

Introduction
The Center for International Meetings on Biology workshop on 'Exchange Factors' provided an up-to-date view of how guanine-nucleotide-exchange factors (GEFs) affect the biochemical pathways that regulate the Ras superfamily of small GTPases, and how these molecules function as part of an interrelated network of regulatory proteins that mediate the biological responses of Ras-like GTPases (Fig. 1). Reflecting the complex nature of GEF functions, the meeting focused on their structure–function relationships, regulation and biological functions. Particular emphasis was placed on a subset of regulators of the Ras and Rho subfamilies that contain catalytic CDC25 homology domains or Dbl homology (DH)/pleckstrin homology (PH) cassettes, respectively (Fig. 2; Quilliam et al., 2002; Zheng, 2001).

The workshop on 'Exchange Factors' was held at Instituto Juan March de Estudios e Investigaciones, Madrid, Spain, during November 4–6, 2002. It was organized by Xose R. Bustelo, J. Silvio Gutkind and Piero Crespo.
Figure 1.
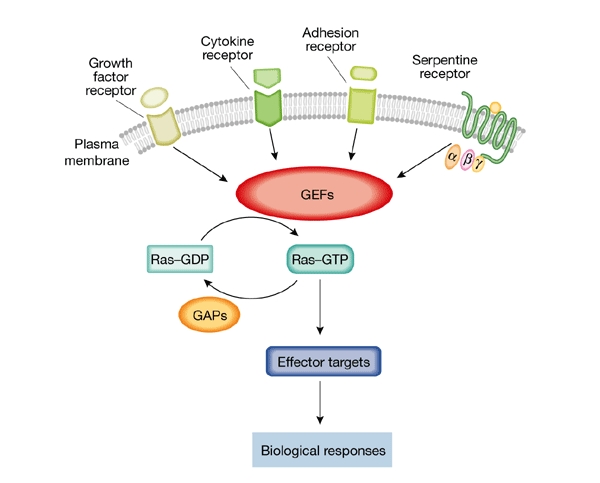
A biochemical model for guanine-nucleotide-exchange factor activation of Ras-like GTPases. The cycle between active (GTP-bound) and inactive (GDP-bound) Ras proteins is regulated by guanine-nucleotide-exchange factors (GEFs) and GTPase-activating proteins (GAPs). Extracellular signals, conveyed through specific cell-surface receptors, modulate GEF activity, which in turn regulates the activity of individual Ras GTPases. The active Ras proteins are able to interact with many effectors, leading to diverse biological responses.
Figure 2.
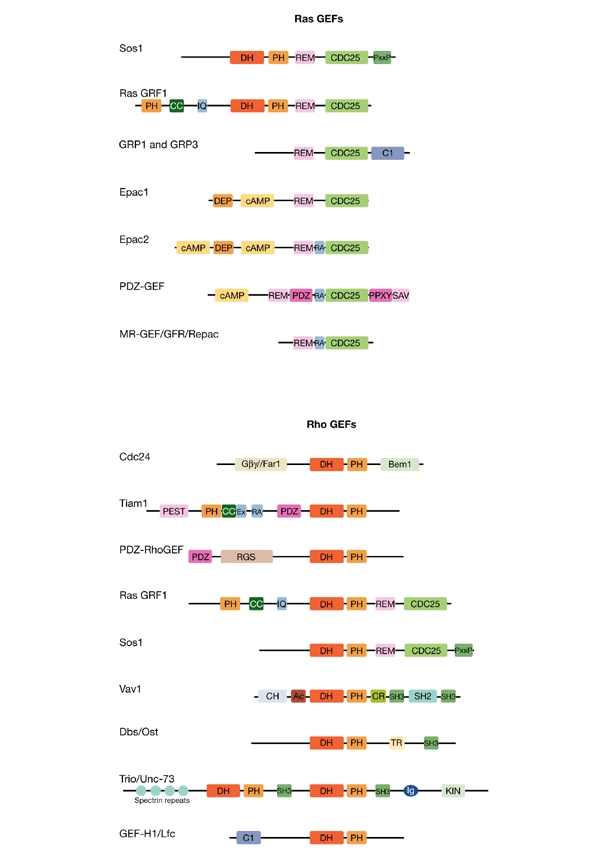
Many guanine-nucleotide-exchange factors are involved in signalling to small GTPases. The multifunctional domain features of the representative guanine-nucleotide-exchange factors (GEFs) discussed at the meeting reflect their diverse biochemical modes of regulation (signal convergence and divergence) and biological function. Due to space limitations, only a subset of these is presented here. GEFs that regulate Ras and Rap proteins are aligned according to their CDC25 domains, whereas GEFs that regulate Rho and Rac proteins are aligned on the basis of their Dbl homology (DH) and pleckstrin homology (PH) domains. Ac, acidic amino-acid-rich motif; Bem1, Bem1-binding domain; cAMP, cyclic-AMP-binding domain; CC, coiled coil; CDC25, Ras GEF catalytic domain; CH, calponin homology; CR, cysteine-rich zinc-butterfly motif; DEP, dishevelled, egl, pleckstrin domain; C1, EF-hand calcium-binding motif; Ex, an uncharacterized region that co-operates with the PH and CC regions of Tiam1; Gβγ/Far-1, G protein Gβγ-subunit /Far1-binding domain; Ig, immunoglobulin-like domain; IQ, calmodulin-binding motif; KIN, serine/threonine protein kinase domain; PDZ, PSD95/Dlg/ZO1 domain; PEST, amino-acid P-, E-, S- and T-rich degradation motif; PPXY, WW-domain-binding sequence; RA, Ras association domain; REM, Ras exchange motif; RGS, regulator of G-protein signalling domain; SAV, carboxy-terminal PDZ-domain-binding sequence; SH2, Src homology 2; SH3, Src homology 3.
Mechanism of GEF reactions
The Ras superfamily of GTPases can be subdivided into the Ras, Rho, Rab, ARF and Ran subfamilies. Whereas the GEFs that regulate each subfamily are structurally distinct, those that regulate members of any given subfamily show a high degree of sequence conservation. Recent structural studies of GEF–GTPase complexes have also revealed mechanistic features that are shared by all GEFs. A. Wittinghofer (Dortmund, Germany) summarized some of the common principles of exchange-factor-catalysed reactions. GEFs typically stimulate the intrinsic release of GDP from the GTPase by ∼1 × 105-fold (Vetter & Wittinghofer, 2001), through interactions both with the switch regions (I and II) and with the phosphate-binding loop of the GTPase. On the basis of studies of Ran regulation by the exchange factor RCC1, Wittinghofer proposed that there are several transition steps during the exchange reaction of Ras proteins, with the guanine ring of GDP being dislodged first, to generate a low-affinity Ras–GDP:GEF complex. Subsequently, the phosphate moiety and an Mg2+ ion are displaced, and a high-affinity Ras–GEF complex is established. Because dislodging the bound Mg2+ ion is not sufficient for GDP dissociation, it seems that the disruption of contacts with the phosphate moiety is the key step required for rapid nucleotide release. According to the principle of micro-reversibility, the entry of GTP into the binding pocket to displace the GEF and create a high-affinity nucleotide complex would have to occur in the reverse order: that is, phosphate first, base second. Y. Zheng (Cincinnati, OH, USA) presented an alternative model for the mechanism of activation of the Rho subfamily GTPase, Rac1, by the Rho-family GEF, Trio. The fact that the Trio-catalysed GEF reaction depends on the nature and concentration of free nucleotides was taken to indicate that the binding of incoming GTP is required for GDP displacement, and that a two-nucleotide–one-G-protein intermediate, GTP–Rac1–GDP, is involved. This interpretation was one of the more extensively debated issues of the workshop.
GEFs for Ras/Rap GTPases
Sos1 is the most widely expressed Ras GEF, and the only one for which a catalytic-domain structure is available. In refining this structure, D. Bar-Sagi (Stony Brook, NY, USA), in collaboration with J. Kuriyan (Berkeley, CA, USA), made the surprising observation that Sos1 binds two molecules of Ras simultaneously. Crystals of an Sos1 fragment that comprised the CDC25 domain and the Ras exchange motif (REM) bound to Ras64A (a Ras mutant that is locked into the GTP-bound state), showed a GTP-dependent association between this GTPase and the REM (Fig. 2), which abuts and stabilizes the CDC25 domain. These contacts alter the position of the hairpin structure that pries open the GTP-binding pocket of Ras, and may thereby alter the enzymatic activity of the CDC25 domain. Although co-precipitation of Ras–GTP with Sos1 has not yet been accomplished, the addition of Ras64A–GTPγS to Sos1 and Ras–GDP in vitro enhances nucleotide exchange, suggesting that Ras–GTP may induce a feedback-activation loop by binding to the Sos1 REM domain.
Structural analysis of a larger Sos1 fragment that included the DH/PH cassette, which encodes GEF activity towards Rac1, showed that this region folds over on the CDC25 domain. This provides a structural rationale for the previously shown amino-terminal auto-inhibition of the Ras GEF activity of Sos1. Interestingly, the REM is also occluded by the DH/PH region, suggesting that occupation of this site by Ras–GTP might overcome autoinhibition.
Several exchange factors for the Ras subfamily member Rap1 were reported to contain a Ras association (RA) domain, similar to those found in Ras effectors (such as Raf and RalGDS), between their REM and CDC25 homology domains (Fig. 2). L. A. Quilliam (Indianapolis, IN, USA) reported that binding of H-Ras–GTP to the Rap GEF, Epac2, promotes the translocation of Epac2 to the plasma membrane, where it can activate a different pool of cellular Rap1. Similar observations have been made for other Rap GEFs, including PDZ-GEF and MR-GEF. Thus, many Ras/Rap GEFs seem to be regulated by association with GTP-loaded Ras family proteins, resulting in increased intrinsic exchange activity and/or subcellular relocalization.
Mammalian cells contain three Ras genes (H-, N- and K-ras) that have overlapping but distinct functions. Ras17N dominant-inhibitory mutants have been used as a tool in our signalling arsenal for more than 15 years. P. Crespo (Madrid, Spain) reported that the inhibitory power and specificity of H-Ras17N, K-Ras17N and N-Ras17N mutants is dependent on their subcellular localization. Whereas H-Ras17N is found in both lipid rafts and in non-rafts, and can therefore antagonize H-Raswt, K-Raswt or N-Raswt in either compartment, K-Ras17N is present only in non-raft membranes, and N-Ras17N only in lipid rafts. Therefore, the K-Ras17N and N-Ras17N mutants can inhibit K-Raswt and N-Raswt, respectively, but have a limited ability to antagonize the activation of their 'cousins' in other membrane compartments. These tools may provide a means to better understand the unique functions of the various Ras isoforms.
Recent work has indicated that Ras resides in the Golgi apparatus, as well as at its previously known location, the plasma membrane (Chiu et al., 2002). But what is Ras doing in the Golgi, and how does this substantial pool become activated? Using the effector construct GFP–Raf-RBD (GFP-tagged Ras-binding domain of Raf) to detect Ras activation, T. Bivona of M. Philips' group (New York, NY, USA) found that after epidermal growth factor stimulation of Cos1 cells, cellsurface Ras–GTP levels peaked rapidly, but Golgi-localized Ras–GTP was maximally activated only after 30 minutes. The activation of intracellular Ras was blocked by a phospholipase C (PLC) inhibitor or by Ca2+ chelation, and growth factors induced the translocation of the Ca2+-activated and diacylglycerol (DAG)-activated GEF, GRP1, from the cytosol to endomembranes (including the Golgi and nuclear envelope). In Jurkat T cells, which are rich in GRP1, stimulation through the T-cell receptor led to rapid activation of Ras at the Golgi, but no activation of plasma-membrane-associated Ras was observed. Further work is required to determine the biological differences between Ras signalling at the plasma membrane and that at the intracellular membranes. M.J. Caloca (Salamanca, Spain) presented similar observations, indicating that activation of Ras at the Golgi is mediated by Ras GRP1, but not by Sos1 or by Ras GRF1.
J.C. Stone (Edmonton, Canada) described the characterization of GRP3 (Fig. 2), a broadspecificity GEF that is regulated by Ca2+ and DAG, and that is abundant in B cells. B-cell stimulation (IgM- or phorbol-ester-mediated) results in extensive phosphorylation of GRP3, suggesting that DAG regulates the activity of this GEF both by direct binding and by inducing its phosphorylation by protein kinase C (PKC). Unlike PKCs, GRPs are not downregulated by prolonged exposure of cells to phorbol ester, but are inhibited by the C1-domain-binding PKC inhibitor, calphostin C. It is assumed that the levels of DAG and Ca2+ dictate the amount and duration of GRP activation. I. Mérida (Madrid, Spain) described how the ability of DAG kinase-α to convert DAG to phosphatidic acid regulated the duration of GRP1-induced Ras activation after T-cell-receptor stimulation.
J.L. Bos (Utrecht, The Netherlands) presented a structural analysis of the N-terminal cyclic AMP (cAMP)-binding region of Epac2. This is the first structure of a cAMP-binding domain to be obtained in the absence of nucleotide binding. Comparison with the structures of nucleotide-bound cAMP-binding domains suggests that a significant conformational change occurs on ligand binding, placing a 'lid' on cAMP to stabilize the binding site of the base. This conformational change was predicted to reorientate the cAMP-binding domain, and to free the catalytic domain from intrasteric inhibition. Indeed, mutation of a Val–Leu–Val–Leu–Glu hinge region between the cAMP-binding and catalytic domains resulted in cAMP-independent (constitutive) activation of Epac2. A surprising twist was described by O. Coso (Buenos Aires, Argentina), who found that Epac1 activity enhances Jun N-terminal kinase (JNK) activation (Fig. 3). This occurs even in the absence of the Epac1 CDC25 domain, and is insensitive to the transfection of cells with the Rap1-17N dominant-negative mutant or with Rap GAP. Further dissection of Epac1 function revealed that a region encompassing its REM is sufficient to promote JNK activity. How this might occur requires further investigation.
Figure 3.
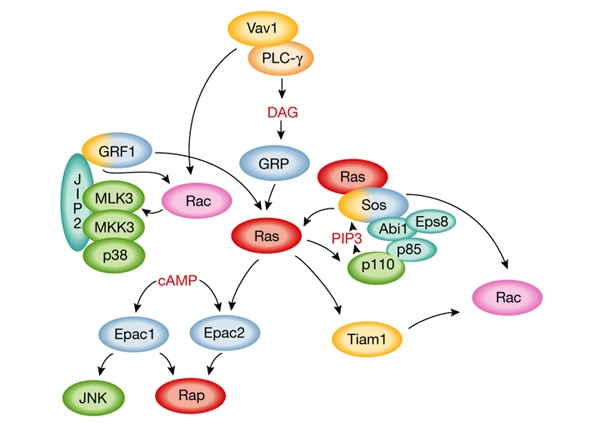
Considerable crosstalk occurs between Ras- and Rac-family regulators. Many of the interconnections that were discussed at the meeting are shown here. Ras-family GTPases are shown in red, Rac in pink, Ras GEFs (guanine-nucleotide-exchange factors) in blue, Rac GEFs in yellow (GRF1 and Sos can act on both as they have both catalytic domains, see Fig 2), adaptor and scaffold proteins in turquoise, kinases in green and lipid-modifying enzymes in orange. p85 and p110 are the subunits of phosphatidylinositol-3-OH-kinase (PI(3)K). cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; PIP3, phosphatidylinositol-3,4,5-trisphosphate.
GEFs for Rho GTPases
Although many extracellular stimuli cause the activation of Rho GTPases by regulating their GEFs, how such diverse stimuli signal to these DH/PH-domain-containing proteins remains unclear. Certainly, several pathways are involved. For example, many signalling events elicited by heterotrimeric G proteins depend on Rho-family GTPases, and different GEFs seem to be used for this by the Gβγ subunit, or by the Gα12 and Gα13 subunits. In the case of Gβγ, P-Rex1 and related GEFs, which are responsive to phosphoinositide stimulation of their PH domains, are involved (H. Welch, Babraham, UK), whereas Gα12 and Gα13 function by binding to regulators of G-protein signalling (RGS)-domain-containing GEFs of the Rho subfamily. J.S. Gutkind (Bethesda, MD, USA) described the efforts of his laboratory to characterize two members of the RGS-containing GEF family, PDZ-RhoGEF (Fig. 2) and LARG. He reported that, in addition to the role of the RGS domains in coupling Gα12 and Gα13 to RhoA activation, the N-terminal PDZ domains of these GEFs also link RhoA to plexin B2, the receptor for semaphorins. The chimaeras made between the nerve growth factor (NGF)-receptor TrkA and plexin B2 enabled NGF to activate RhoA through the sequestration of PDZ-RhoGEF. Thus, these GEFs seem to have dual roles, mediating signalling to RhoA GTPase both by G-protein-coupled receptors and by plexin-family semaphorin receptors.
The regulatory mechanisms of DH-domain-containing GEFs seem to be diverse, and include phosphorylation and lipid or protein intramolecular interactions. One of the better characterized mechanisms is the phosphorylation of the Vav GEFs (Vavs 1–3) by Src family tyrosine kinases. Structural studies by M.K. Rosen's laboratory (Dallas, TX, USA) have revealed previously that an N-terminal extension of Vav1 forms a short α-helix, which occupies the GTPase-binding site of the DH domain and maintains an autoinhibitory conformation (Aghazadeh et al., 2000). On phosphorylation of Tyr174 by Src or related kinases, the α-helix becomes unstructured, relieving this autoinhibition. But how can Src gain access to this buried tyrosine in the auto-inhibited structure? Rosen proposed a solution based on recent nuclear magnetic resonance spectroscopy data that revealed that, in addition to Tyr174, two adjacent residues, Tyr142 and Tyr160, also function as good substrates for Src kinases. These phosphotyrosines are located in consensus binding sites for Src-family SH2 (Src homology 2) domains, and may bring the kinase into close proximity with the Tyr174 residue of Vav1. This initial contact of the kinase with the point of access to the helix could, therefore, convert the potentially slow phosphorylation of Tyr174 to an intramolecular process, thus overcoming the kinetic barrier posed by the inaccessibility of the helix. Such a mechanism of 'access-point control' to alleviate autoinhibitory regulation may also exist in other GEF family members.
G. Bokoch (La Jolla, CA, USA) reported that the Rho GEF, GEF-H1, which associates with microtubules, is subject to at least two modes of regulation: microtubule depolymerization and phosphorylation by the Rac/CDC42 effector, PAK1 kinase. Targeting of GEF-H1 to microtubules suppresses its activation of Rho, whereas phosphorylation of residue Ser885 by PAK1 may affect its ability to recruit additional signalling partners, including the adaptor protein, band 14-3-3. Therefore, this GEF may couple changes in microtubule integrity to Rho-regulated actin structure, and co-ordinate Rho signalling events with those mediated by Rac/CDC42.
With few exceptions, the PH domains of Dbl family GEFs are found immediately carboxy-terminal to their DH domains (Fig. 2). Previous case studies of the Dbl, Dbs and Lbc GEFs have indicated that one role of the PH domain is to recruit the catalytic DH domain to the plasma membrane and/or actin structures, where some Rho substrates are thought to reside. C.J. Der (Chapel Hill, NC, USA) presented evidence that the PH domain of Dbs is crucial for the intrinsic catalytic activity of DH, as well as being involved in phospholipid binding and membrane association. Using a genetic approach in Caenorhabditis elegans, T.J. Kubiseski (Toronto, Canada) showed that mutations in the PH domain of the Rac GEF, UNC-73, that impair phospholipid binding, but not GEF catalytic activity, failed to rescue the axon-guidance phenotype caused by deletion of the unc-73 gene. This helps to establish the significance of the PH domain in vivo. Interestingly, Der also showed that amino-acid sequences C-terminal to the DH/PH module in the Rho GEF, Ect2, which do not share detectable homology with known proteins, are required for the substrate specificity of Ect2 in cells. This study supports previous work on Vav3 (Movilla & Bustelo, 1999), suggesting that residues outside the core DH/PH domains may be involved in Rho GTPase recognition and/or specification.
The intracellular localization of GEFs may also be important for their regulation by means other than the relief of their autoinhibition. In the budding yeast Saccharomyces cerevisiae, Cdc24 seems to be the only GEF for Cdc42, and is required for several cellular functions, including the mating response and polarized growth. It has been shown previously that Cdc24 is located in the nucleus before pheromone stimulation, and is recruited to the site of polarization by an adaptor molecule, Far1. It is therefore of interest that a mammalian Rho GEF, Net1, may also be regulated by nuclear sequestration (A. Schmidt, London, UK). D.I. Johnson (Burlington, VT, USA) reported that during the mitotic cell cycle, Cdc24 colocalizes with Cdc42 at sites of polarized growth. This localization is mediated by two distinct regions at the C terminus of Cdc24; one is required for specific targeting, the other for efficient anchoring in a cytoskeletal complex that includes the Bem1 scaffold protein, the Rsr1/Bud1 GTPase, and a novel transmembrane protein, YGR221C. It remains to be seen whether the requirement of both targeting and anchoring functions applies to the mammalian counterparts of Cdc24.
Crosstalk between Ras and Rho
In addition to their Ras-activating CDC25 homology domains, Sos and Grf also contain a Rac-activating DH/PH module (Fig. 2). Work presented by L.A. Feig (Boston, MA, USA) suggests that Grf1 not only generates Rac–GTP, but also promotes the interaction of Rac with specific downstream effectors (Fig. 3). His laboratory recently published the finding that the N-terminal regulatory region (encompassing PH/coiled-coil/IQ domains) of Grf1 binds the JIP2 scaffold protein that links the Rac target MLK3 to the downstream kinases, MKK3 and p38 (Buchsbaum et al., 2002). The Grf1 N terminus was also found to associate with JIP1 to promote JNK activation with spinophilin/neurabin2, which binds the Rac target S6 kinase, and with a fourth Grf scaffold that is connected to Rac signalling. Interestingly, the Rac GEF, Tiam1, which has an N-terminal PH/coiled-coil/Ex (an undefined region that cooperates with the PH and CC domains) region similar to the PH/coiled-coil/IQ region of Grf1 (Fig. 2), also binds to each of these scaffold proteins. Thus, Sos1, Grf1 and Tiam1 may all contribute to Rac signalling specificity by binding to a specific scaffold protein. The DH and IQ domains of Ras Grf1 were reported, by P. Crespo, to be important in regulating the activation of H-Ras in different subcellular locations.
Although Sos1 has been shown to be a Rac GEF in cells, how its exchange activity is regulated remains unclear, as Sos1 by itself does not show detectable Rac-GEF activity in vitro. One possibility, raised by M. Innocenti (Milan, Italy), is that Sos1 and the adaptor proteins Eps8 and Abi1 might form a Rac-activating complex that also is able to recruit phosphatidylinositol-3-OH-kinase (PI(3)K) through an interaction between the p85 subunit of PI(3)K and Abi1 (Fig. 3). p85 recruitment to the Sos complex, and phosphatidylinositol trisphosphate, which is the main product of PI(3)K, are indispensable for the activation of Rac and Rac-mediated actin remodelling.
X. Bustelo (Salamanca, Spain) described a novel mechanism for Vav1-mediated crosstalk between the Rho and Ras pathways. The activation of Rac by Vav1 was shown to cause the translocation of one of the Ras exchange factors, GRP1/CalDAG-GEFII, to peripheral cellular structures that are enriched in F-actin, resulting in an increase in Ras–GTP levels. This pathway was also shown to depend on the integrity of the PLC-γ pathway, which might itself be regulated separately by a receptor tyrosine kinase or by Vav1 (Fig. 3). Such crosstalk may be important in lymphoid cell development, as Ras activation cannot occur in the absence of Vav1.
Mammalian genetic studies of GEFs
Targeted disruption of the Ras GEFs, Grf1 and Grf2, showed previously that both genes (even in the double knockout) are dispensable for mouse growth and development (Fernández-Medarde et al., 2002). However, Grf1-null mice have a learning defect, presumably due to defects in Ras or Rac activation. L.A. Feig reported that neurons from Grf2−/− or, more significantly, from Grf1−/−/Grf2−/− mice, showed attenuated activation of extracellular regulated kinases (ERKs) on K+-induced depolarization of Ca2+ channels. This provides support for the involvement of Grf/Ras/ERK signalling in memory. E. Santos' group (Salamanca, Spain) reported a novel phenotype for the Grf1 knockout. The gene is normally expressed in the nervous system, but it is also expressed in β-cells of the pancreatic islet. Interestingly, Grf1-deficient mice had reduced body size (not 'runted'), hypoinsulinaemia and glucose intolerance, associated with a significant reduction of the β-cell mass in the pancreas. The reduction in circulating insulin did not reflect defective glucose sensing or insulin production, but resulted from impaired β-cell proliferation and/or neogenesis. The islet β-cells also showed attenuated signalling in response to the kinases ERK and Akt. The observed phenotype resembles preclinical type 2 diabetes, but the knockout animals do not develop diabetes, suggesting that compensatory mechanisms are able to correct the effects of this gene deletion. An interesting twist, described by R.R. Mattingly (Detroit, MI, USA) was the finding that Grf1 is regulated in vivo by phosphorylation. Using a phosphospecific antibody for a site required for full Grf activation (Ser898 of rat Grf1), he found that Grf1 is phosphorylated specifically in the dendrites of rat cortical neurons.
Extensive cellular studies have implicated Rho family GTPases as essential downstream components of Ras-induced transformation and invasion. However, until recently, there was little direct evidence connecting Ras and Rho GTPases to tumorigenesis in vivo. J. Collard's group (Amsterdam, The Netherlands) showed that deletion of the Tiam1 gene, which encodes a Rac-specific GEF, leads to a reduction in the number of Ras-induced skin tumours (Malliri et al., 2002). In fact, both tumour initiation and promotion seem to be dependent on the gene dosage of Tiam1, but in the case of tumour promotion, the Tiam1 deficiency acts positively, that is, it results in a higher frequency of conversion to malignancy. These studies provide convincing evidence that Rho misregulation, specifically due to GEF deficiency, can result in cancer progression in an animal model.
One of the better-understood subfamilies of GEFs, at the level of mammalian genetics, is the Vav family. Vav1 deficiency in mice causes developmental failure in T cells and B cells, and the distortion of many signalling events that are mediated by T-cell or B-cell receptors (TCRs and BCRs). V.L.J. Tybulewicz (London, UK) described the defective phosphorylation of the Tec-family kinases, Itk and Tec, in the CD4+CD8+ double-positive T cells of a Vav1-knockout mouse, which probably resulted from reduced PI(3)K activation. This failure of Tec kinase activation might account for the previously observed defective PLC-γ activation in Vav1−/− cells. Conversely, Vav1−/− CD4+CD8+ double-positive thymocytes show selective abnormalities in actin-dependent events. These include thymocyte polarization and the formation of conjugates with antigen-presenting cells (APCs) on exposure to the agonist, peptide-loaded APCs. Tybulewicz concluded that Vav1 converts signals, probably through Rac-related G proteins, to a subset of cytoskeleton-dependent events at the immunological synapse. M. Turner (Cambridge, UK) reported on the unique, as well as the overlapping, roles that Vav proteins have in B-cell signalling, as determined by studies of Vav2−/− and Vav1−/−/Vav2−/− mice. In particular, he highlighted the possible involvement of Vav1 and Vav2 proteins in BCR-induced Ca2+ influx and the activation of Bruton's tyrosine kinase (Btk). Finally, through studies of mice that congenitally lack all three Vav proteins, W. Swat's laboratory (St Louis, MO, USA) showed that, although the Vav family is indispensable for the development of T and B lymphocytes and for the function of the adaptive immune system, members of this family may have redundant as well as non-redundant functions in several haematopoietic lineages, including osteoclasts. Interestingly, unlike Vav1 and Vav2, Vav3 is expressed at extremely low levels in most tissues, except osteoclasts, and its expression pattern seems to be regulated during the cell cycle.
New tools for playing with GEFs
Recent mechanistic insights into how small G proteins are activated by GEFs, and the apparent significance of these events to human disease, has resulted in the development of several novel biological and pharmacological tools. Work from the laboratories of J. Galán (New Haven, CT, USA) and K. Aktories (Freiburg, Germany) has shown that many bacteria have evolved mechanisms to mimic or disable the regulatory or effector functions of Rho-family GTPases. These include the convergent evolution of novel Rho GAPs and GEFs (Galan, 2001), and an arsenal of toxins that disable or activate many Rho-family GTPases by covalent modifications, including ADP ribosylation, glucosylation, proteolytic cleavage and deamidation (Lerm et al., 2000). Studies of these bacterial toxins and regulators provide both valuable insights into Rho regulation and pharmacological tools with which to manipulate the activity of small G proteins. For example, increased proteosomal degradation of Rac1 after its activation by cytotoxic necrotizing factor (CNF) points to an alternative inactivation mechanism for permanently active Rho GTPases.
L. Renault (Gif-sur-Yvette, France) presented a preliminary analysis of the basis for the effects of a previously identified Arf GTPase inhibitor, brefeldin A (BFA). He concluded that the residues of the Arf GEF Sec7 domain that determine BFA sensitivity are also those that are required for the local movement of two of its subdomains, and that this flexibility is important for the inhibition mechanism. Y. Zheng described his group's efforts towards developing a first generation Rac-specific inhibitor. Their strategy is based on information from structure–function analysis of Rho GTPase activation by GEFs. Prediction by structural simulation led to the discovery of a small-molecule chemical compound that can competitively block Trio-mediated activation of Rac in vitro and in cells. Application of this compound to human prostate cancer cells in which the PTEN tumour suppressor is deleted and Rac activity is elevated, yielded promising results, with the suppression of Rac activity and the reversal of tumour-cell transformation and invasion. Along similar lines, J. Bos showed that, based on the regulatory mechanism of the Epac GEFs, his group has successfully developed a cAMP analogue that efficiently activates Epac1 and Epac2, but not protein kinase A (PKA). A key difference between the sequences of the cAMP-binding domains of these Epacs and those in other proteins, including the RI and RII regulatory subunits of cAMP-dependent PKA, is the absence of a highly conserved glutamate, corresponding to residue 200 of RIα. This difference provided the opportunity to design cAMP analogues that bind to the Epacs, but not to PKA. 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8CPT-2Me-cAMP) was found to have an ∼100 times higher affinity for Epac1 than for either RI or RII. As cAMP, but not 8CPT-2Me-cAMP, induced phosphorylation of ERK, it would seem that cAMP-induced ERK activation is independent of Rap1.
Conclusion
It is becoming increasingly clear that the activation mechanisms of small GTPases are almost as diverse as the number of GEFs that exist in nature. This workshop on exchange factors illustrated nicely the individuality of the regulatory mode and function of each GEF member, as well as the common themes shared by closely related siblings, or even evolutionarily diverse distantly related cousins. Many issues, including how various intracellular signals converge on the GEFs for their tight regulation, what signals would diverge from their cognate small G-protein substrates, and where each GEF resides in the wider context of signalling networks, need to be further tackled in this field. The temporal and spatial regulation of GEFs will be key areas of investigation in the years to come.
Acknowledgments
We thank the Instituto Juan March for being a warm host to the workshop and are grateful to many attendees for helpful critiques.
References
- Aghazadeh B., Lowry W.E., Huang X.-Y. & Rosen M.K. (2000) Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell, 102, 625–633. [DOI] [PubMed] [Google Scholar]
- Buchsbaum R.J., Connolly B.A. & Feig L.A. (2002) Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol. Cell. Biol., 22, 4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu V.K., Bivona T., Hach A., Sajous J.B., Silletti J., Wiener H., Johnson R.L., Cox A.D. & Philips M.R. (2002) Ras signalling on the endoplasmic reticulum and the Golgi. Nature Cell Biol., 4, 343–350. [DOI] [PubMed] [Google Scholar]
- Fernández-Medarde A., Esteban L.M., Núñez A., Porteros A., Tessarollo L. & Santos E. (2002) Targeted disruption of Ras-Grf2 shows its dispensability for mouse growth and development. Mol. Cell. Biol., 22, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan J.E. (2001) Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell. Dev. Biol., 17, 53–86. [DOI] [PubMed] [Google Scholar]
- Lerm M., Schmidt G. & Aktories K. (2000) Bacterial protein toxins targeting rho GTPases. FEMS Microbiol. Lett., 188, 1–6. [DOI] [PubMed] [Google Scholar]
- Malliri A., van der Kammen R.A., Clark K., van der Valk M., Michiels F. & Collard J.G. (2002) Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature, 417, 867–871. [DOI] [PubMed] [Google Scholar]
- Movilla N. & Bustelo X.R. (1999) Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell. Biol., 19, 7870–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quilliam L.A., Rebhun J.F. & Castro A.F. (2002) A growing number of guanine nucleotide exchange factors is responsible for activation of Ras family GTPases. Prog. Nucleic Acid Res. Mol. Biol., 71, 391–444. [DOI] [PubMed] [Google Scholar]
- Vetter I.R. & Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science, 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Zheng Y. (2001) Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci., 26, 724–732. [DOI] [PubMed] [Google Scholar]