

Abstract
Mesenchymal stem cells have the potential to differentiate into various cell lineages, including adipocytes and osteoblasts. The induction of adipocyte differentiation by glucocorticoids (GCs) not only causes the accumulation of fat cells in bone marrow, but also depletes the supply of osteoblasts for new bone formation, thus leading to osteoporosis. We have shown that a GC-induced leucine-zipper protein (GILZ) antagonizes adipocyte differentiation. GILZ binds to a tandem repeat of CCAAT/enhancer-binding protein (C/EBP) binding sites in the promoter of the gene encoding peroxisome-proliferator-activated receptor-γ2 (PPAR-γ2), and inhibits its transcription as a sequence-specific transcriptional repressor. We have also shown that ectopic expression of GILZ blocks GC-induced adipocyte differentiation. Furthermore, adipogenic marker genes (for example, those encoding PPAR-γ2, C/EBP-α, lipoprotein lipase and adipsin) are also inhibited by GILZ. Our results reveal a novel GC antagonistic mechanism that has potential therapeutic applications for the inhibition of GC-induced adipocyte differentiation.
Introduction
The steroid hormone glucocorticoids (GCs) have a crucial function in regulating the transcription of several genes. These hormones are effective anti-inflammatory and immunosuppressive agents, and are widely used for the control of chronic lung disease, rheumatoid arthritis and asthma, and for preventing transplant rejection (Manolagas & Weinstein, 1999; Nishimura & Ikuyama, 2000; Lane, 2001). However, long-term use of GCs adversely affects body structure and function. Excess exposure increases osteoclastogenesis, inhibits osteoblastogenesis and promotes osteoblast and osteocyte apoptosis. These pathological conditions ultimately result in osteoporosis and bone fractures in as many as 50% of affected individuals (Reid, 2000; Toth & Tulassay, 2000; Walsh et al., 2001). Another detrimental effect of GCs on bone metabolism is the accumulation of adipocytes in bone marrow. Hypertrophy and hyperplasia of fat cells in bone marrow result in increased blood pressure and collapse of the sinusoids in the femoral head. This ultimately leads to increased osteonecrosis. Paradoxically, GCs are required for in vitro bone nodule formation and mineralization, as well as bone marrow mesenchymal stem-cell proliferation and osteogenic differentiation (Bellows et al., 1987; Shalhoub et al., 1992; Lian & Stein, 1993; Jaiswal et al., 1997; Bellows et al., 1998). The molecular mechanisms governing the apparently diametric actions of GCs are not clear.
GCs induce adipogenesis by activating the peroxisome-proliferator-activated receptor-γ2 (PPAR-γ2). We have shown that GC-induced adipogenesis of human bone marrow stromal stem cells is a cascade reaction, in which GC induces the expression of CCAAT/enhancer-binding protein-δ (C/EBP-δ). C/EBP-δ then binds to the PPAR-γ2 promoter and transactivates its expression, after which PPAR-γ2 initiates the adipocyte differentiation programme (Shi et al., 2000).
Here, we have identified a GC-induced leucine-zipper protein (GILZ), which specifically binds to a tandem repeat of C/EBP sites in the PPAR-γ2 promoter. GILZ was first identified as a transcription factor involved in the regulation of T-cell apoptosis (D'Adamio et al., 1997). We show that GILZ functions as a sequencespecific transcriptional repressor and antagonizes GC-induced adipocyte differentiation.
RESULTS
We have previously shown that GC-induced PPAR-γ2 gene expression is mediated by C/EBP-δ (Shi et al., 2000). We observed that a novel GC-induced nuclear protein binds specifically to a 40-bp DNA fragment, containing a tandem repeat of the C/EBP binding sites, in the −311 to −351 region of the PPAR-γ2 promoter (Fig. 1A; Fig. 1B, lane 3). This nuclear protein is a de novo product, as it is not present in cells that are not treated with dexamethasone (Dex) (Fig. 1B, lane 2), or in cells treated simultaneously with Dex and cycloheximide, which is a protein synthesis inhibitor (Fig. 1B, lane 4). Each of the single C/EBP sites was also examined for nuclear protein binding activity in a gelshift assay. Interestingly, the novel nuclear protein did not bind to either of the C/EBP elements (Fig. 1B, lanes 10 and 13) or to a consensus C/EBP binding site (Fig. 1B, lane 7). Lanes 1, 5, 8 and 11 (Fig. 1B) contained no protein. Lanes 2, 6, 9 and 12 (Fig. 1B) contained nuclear proteins from untreated cells.
Figure 1.
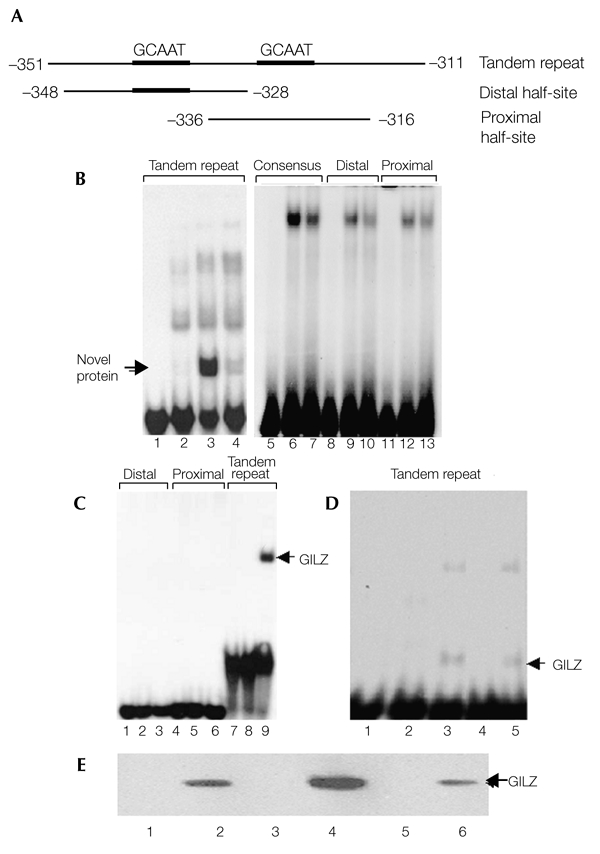
The novel glucocorticoid-induced nuclear protein that binds to the peroxisome-proliferator-activated receptor-γ2 promoter is the glucocorticoid-induced leucine-zipper protein, GILZ. (A) Representation of peroxisome-proliferator-activated receptor-γ2 (PPAR-γ2) promoter fragments used as probes. (B) Electrophoretic mobility-shift assays, showing the binding of a glucocorticoid (GC)-induced nuclear protein to the tandem repeat of CCAAT/enhancer-binding protein (C/EBP) binding sites in the PPAR-γ2 promoter. Nuclear proteins from untreated (lanes 2, 6, 9 and 12), dexamethasone (Dex)-treated (lanes 3, 7, 10 and 13) or Dex/cycloheximide-treated (lane 4) C3H10T1/2 cells were incubated with the labelled probes shown in (A) and a high-affinity consensus C/EBP binding site from the C/EBP-α promoter (lanes 5–7). Lanes 1, 5, 8 and 11 contained no protein. (C) Electrophoretic mobility-shift assays, showing the specific binding of the glutathione-S-transferase (GST)–GILZ fusion protein to the tandem repeats of C/EBP binding sites. Lanes 1, 4 and 7 contained no protein. Lanes 2, 5 and 8 contained 5 μg of GST. Lanes 3, 6 and 9 contained 5 μg of the GST–GILZ fusion protein. (D) Overexpressed GILZ binds to a tandem-repeat probe. COS1 cells were transfected with empty pcDNA3 or with the pcDNA3–GILZ expression plasmid, and whole-cell lysates were incubated with a labelled tandem-repeat probe and used in electrophoretic mobility-shift assays. Lane 1, no protein. Lanes 2 and 3, nuclear protein from untreated and Dex-treated C3H10T1/2 cells, respectively. Lanes 4 and 5, whole-cell lysates from pcDNA3 and pcDNA3–GILZ-transfected COS1 cells. (E) Western blot analysis using an anti-GILZ antibody to detect proteins from the following samples: cell lysates from C3H10T1/2 cells, either untreated (lane 1) or treated with Dex (lane 2); COS1 cell lysates transfected with an empty pcDNA3 vector (lane 3) or with the pcDNA3–GILZ expression plasmid (lane 4); and nuclear proteins from C3H10T1/2 cells, either untreated (lane 5) or treated with Dex (lane 6).
We then attempted to identify and clone this GC-induced DNA-binding protein. The results of the gel-shift assays suggest that the molecular weight of the DNA-binding protein is small. Among known GC-induced proteins, GILZ, which has a molecular weight of 14 kDa, was recently identified as a transcription factor involved in the regulation of T-cell apoptosis. To determine if GILZ is the nuclear protein that binds to the C/EBP tandem DNA-binding site, we made a glutathione-S-transferase (GST)–GILZ fusion protein expression plasmid. The fusion protein was produced in bacteria and purified by affinity chromatography. The purified GST–GILZ protein was incubated with three DNA probes, in the same way as for the gel-shift assays described above. GST–GILZ has a binding pattern identical to that of the previously identified novel nuclear protein (Fig. 1C). GILZ binds specifically to the 40-bp tandem repeat of the C/EBP binding sites (Fig. 1C, lane 9), but not to the distal (Fig. 1C, lane 3) or proximal (Fig. 1C, lane 6) halfsites, as does the unknown nuclear protein. To determine whether the GC-induced nuclear protein is GILZ, we overexpressed GILZ in COS1 cells and carried out a gel-shift assay with whole-cell lysates. A shifted band of the expected molecular mass was observed in the lysate from GILZ-overexpressing cells (Fig. 1D; compare lane 5 to lane 3, containing the nuclear protein from Dex-treated cells), but not in the lysate from control COS1 cells (Fig. 1D, lane 4) or from untreated C3H10T1/2 cells (Fig. 1D, lane 2). These results suggest that GILZ is the novel, GC-induced DNA-binding protein. To confirm this, we carried out western blot analyses using an anti-GILZ antibody on whole-cell lysates from the following: C3H10T1/2 cells, either untreated or treated with Dex (Fig. 1E, lanes 1 and 2, respectively); COS1 cells transfected with either an empty pcDNA3 plasmid (Fig. 1E, lane 3) or with a GILZ plasmid (Fig. 1E, lane 4); and nuclear extracts from C3H10T1/2 cells, either untreated or treated with Dex (Fig. 1E, lanes 5 and 6, respectively). As expected, the anti-GILZ antibody recognized proteins from both the Dex-treated C3H10T1/2 whole-cell lysates (Fig. 1E, lane 2) and from nuclear extracts (Fig. 1E, lane 6), that have the same molecular mass as GILZ (Fig. 1E, lane 4). By contrast, this protein is not present in untreated cells (Fig. 1E, lanes 1 and 5) or cells transfected with an empty vector (Fig. 1E, lane 3). These data show that GILZ is the Dex-induced nuclear protein, and that it specifically binds to the C/EBP tandem DNA-binding site.
To determine whether GILZ messenger RNA and protein expression are regulated in mesenchymal cells, we treated C3H10T1/2 cells with Dex, and harvested total cellular RNA and protein. Northern blot analysis showed that the expression of GILZ mRNA is induced by Dex (Fig. 2A). The expression level of the GILZ mRNA peaked 6 h after treatment with Dex, and began to decrease within 20 h. It returned to pre-induction levels at about 24 h. The expression of GILZ protein showed a similar pattern to that of GILZ mRNA (Fig. 2C). To determine whether GILZ is induced by Dex in pre-adipocytes, we treated 3T3-L1 cells with Dex and carried out western blot analysis. Similar to its expression in C3H10T1/2 cells, GILZ protein was induced within 3 h, and started to decline after 24 h of Dex treatment (Fig. 2D). The expression of C/EBP-δ in response to Dex treatment was also examined. Western blot analysis shows that C/EBP-δ expression is also transiently induced by Dex, and its induction occurs slightly later than that of GILZ (Fig. 2E). These results show that GC can induce GILZ expression in different cells.
Figure 2.
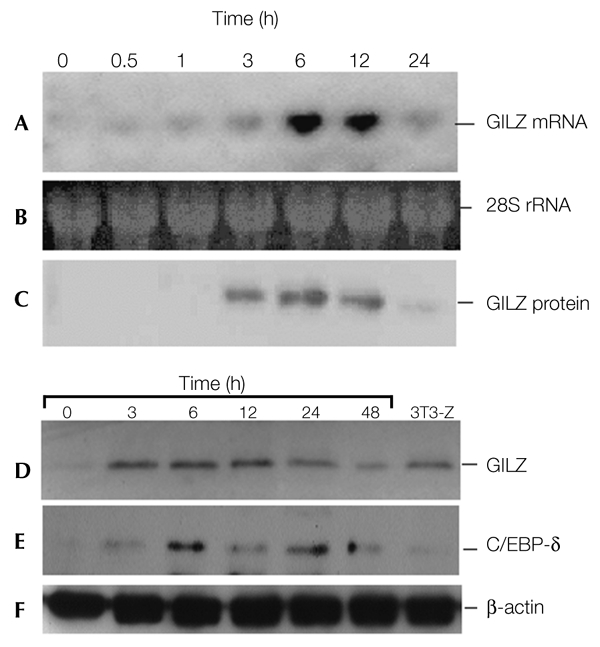
Glucocorticoid-induced leucine-zipper protein messenger RNA and protein expression are induced by glucocorticoid in a time-dependent manner. (A–C) C3H10T1/2 cells were treated with dexamethasone (Dex), and total RNA and protein were prepared at the time points indicated. Levels of glucocorticoid-induced leucine-zipper protein (GILZ) mRNA (A) and protein (C) expression were determined by northern and western blot analyses, using a randomly primed GILZ complementary DNA probe and an anti-GILZ antibody, respectively. Equal loading of lanes is shown by ethidium bromide staining of 28S ribosomal RNA (B). (D–F) 3T3-L1 cells were treated with 100 nM of Dex, and the whole-cell lysates were prepared at the time points indicated. The levels of GILZ protein (D) were determined by western blot analysis using an anti-GILZ antibody. A sample from a stable cell line (3T3-Z) was also included to show the expression level of GILZ protein in these cells. (E) The same membrane as in (D) was stripped and re-probed with anti-CCAAT/enhancer-binding protein-δ (C/EBP-δ) antibody to show the levels of C/EBP-δ expression obtained in response to stimulation with Dex. Equal loading of lanes is shown by assaying levels of β-actin expression (F).
We then examined whether GILZ has a role in regulating PPAR-γ2 transcription. Different amounts of a GILZ expression plasmid (pcDNA3–GILZ) were co-transfected into C3H10T1/2 cells with the PPAR-γ2-promoter–luciferase reporter construct, 615-Luc. GILZ inhibited PPAR-γ2 promoter activity in a dose-dependent manner (Fig. 3A). C/EBP-δ has been shown to bind to tandem C/EBP sites and activate PPAR-γ2 expression. Therefore, the effect of GILZ on C/EBP-δ-mediated PPAR-γ2 transcription was also examined. 615-Luc was co-transfected with C/EBP-δ, GILZ, or both into C3H10T1/2 cells. C/EBP-δ-mediated PPAR-γ2 promoter activity was significantly reduced by expression of GILZ (Fig. 3B). However, deletion of a 22-nucleotide fragment containing the tandem repeat of C/EBP binding sites in the PPAR-γ2 promoter (615-Luc-del) abolished the GILZ inhibition effect (Fig. 3C). Histone acetylation is one of the main chromatin modifications that are important in the transcriptional regulation of genes, and hypoacetylation of histones leads to histone deacetylase (HDAC)-mediated repression of gene expression (Grunstein, 1997; Kuo & Allis, 1998; Mannervik et al., 1999). To characterize the molecular mechanisms determining HDAC-mediated transcriptional repression by GILZ, we examined whether HDACs interact with GILZ. A GILZ construct, tagged with the Flag epitope (Flag–GILZ) and haemagglutinin (HA)-tagged HDAC constructs (HA–HDAC-1, -3, -4, -5 and -6) were co-transfected into COS1 cells. The cell extracts were immunoprecipitated using an anti-Flag antibody, and the immunoprecipitates were analysed by western blotting using an anti-HA antibody. HDAC1, but not the other HDACs (data not shown), co-immunoprecipitated with GILZ (Fig. 3D, upper panel, lane 3), suggesting that GILZ recruits class I HDACs to inhibit transcriptional activity. The interaction of SMAD6C and HDAC1 was used as a positive control (Fig. 3D, upper panel, lane 4). We also carried out chromatin immunoprecipitation (ChIP) assays to test the in vivo binding of GILZ to the PPAR-γ2 promoter. As for the in vitro electrophoretic mobilityshift assays shown in Fig. 1, ChIP assays showed that GILZ and C/EBP-δ can bind either separately, or as a complex of GILZ and C/EBP-δ, to the PPAR-γ2 promoter in vivo (Fig. 3E). These data suggest that GILZ is a transcriptional repressor, and that it inhibits C/EBP-δ-mediated PPAR-γ2 expression by interfering with the transcriptional function of C/EBP-δ.
Figure 3.
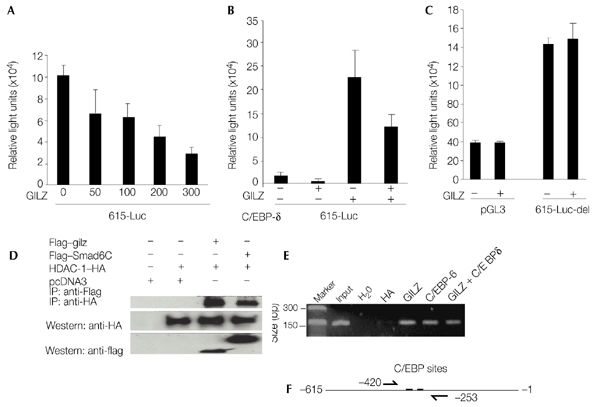
The glucocorticoid-induced leucine-zipper protein represses CCAAT/enhancer-binding-protein-δ-mediated transcription of the gene encoding peroxisome-proliferator-activated receptor-γ2. (A,B) A wild-type peroxisome-proliferator-activated receptor-γ2 (PPAR-γ2) promoter–luciferase reporter (615-Luc) or (C) a mutant, which has a 22-nucleotide deletion of the region containing the tandem repeat of the CCAAT/enhancer-binding protein (C/EBP) binding sites (615-Luc-del), were co-transfected with a glucocorticoid-induced leucine-zipper protein (GILZ) expression vector, or with both GILZ and C/EBP-δ expression vectors, into C3H10T1/2 cells. The cells were incubated for 48 h before luciferase activities were measured. The empty luciferase reporter vector, pGL3-basic (pGL3), was used as a control (C). The experiment in panel (A) was performed in 12-well plates with 400 ng per well of reporter plasmid; and 24-well plates were used in (B) and (C) with 200 ng of reporter plasmid and 100 ng of the pcDNA3–GILZ plasmid. MSV–EBP-δ (C/EBP-δ) plasmid (50 ng) was used in (B). The values were normalized to a Renilla luciferase internal control. Representative examples of results from many experiments, carried out in triplicate, are shown. (D) GILZ interacts with histone deacetylase 1 (HDAC1). Flag-epitope-tagged GILZ and haemagglutinin (HA)-tagged HDAC1 expression plasmids were co-transfected into COS1 cells. Cell lysates were immunoprecipitated using an anti-Flag antibody, and the immunoprecipitates were analysed by western blotting using an anti-HA antibody (upper panel). The interaction of SMAD6C and HDAC1 is shown as a positive control (upper panel, lane 4). The two lower panels show western blots of cell lysates, indicating the relative expression levels of the corresponding proteins. (E) Chromatin immunoprecipitation (ChIP) assay indicating the in vivo interaction between GILZ and C/EBP-δ at the PPAR-γ2 promoter region. 3T3-L1 cells were treated with 100 nM of dexamethasone for 12 h, after which the ChIP assays were carried out (see Methods) using anti-HA, anti-GILZ and anti-C/EBP-δ antibodies, as indicated. (F) Location of primers from the PPAR-γ2 promoter region used in the ChIP assay in (E) to amplify target DNA by PCR. The size of the predicted PCR product was 167 bp.
To examine the role of GILZ in GC-induced adipocyte differentiation, we established stable GILZ transfectants in the 3T3-L1 pre-adipocyte cell line. Eight independent GILZ-expressing transfectants were selected, along with one control cell line that contains an empty pcDNA3 vector (3T3-C). The expression of the GILZ protein was confirmed by western blot analysis (Fig. 4A). Because each of the eight stable transfectants express the GILZ protein at different levels, several cell lines were used for this study. The results shown here were obtained using line 8 (3T3-Z8). 3T3-C and 3T3-Z8 cells were grown to confluence, and a standard adipocyte differentiation protocol was carried out on these cells (Christy et al., 1989; Yeh et al., 1995). Adipocyte differentiation was examined by Oil-Red O (Sigma) staining of the lipid droplets in the cells. Adipocyte differentiation was blocked in 3T3-Z8 cells overexpressing GILZ. By contrast, more than 80% of the cells from the control line (3T3-C) differentiated into adipocytes (Fig. 4B, compare b with d). Similar results were seen for other GILZ stable transfectant lines. Cells grown in normal growth media had no lipid accumulation (Fig. 4B, compare a with c), suggesting that overexpression of GILZ blocks 3T3-L1 preadipocytes from differentiating into adipocytes under adipocyte-inducing conditions. We also established the stable expression of GILZ in C3H10T1/2 cell lines. Treatment of these cell lines with Dex resulted in lipid droplet accumulation in control cells (10T1/2-C) that contained an empty pcDNA3 vector, but not in GILZ-expressing cells (10T1/2-Z) (Fig. 4C, D). Lipid accumulation was not observed when either cell line was cultured in standard growth media. These results show that GILZ functions as a transcriptional repressor in the adipogenesis pathway, a result consistent with its inhibitory effect on PPAR-γ2 expression (Fig. 3).
Figure 4.
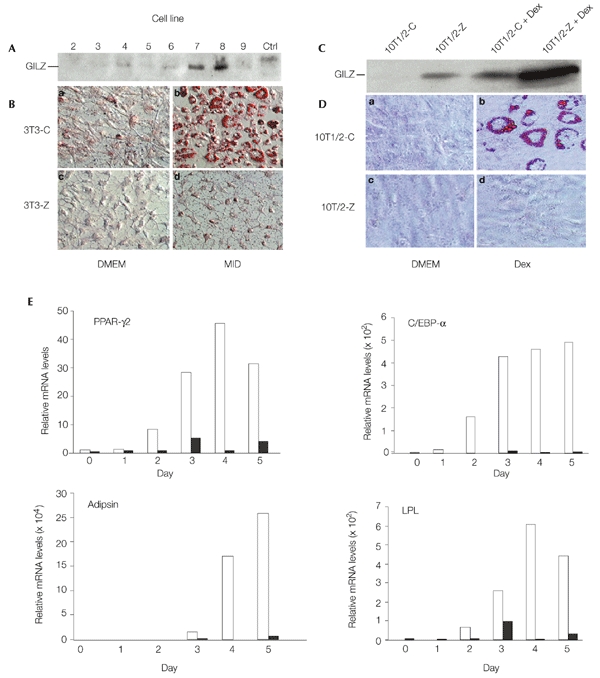
The glucocorticoid-induced leucine-zipper protein inhibits adipocyte differentiation of 3T3-L1 and C3H10T1/2 cells. (A) Western blot analysis of glucocorticoid-induced leucine-zipper protein (GILZ) expression in 3T3-L1 stable cell lines. Lysates from 3T3-Z (lanes 2–9) and 3T3-C (Ctrl) cells were analysed by western blotting using an anti-GILZ antibody. (B) The control (3T3-C) and GILZ-expressing (3T3-Z) cells were cultured in DMEM (a, c) or adipocyte induction medium (MID) containing isobutylmethylxanthine (0.5 mM), insulin (10 μg ml−1) and dexamethasone (Dex; 1 μM) (b, d). The cells were stained with Oil-Red O 5 days after induction. The red colour indicates the lipid droplets in adipocytes. (C) Western blot analysis of GILZ expression, carried out as for (A), in C3H10T1/2 stably transfected cells. Lanes 3 and 4 show the levels of GILZ in cells treated with Dex. (D) Control (10T1/2-C) and GILZ-expressing (10T1/2-Z) cells were cultured in DMEM (a, c) or treated with 1 μM Dex (b, d) for 12 days, and were then stained as in (B). (E) Real-time RT–PCR (reverse transcription followed by PCR) analysis of adipocyte marker gene expression during adipocyte differentiation. An adipocyte differentiation procedure was carried out on stably transfected 3T3-C (empty bars) and 3T3-Z (filled bars) cell lines. Total RNAs were isolated at the timepoints indicated, and messenger RNA levels for PPAR-γ2, C/EBP-α, adipsin and lipoprotein lipase (LPL) were determined by real-time RT-PCR. The levels of mRNA were normalized to that of β-actin, and the values from 3T3-C cells (control) for each gene at day 0 were set as 1. Two independent experiments were carried out, and one representative experimental result is shown. Note that the scales used to show mRNA levels vary for the different genes analysed.
We then examined the effect of GILZ on endogenous PPAR-γ2, C/EBP-α, lipoprotein lipase (LPL) and adipsin mRNA expression during adipocyte differentiation by real-time RT–PCR (reverse transcription followed by PCR) using the primers shown in Table 1. An adipocyte differentiation protocol was carried out on 3T3-C and 3T3-Z8 stable transfectants. Total RNA was harvested at the time-points indicated in Fig. 4E, and real-time RT–PCR was carried out. The results showed that the levels of PPAR-γ2, C/EBP-α, LPL and adipsin mRNAs were significantly inhibited in GILZ-expressing cells during the differentiation programme compared with mRNA levels in control cells (Fig. 4E). The levels of expression of these adipocyte marker genes in control cells, however, are consistent with the well-documented expression patterns of these genes during adipocyte differentiation (Christy et al., 1989; Wabitsch et al., 2001); that is, their levels increase as the cells differentiate towards the terminal adipocyte state. More than 80% of the cells in the control group became adipocytes 4 days after induction. These results show that GILZ is a transcriptional repressor of adipogenesis, and that this inhibition by GILZ occurs through downregulation of key genes expressed during the adipocyte differentiation programme.
Table 1.
Primers used for real-time reverse transcription–PCR analysis
| Gene product | Primer sequence |
|---|---|
| PPAR-γ2 | 5′-TTTTCCGAAGAACCATCCGAT-3′ (F) |
| 5′-ACAAATGGTGATTTGTCCGTTG-3′ (R) | |
| C/EBP-α | 5′-GATAAAGCCAAACAACGCAACG-3′ (F) |
| 5′-CTAGAGATCCAGCGACCCGAA-3′ (R) | |
| Lipoprotein lipase | 5′-TTAACTACCCCCTAGACAACGTCCA-3′ (F) |
| 5′-AAGAGATGAATGGAGCGCTCG-3′ (R) | |
| Adipsin | 5′-AGACCCCTACCCTTGCAATACG-3′ (F) |
| 5′-TGTTACCATTTGTGATGTTTTCGATC-3′ (R) | |
| GILZ | 5′-GCTGCACAATTTCTCCACCT-3′ (F) |
| 5′-GCTCACGAATCTGCTCCTTT-3′ (R) | |
| β-actin | 5′-AACACCCCAGCCATGTACGTAG-3′ (F) |
| 5′-GTGTTGGCATAGAGGTCTTTACGG-3′ (R) |
C/EBP-δ, CCAAT/enhancer-binding-protein-δ; F, forward primer (5′–3′); PPAR-γ, peroxisome-proliferator-activated receptor-γ; R, reverse primer (5′–3′).
DISCUSSION
GILZ has been identified as a GC-induced protein, and protects T cells from activation-induced apoptosis. We provide evidence to show that GILZ antagonizes GC function by inhibiting GC-induced adipogenesis. PPAR-γ2 is a key regulator of adipogenesis, and PPAR-γ2 transcription is activated by C/EBP-δ through the direct binding of C/EBP-δ to the PPAR-γ2 promoter. By contrast, GILZ binds to the PPAR-γ2 promoter element and inhibits C/EBP-δ-mediated transcription. Both C/EBP-δ and GILZ are transcriptionally activated by GCs, but whereas C/EBP-δ induces PPAR-γ2 expression, GILZ represses it. Recent studies have also shown that GILZ functions as a transcriptional repressor, and inhibits gene expression through direct association with nuclear factor-κB (Ayroldi et al., 2001; Riccardi et al., 2001) and activating protein 1 (Mittelstadt & Ashwell, 2001). Taken together, these results suggest that GILZ functions as an antagonist in the GC negative-feedback circuit.
GILZ belongs to the leucine-zipper family of transcription factors. Our gel-shift results show that GILZ binds specifically to tandem C/EBP sites, but not to a single C/EBP element. These results also suggest that GILZ interacts with other nuclear factors, or itself, to form a complex when bound to the DNA element. There are three members of the C/EBP family of transcription factors, C/EBP-α, C/EBP-β and C/EBP-δ, that are implicated in the induction of adipocyte differentiation. These factors are able to form homodimers, or heterodimers, with other family members, when bound to DNA. It would be interesting to identify the factors that form complexes with GILZ, to gain a better understanding of the mechanism that regulates GILZ-mediated transcription. One would predict that these factors should be members of the leucine-zipper family of transcription factors. For example, ectopic expression of C/EBP-homologous protein 10 (CHOP10), a member of the C/EBP family, also inhibits adipocyte differentiation in 3T3-L1 pre-adipocyte cells (Batchvarova et al., 1995). It forms heterodimers with other C/EBP members (for example, C/EBP-α and C/EBP-β), and functions as a dominant-negative inhibitor. CHOP10 only forms heterodimers with other C/EBPs on a subset of C/EBP sites (Ubeda et al., 1996). It is also possible that GILZ only heterodimerizes with C/EBPs at specific C/EBP sites that we have not tested.
In this study, we showed that GILZ, which is transcriptionally induced by GCs, inhibits the transcription of the PPAR-γ2 gene and blocks adipocyte differentiation. We have also shown that GILZ is a sequence-specific DNA-binding factor that binds to a C/EBP tandem DNA element in the PPAR-γ2 promoter. GILZ functions as a transcriptional repressor by interfering with the functions of C/EBP transcription factors, and possibly by recruiting HDACs to target-gene promoters. As well as the inhibition of PPAR-γ2 transcripts, the mRNA levels of other key adipogenic regulators (for example, C/EBP-α) and the downstream adipocyte differentiation marker genes (for example, LPL and adipsin) are all inhibited in GILZ-expressing 3T3-L1 cells.
METHODS
Cell culture and differentiation.
C3H10T1/2 and 3T3-L1 cells (American Type Culture Collection) were maintained in DMEM with 10% FBS. Adipocyte differentiation and staining protocols were carried out as described previously (Shi et al., 2000).
RNA purification, northern blot analysis and real-time reverse transcription–PCR.
Cells were treated as indicated in the figures. RNA isolation and nothern blot analysis were carried out as described previously (Shi et al., 2000). PPAR-γ2, C/EBP-α, LPL and adipsin mRNA expression was determined by reverse transcription, followed by real-time TaqMan (Applied Biosystems) PCR analysis in accordance with the manufacturer's instructions. The sequences of the primers used are shown in Table 1.
Western blot analysis.
Equal amounts of total cell lysates were separated by SDS–polyacrylamide gel electrophoresis, transferred onto a membrane, probed with polyclonal anti-GILZ (custom-made by Strategic Biosolutions) or anti-C/EBP-δ (Santa Cruz) antibodies, and detection was carried out using an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech) as described previously (Shi et al., 2001).
Chromatin immunoprecipitation assays.
ChIP assays were carried out as described in Shang et al. (2000), except that the cells were treated with 100 nM Dex for 12 h before crosslinking with 1% formaldehyde. The primers used in the ChIP assays were 5′-GGCCAAATACGTTTATCTGGTG-3′ (forward) and 5′-TCACTGTTCTGTGAGGGGC-3′ (reverse). The size of the predicted PCR product is 167 bp.
Expression and purification of the GST–GILZ fusion protein.
To make the GST–GILZ fusion protein, the coding region of the gene encoding GILZ was amplified by PCR from the pcDNA3–GILZ plasmid and inserted in-frame into pGEX-5X-1 (Amersham Pharmacia Biotech) between the EcoRI and XhoI sites. The GST–GILZ fusion protein was purified as described previously (Shi et al., 2000).
Electrophoretic mobility-shift assays.
C3H10T1/2 cells were treated with 1 μM Dex for 12 h. The preparation of nuclear extracts, the DNA probes used and the experimental procedures have been described previously (Shi et al., 2000).
Plasmid constructs and luciferase assays.
The PPAR-γ-promoter–luciferase reporter construct, 615-Luc, transfection procedures and luciferase activity assays have been described previously (Shi et al., 2000).
Supplementary information are available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor805-s1.mov).
Supplementary Material
supplementary information
Acknowledgments
We thank C. Reccardi for pcDNA3–GILZ and S.L. McKnight for the MSV–EBP-α, -β, and -δ expression plasmids. This work was supported in part by a grant from the American Arthritis Foundation (to X.S.), and by US Department of Defense grant 527400 (to X.C.).
References
- Ayroldi E., Migliorati G., Bruscoli S., Marchetti C., Zollo O., Cannarile L., D'Adamio F. & Riccardi C. (2001) Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor κB. Blood, 98, 743–753. [DOI] [PubMed] [Google Scholar]
- Batchvarova N., Wang X.Z. & Ron D. (1995) Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153). EMBO J., 14, 4654–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows C.G., Aubin J.E. & Heersche J.N. (1987) Physiological concentrations of glucocorticoids stimulate formation of bone nodules from isolated rat calvaria cells in vitro. Endocrinology, 121, 1985–1992. [DOI] [PubMed] [Google Scholar]
- Bellows C.G., Ciaccia A. & Heersche J.N. (1998) Osteoprogenitor cells in cell populations derived from mouse and rat calvaria differ in their response to corticosterone, cortisol and cortisone. Bone, 23, 119–125. [DOI] [PubMed] [Google Scholar]
- Christy R.J., Yang V.W., Ntambi J.M., Geiman D.E., Landschulz W.H., Friedman A.D., Nakabeppu Y., Kelly T.J. & Lane M.D. (1989) Differentiation-induced gene expression in 3T3-L1 preadipocytes: CCAAT/enhancer binding protein interacts with and activates the promoters of two adipocytespecific genes. Genes Dev., 3, 1323–1335. [DOI] [PubMed] [Google Scholar]
- D'Adamio F., Zollo O., Moraca R., Ayroldi E., Bruscoli S., Bartoli A., Cannarile L., Migliorati G. & Riccardi C. (1997) A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity, 7, 803–812. [DOI] [PubMed] [Google Scholar]
- Grunstein M. (1997) Histone acetylation in chromatin structure and transcription. Nature, 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Jaiswal N., Haynesworth S.E., Caplan A.I. & Bruder S.P. (1997) Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J. Cell. Biochem., 64, 295–312. [PubMed] [Google Scholar]
- Kuo M.H. & Allis C.D. (1998) Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays, 20, 615–626. [DOI] [PubMed] [Google Scholar]
- Lane N.E. (2001) An update on glucocorticoid-induced osteoporosis. Rheum. Dis. Clin. North Am., 27, 235–253. [DOI] [PubMed] [Google Scholar]
- Lian J.B. & Stein G.S. (1993) The developmental stages of osteoblast growth and differentiation exhibit selective responses of genes to growth factors (TGF-β1) and hormones (vitamin D and glucocorticoids). J. Oral Implantol., 19, 95–105. [PubMed] [Google Scholar]
- Mannervik M., Nibu Y., Zhang H. & Levine M. (1999) Transcriptional coregulators in development. Science, 284, 606–609. [DOI] [PubMed] [Google Scholar]
- Manolagas S.C. & Weinstein R.S. (1999) New developments in the pathogenesis and treatment of steroid-induced osteoporosis. J. Bone Miner. Res., 14, 1061–1066. [DOI] [PubMed] [Google Scholar]
- Mittelstadt P.R. & Ashwell J.D. (2001) Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J. Biol. Chem., 276, 29603–29610. [DOI] [PubMed] [Google Scholar]
- Nishimura J. & Ikuyama S. (2000) Glucocorticoid-induced osteoporosis: pathogenesis and management. J. Bone Miner. Metab., 18, 350–352. [DOI] [PubMed] [Google Scholar]
- Reid I.R. (2000). Glucocorticoid-induced osteoporosis. Baillieres Best Pract. Res. Clin. Endocrinol. Metab., 14, 279–298. [DOI] [PubMed] [Google Scholar]
- Riccardi C., Bruscoli S., Ayroldi E., Agostini M. & Migliorati G. (2001) GILZ, a glucocorticoid hormone induced gene, modulates T lymphocyte activation and death through interaction with NF-κB. Adv. Exp. Med. Biol., 495, 31–39. [DOI] [PubMed] [Google Scholar]
- Shalhoub V., Conlon D., Tassinari M., Quinn C., Partridge N., Stein G.S. & Lian J.B. (1992) Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J. Cell. Biochem., 50, 425–440. [DOI] [PubMed] [Google Scholar]
- Shang Y., Hu X., DiRenzo J., Lazar M.A. & Brown M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell, 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Shi X.M., Blair H.C., Yang X., McDonald J.M. & Cao X. (2000) Tandem repeat of C/EBP binding sites mediates PPARγ2 gene transcription in glucocorticoid-induced adipocyte differentiation. J. Cell. Biochem., 76, 518–527. [DOI] [PubMed] [Google Scholar]
- Shi X., Bai S., Li L. & Cao X. (2001) Hox-a9 represses transforming growth factor-β-induced osteopontin gene transcription. J. Biol. Chem., 276, 850–855. [DOI] [PubMed] [Google Scholar]
- Toth M. & Tulassay Z. (2000) Glucocorticoid-induced osteoporosis. Orvosi Hetilap., 141, 219–223. (In Hungarian.) [PubMed] [Google Scholar]
- Ubeda M., Wang X.Z., Zinszner H., Wu I., Habener J.F. & Ron D. (1996) Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell. Biol., 16, 1479–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wabitsch M., Brenner R.E., Melzner I., Braun M., Moller P., Heinze E., Debatin K.M. & Hauner H. (2001) Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. Relat. Metab. Disord., 25, 8–15. [DOI] [PubMed] [Google Scholar]
- Walsh L.J., Wong C.A., Oborne J., Cooper S., Lewis S.A., Pringle M., Hubbard R. & Tattersfield A.E. (2001) Adverse effects of oral corticosteroids in relation to dose in patients with lung disease. Thorax, 56, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh W.C., Cao Z., Classon M. & McKnight S.L. (1995) Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev., 9, 168–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information