | 123 | 163 | 215 | 115 | 125 | 133 | 161 | x | |||||||||||||||||||||
| MWF | 148 | 147 | 172 | 166 | 136 | 166 | 111 | 167 | 209 | 222 | 135 | 148 | 185 | 133 | 164 | 158 | 115 | 134 | 148 | 136 | 163 | x | ||||||||||||||||||||
| MNS | 151 | 167 | 178 | 173 | 158 | 186 | 122 | 169 | 239 | 250 | 155 | 194 | 210 | 128 | 176 | 166 | 123 | 132 | 156 | 141 | 159 | 137 | x | |||||||||||||||||||
| NAR | 147 | 169 | 184 | 212 | 168 | 177 | 145 | 177 | 233 | 249 | 146 | 155 | 197 | 134 | 193 | 188 | 137 | 166 | 122 | 170 | 153 | 164 | 151 | x | ||||||||||||||||||
| OM | 127 | 161 | 153 | 170 | 120 | 158 | 143 | 150 | 216 | 222 | 154 | 138 | 176 | 125 | 147 | 183 | 125 | 156 | 127 | 156 | 139 | 143 | 144 | 147 | x | |||||||||||||||||
| PAR | 140 | 182 | 168 | 166 | 149 | 158 | 133 | 175 | 225 | 227 | 155 | 138 | 167 | 136 | 159 | 169 | 128 | 136 | 127 | 151 | 159 | 133 | 160 | 150 | 162 | x | ||||||||||||||||
| PVG | 95 | 164 | 152 | 153 | 153 | 181 | 129 | 184 | 252 | 263 | 170 | 151 | 164 | 110 | 150 | 148 | 142 | 175 | 145 | 143 | 196 | 146 | 161 | 172 | 160 | 147 | x | |||||||||||||||
| R33 | 155 | 198 | 183 | 213 | 169 | 198 | 186 | 177 | 261 | 257 | 173 | 210 | 204 | 142 | 184 | 223 | 183 | 209 | 164 | 185 | 196 | 191 | 187 | 182 | 159 | 173 | 189 | x | ||||||||||||||
| RP | 146 | 159 | 171 | 171 | 132 | 164 | 109 | 186 | 216 | 230 | 113 | 161 | 176 | 138 | 175 | 153 | 119 | 141 | 134 | 141 | 166 | 108 | 157 | 136 | 147 | 132 | 139 | 166 | x | |||||||||||||
| SD | 121 | 154 | 156 | 177 | 153 | 160 | 129 | 118 | 233 | 247 | 149 | 134 | 174 | 100 | 148 | 184 | 131 | 138 | 135 | 125 | 145 | 144 | 141 | 149 | 103 | 133 | 130 | 147 | 138 | x | ||||||||||||
| SD2 | 95 | 116 | 134 | 150 | 117 | 130 | 122 | 85 | 219 | 220 | 117 | 96 | 145 | 104 | 115 | 168 | 109 | 115 | 92 | 99 | 110 | 118 | 121 | 107 | 90 | 121 | 109 | 126 | 109 | 16 | x | |||||||||||
| SHR | 159 | 212 | 166 | 186 | 179 | 175 | 168 | 204 | 264 | 275 | 188 | 178 | 180 | 135 | 189 | 205 | 156 | 176 | 173 | 202 | 188 | 180 | 200 | 184 | 182 | 177 | 192 | 207 | 187 | 185 | 139 | x | ||||||||||
| SR | 129 | 171 | 172 | 170 | 160 | 161 | 147 | 138 | 235 | 244 | 163 | 131 | 170 | 120 | 172 | 174 | 146 | 169 | 156 | 157 | 175 | 150 | 160 | 164 | 136 | 117 | 134 | 166 | 160 | 60 | 64 | 184 | x | |||||||||
| SS | 114 | 145 | 167 | 183 | 142 | 150 | 139 | 111 | 249 | 253 | 149 | 152 | 175 | 117 | 161 | 186 | 136 | 161 | 121 | 134 | 156 | 146 | 153 | 128 | 130 | 143 | 145 | 145 | 144 | 69 | 46 | 191 | 83 | x | ||||||||
| WAG | 120 | 105 | 156 | 147 | 143 | 158 | 115 | 160 | 197 | 200 | 96 | 125 | 164 | 94 | 136 | 151 | 110 | 132 | 127 | 96 | 145 | 120 | 121 | 129 | 140 | 126 | 128 | 154 | 108 | 118 | 84 | 181 | 119 | 110 | x | |||||||
| WC | 140 | 164 | 158 | 155 | 129 | 180 | 92 | 188 | 214 | 232 | 151 | 177 | 177 | 125 | 158 | 157 | 131 | 160 | 156 | 145 | 171 | 87 | 157 | 179 | 152 | 158 | 138 | 213 | 126 | 153 | 122 | 195 | 170 | 157 | 126 | x | ||||||
| WF | 155 | 183 | 195 | 211 | 162 | 164 | 152 | 183 | 266 | 274 | 176 | 173 | 193 | 129 | 175 | 224 | 120 | 123 | 133 | 156 | 50 | 180 | 158 | 148 | 141 | 155 | 196 | 189 | 157 | 141 | 97 | 201 | 179 | 135 | 149 | 186 | x | |||||
| WIST | 101 | 116 | 116 | 113 | 98 | 101 | 112 | 93 | 160 | 162 | 107 | 91 | 119 | 83 | 114 | 133 | 82 | 86 | 82 | 110 | 95 | 89 | 94 | 85 | 105 | 101 | 96 | 115 | 100 | 83 | 51 | 112 | 71 | 67 | 76 | 114 | 91 | x | ||||
| WKY | 169 | 208 | 198 | 213 | 164 | 188 | 168 | 208 | 264 | 276 | 208 | 181 | 205 | 128 | 170 | 228 | 180 | 210 | 165 | 229 | 206 | 200 | 220 | 195 | 189 | 197 | 208 | 205 | 194 | 196 | 148 | 115 | 196 | 189 | 183 | 216 | 203 | 101 | x | |||
| wild3 | 137 | 181 | 153 | 177 | 162 | 164 | 134 | 149 | 175 | 194 | 156 | 173 | 153 | 111 | 172 | 169 | 162 | 182 | 156 | 184 | 173 | 171 | 160 | 149 | 134 | 140 | 161 | 154 | 165 | 161 | 120 | 152 | 160 | 152 | 140 | 158 | 178 | 108 | 163 | x | ||
| wild2 | 197 | 233 | 207 | 223 | 182 | 213 | 194 | 187 | 256 | 268 | 203 | 221 | 211 | 181 | 227 | 218 | 204 | 243 | 180 | 234 | 220 | 213 | 210 | 203 | 177 | 198 | 219 | 202 | 214 | 210 | 163 | 190 | 208 | 205 | 208 | 213 | 210 | 141 | 210 | 52 | x | |
| wild1 | 334 | 414 | 368 | 406 | 329 | 405 | 339 | 372 | 520 | 551 | 369 | 400 | 395 | 315 | 392 | 397 | 386 | 446 | 338 | 415 | 410 | 413 | 391 | 403 | 331 | 352 | 377 | 371 | 387 | 373 | 321 | 334 | 375 | 388 | 372 | 404 | 410 | 280 | 357 | 134 | 157 | x |
The matrix is built from genotyping data of 960 polymorphisms in 36 strains and three wild individuals. Two inbred strains are represented by two substrains (BN and DA) and outbred SD is represented by two individuals from different stocks. Sets of polymorphisms, including a graphical representation, can be retrieved from [24].