

Abstract
There is a pressing need to develop methods to engineer small-calibre arteries for bypass surgery. We hypothesized that the rate-limiting step that has thwarted previous attempts to engineer such vessels from non-neonatal tissues is the limited proliferative capacity of smooth muscle cells (SMCs), which are the main cellular component of these vessels. Ectopic expression of the human telomerase reverse transcriptase subunit (hTERT) has been shown recently to extend the lifespan of certain human cells. We therefore introduced hTERT into human SMCs and found that the resulting cells proliferated far beyond their normal lifespan but retained characteristics of normal control SMCs. Importantly, using these non-neonatal SMCs, we were able to engineer mechanically robust human vessels, a crucial step towards creating arteries of clinical value for bypass surgery.
Introduction
In the USA, 1.4 million patients per year undergo operations requiring arterial prostheses (Langer & Vacanti, 1993; Vacanti & Langer, 1999). Annually, ∼100,000 patients require vascular bypass of small-calibre arteries but have no usable autologous arteries or veins for grafting (Kempczinski, 2000; Williams, 2000). Hence, there is a pressing need for autologous tissue-engineered blood vessels to treat atherosclerotic disease (Lefkowitz & Willerson, 2001; Niklason, 1999).
Small-calibre blood vessels consist of three cell layers: an inner monolayer of endothelial cells (ECs), a thick, dense middle layer of smooth muscle cells (SMCs), and a loosely organized outer fibroblast layer. We have engineered arteries with an internal monolayer of ECs and a thick muscular layer of SMCs from cows and pigs that were strong enough for implantation and were functional in vivo after at least one month of observation (Niklason et al., 1999, 2001). Disappointingly, attempts to translate this approach using non-neonatal human cells to provide a viable therapy for hundreds of thousands of bypass patients, who are deemed untreatable at present, have all failed (Niklason, 1999). The only success came from the use of umbilical-cord-derived (neonatal) SMCs and ECs (L'Heureux et al., 1998), which is impractical for growing autologous vessels for patients. To engineer autologous human vessels that resist immunologic rejection, techniques must be developed to generate arteries from non-neonatal human cells.
In the aforementioned arteries that were successfully engineered, the SMCs that comprise most of the arteries had a long lifespan in culture. SMCs divide a finite number of times before undergoing growth arrest in a state known as senescence. This is largely due to the progressive erosion of chromosome-capping telomeres with each cell division, which ultimately leads to a critically short telomere length that signals senescence (Bierman, 1978; Bonin et al.,1999; Sedivy, 1998). SMCs must proliferate for at least 45–60 population doublings (PDs) to produce a mechanically robust artery in vitro. Bovine, porcine and human fetal and neonatal cells have long telomeres, and correspondingly divide extensively in culture (Kozik et al., 1998; Allsopp et al., 1992; and data not shown); long enough, in fact, to form arteries in vitro. However, non-neonatal human SMCs that would be used clinically for tissue engineering can proliferate in vitro for only 10–30 PDs before undergoing senescence (Bierman, 1978; Bonin et al., 1999). The limited lifespan of non-neonatal SMCs may, therefore, be the rate-limiting step in constructing autologous human arteries in vitro.
Most immortal human cells overcome telomere shortening by the de novo elongation of telomeres by the enzyme telomerase (Collins & Mitchell, 2002). In humans, this enzyme is minimally composed of the telomerase RNA subunit (hTR) and the telomerase reverse transcriptase subunit (hTERT; Nakamura & Cech, 1998). Most somatic cells express hTR, but not hTERT, and hence lack telomerase activity and are therefore mortal (Collins & Mitchell, 2002). However, ectopic expression of hTERT in somatic cells restores telomerase activity, arrests telomere shortening and, in a subset of cell types, overcomes senescence (Bodnar et al., 1998; Vaziri & Benchimol, 1998; Harley, 2002). If expression of hTERT in non-neonatal SMCs could extend their proliferative capacity, such cells could be used to construct robust arteries, which could ultimately be used in bypass surgery. Here, we show for the first time that human arteries can indeed be reconstructed in vitro using non-neonatal SMCs that have had their lifespans extended by ectopic hTERT expression.
Results and Disussion
hTERT extends the lifespan of smooth-muscle cells
Normal SMCs, which were isolated from aortic tissue of a child donor, were infected with control or hTERT retroviral vectors to generate stably infected polyclonal populations. Telomerase activity was restored in the hTERT-expressing SMCs (hTERT-SMCs), but not in uninfected or vector-infected control SMCs (Fig. 1A). The reactivation of telomerase activity also arrested telomere shortening and extended the lifespan of hTERTsMCs. Southern blot analysis using a telomeric probe showed that the telomere-containing terminal restriction fragments of uninfected and vector-infected SMCs decreased over time (Fig. 1B), arresting at a length of 6–8 kb, consistent with the length of telomeres of other senescent cells (Harley et al., 1994). Correspondingly, these cells had a finite lifespan, reaching senescence after ∼37 PDs (Fig. 1C). By contrast, the length of telomeres of hTERTsMCs increased with time (Fig. 1B), and these cells proliferated over 100 PDs, more than twice the lifespan of the control vector-infected SMC cultures, and long enough to theoretically engineer a robust blood vessel (L'Heureux et al., 1998; Niklason et al., 1999, 2001; Fig. 1C). Beyond 37 PDs, the proliferation rate of hTERTsMCs remained constant and identical to that of pre-senescent uninfected and vector-infected SMCs.
Figure 1.

Telomerase activity in smooth-muscle cells increases lifespan and extends telomeres. (A) Stable infection of smooth-muscle cells (SMCs) with a retroviral vector encoding the human telomerase reverse transcriptase subunit (hTERT) yielded hTERT-SMCs (hT) that were positive for telomerase activity, whereas control uninfected SMCs (un) and vector-infected SMCs (v) were negative. Human embryonic kidney cells that expressed hTERT (Armbruster et al., 2001) were used as a positive control (Ctl). (B) Southern hybridization shows telomere erosion in control uninfected (un) and vector-infected (v) SMCs, whereas hTERTsMCs extend telomeres for at least 80 population doublings (PDs). (C) Control uninfected (un) and vector-infected (v) SMCs senesced at PD 37, whereas hTERT-SMCs grew until at least PD 100. The asterisk indicates the PCR internal control. HI, heat inactivated.
hTERT-SMCs are phenotypically normal
As observed in other cell types (Harley, 2002), ectopic expression of hTERT in SMCs had no measurable effect on the differentiated status of the cells. Late-passage (> PD 100) hTERTsMCs retained a normal morphology, having striated cell bodies and forming dense muscle-like sheets of cells, which was similar to normal, pre-senescent SMCs (data not shown). Late-passage hTERT-SMCs also showed a differentiated phenotype, identical to control SMCs, retaining the expression of proteins characteristically expressed in SMCs (Owens, 1995), such as calponin (an intermediate differentiation marker), smooth-muscle myosin-heavy-chains (SM–MHCs; an advanced differentiation marker) and tropoelastin (an extracellular matrix protein; Fig. 2A).
Figure 2.
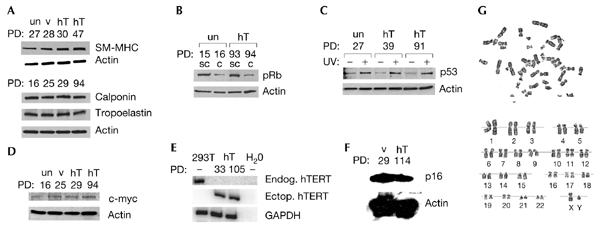
hTERT-SMCs are phenotypically normal. Smooth muscle cells (SMCs) were stably infected with a retroviral vector encoding the hTERT human telomerase reverse transriptase subunit (hT). As controls, SMCs were also stably infected with a control retroviral vector (v) or were left uninfected (un). Both late-passage hTERT-infected SMCs and the control SMCs (A) maintained protein expression of the differentiation markers calponin and smooth-muscle myosin heavy chain (SM-MHC) and the extracellular matrix protein tropoelastin, (B) downregulate hyperphosphorylated retinoblastoma protein (pRb) in high-density cultures, (C) upregulate p53 protein in response to DNA damage by ultraviolet irradiation, (D) did not overexpress c-MYC protein, (E) lacked endogenous hTERT messenger RNA and (F) expressed p16 protein. (G) SMCs stably infected with a retroviral vector encoding hTERT also had a normal karyotype. For immunoblot or RT–PCR (PCR after reverse transcription) analysis, actin or glyceraldehyde-3-phosphate dehydrogenase (GADPH) served as a loading control, respectively. 293T cells served as a positive control and water as a negative control in RT–PCR assays for endogenous hTERT expression. C, confluent cultures; ectop. hTERT, ectopic hTERT; endog. hTERT, endogenous hTERT; PDs, population doublings; SC, subconfluent cultures; UV+ or −, treatment with or without ultraviolet irradiation.
Telomerase is activated in most cancer cells (Shay & Bacchetti, 1997). hTERT expression in normal human cells, in combination with oncogenic Ras and the early region of SV40 (simian virus 40) can also generate a malignant phenotype (Elenbaas et al., 2001; Hahn et al., 1999; Lundberg et al., 2002; Rich et al., 2001). Although the ectopic expression of hTERT in several normal human cell types does not induce neoplastic alterations (Jiang et al., 1999; Morales et al., 1999; Yang et al., 1999; Yang et al., 2001), we nevertheless examined whether hTERTsMCs showed transformed phenotypes. Normal cells can prevent entry into S phase by dephosphorylating the retinoblastoma protein (pRb), whereas many tumour cells escape this block by keeping pRb constitutively hyperphosphorylated (Lundberg & Weinberg, 1999). In hTERTsMCs and uninfected SMCs at high cell density, hyperphosphorylated pRb was downregulated (Fig. 2B). Normal cells also upregulate the p53 protein to halt proliferation in response to DNA damage, whereas many tumour cells lose this response (Lundberg & Weinberg, 1999). DNA damage due to ultraviolet irradiation stimulated the upregulation of p53 in hTERTsMCs, similar to controls (Fig. 2C). Although a recent report showed upregulation of the proto-oncogene c-MYC in one hTERT-expressing cell line (Wang et al., 2000), late-passage hTERTsMCs expressed c-MYC at levels similar to controls (Fig. 2D). In addition, endogenous hTERT messenger RNA was not detected by RT–PCR (PCR after reverse transcription; Fig. 2E), and p16, a tumoursuppressor gene that is often silenced to permit extended proliferation (Drayton & Peters, 2002), was detectable in both control SMCs and late-passage hTERTsMCs (Fig. 2F). This supports the conclusion that hTERTsMCs are immortal due to ectopic hTERT expression, and not because of mutations in endogenous genes. Indeed, late-passage hTERT-SMCs remained polyclonal (data not shown), and 20 late-passage hTERT-SMCs that were analysed by conventional cytogenetic methods had a normal complement of chromosomes (46,XY) and lacked any obvious cytogenetic aberrations (Fig. 2G). Last, late-passage hTERTsMCs, like uninfected SMCs, failed to grow in an anchorage-independent manner in soft agar; such growth is a transformed phenotype that is characteristic of cancer cells (data not shown). Therefore, SMCs that express hTERT are immortal, but, according to several criteria, do not show signs of being transformed or abnormal.
Robust vessels engineered in vitro using hTERT-SMCs
To determine whether hTERT expression would enable SMCs to proliferate sufficiently for use in human vascular tissue engineering, blood vessels were cultured using a biomimetic system as previously described (Niklason et al., 1999, 2001). Uninfected or hTERT-infected SMCs were seeded onto tubular scaffolds of degradable polyglycolic acid (PGA) mesh, and were cultured under pulsatile pressure in bioreactors. Four human vessels were grown each from uninfected and hTERT-infected SMCs. After seven weeks, hTERTsMC blood vessels were seeded luminally with human umbilical vein endothelial cells (HUVECs). HUVECs were used because such cells are readily available and because adult endothelial cells are not a limitation in creating an engineered human artery, due to the few required divisions for these cells (Niklason et al., 1999, 2001). The adhesion and phenotype of the HUVECs were verified by the presence of a confluent monolayer of von Willebrand Factor (vWF)-positive cells in the lumen (Fig. 3E). Vessels generated from unifected SMCs could not be seeded with ECs, as they were extremely fragile.
Figure 3.

Telomerase enables robust vascular tissue engineering. Haematoxylin–eosin staining of smooth-muscle cells (SMCs) uninfected (A) or stably infected with a retrovirus encoding the hTERT human telomerase reverse transcriptase subunit (B). TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labelling) staining of vessels derived from (C) uninfected SMCs shows widespread cell death seen as dark-staining nuclei in contrast to (D) vessels engineered with SMCs expressing hTERT. (E) Immunostaining for von Willebrand Factor shows a confluent monolayer of endothelial cells lining the lumen (*) of an hTERT-SMC vessel. (F) Immunoblots showing that all four hTERT-SMC vessels engineered maintain expression of protein characteristics of differentiated SMCs, and this expression is similar to that seen in vessels derived from uninfected SMCs. Scale bars, 0.5 mm (A,B); 100 µm (C–E). Actin was used as a loading control. SM-MHC, smooth-muscle myosin heavy chain.
The physical appearance of hTERT vessels was markedly improved compared with control SMC vessels (Fig. 3A,B). The wall thicknesses of hTERT vessels (Table 1) were significantly greater than for controls (p < 0.01), and were similar to that of the human saphenous vein (Caro et al., 1978). hTERT vessels also had a cell density (135 ± 21 SMCs ml−1; Table 1) similar to that of the human saphenous vein (164 ± 31 SMCs ml−1; unpublished results). hTERT vessels had significantly higher rupture strengths than control vessels (356 ± 32 mm Hg for hTERT compared with 59 ± 28 mm Hg for controls; p < 0.0005; Table 1). The increased wall thickness was probably resposible for the increased strength of the hTERT vessels. hTERT expression thus enabled the culture of human arteries that were architecturally and mechanically superior to the few vessels that we managed to engineer from unifected SMCs.
Table 1.
Physical characteristics of engineered human blood vessels
| Physical characteristic | Uninfected SMCsa | hTERT-SMCsa |
|---|---|---|
| Wall thickness (μm) | 181 ± 75 | 360 ± 76b |
| Cell density (× 106 SMCs ml−1) | 118 ± 26 | 135 ± 42 |
| Rupture strength (mm Hg) | 59 ± 55 | 356 ± 64c |
| Collagen (% dry weight) | 5.1 ± 2.2 | 7.4 ± 1.5 |
aValues are the mean ± s.e.m. of the four samples. hTERTsMCs, vessels generated using smooth-muscle cells (SMCs) infected with a human telomerase reverse transcriptase (hTERT) encoding retroviral vector; uninfected SMCs, vessels generated using unifected SMCs.
bp < 0.01;
cp < 0.0005.
In engineered tissues, telomerase expression significantly improved cellular viability. Apoptosis in the engineered arteries was evaluated by terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL; Figs. 3C,D). hTERTsMC vessels had 11 ± 2% positive nuclei, compared with 53 ± 11% positive nuclei for controls (p < 0.05), suggesting that the more robust hTERT-SMC vessels did not undergo the cell death that was characteristic of the weaker control vessels. Immunostaining for proliferating cell nuclear antigen (PCNA) showed that hTERT-SMC vessels undergo similar cellular proliferation to uninfected SMC vessels after seven weeks of culture (6.6 ± 1.5% compared to 27 ± 13% positive nuclei, respectively; p was not significant), suggesting that no inappropriate growth occurred in the vessels, which further indicated that a non-transformed phenotype was maintained. Last, hTERT-SMCs in vessels maintained a differentiated phenotype. hTERT vessels and control vessels showed similar levels of expression of calponin, SM-MHC, tropoelastin and collagen (Fig. 3F; Table 1). Thus, hTERTsMCs maintained a viable and differentiated phenotype over prolonged vessel-culture periods.
Speculation
By exploiting telomerase expression in human SMCs, we have produced the first tissue-engineered, non-neonatal human arteries that could potentially be used for surgical applications. Importantly, the lifespan-extended SMCs maintained a differentiated, non-malignant phenotype over prolonged culture periods. As vascular pathology, donor age and cell source probably contribute to the variable replicative ability of adult human SMCs (Bierman, 1978; Bonin et al., 1999; Chang & Harley, 1995), telomerase expression could normalize the replicative capacity of donor cells for vascular tissue engineering. Our work provides a foundation for creating such arteries from older donors, and for exploring the possibility of using autologous vascular cells. In addition, hTERTsMCs and in vitro conditions could be manipulated to generate even stronger vessels. Manipulating telomerase expression therefore has viable therapeutic potential for use in bypass surgery, and by inference, may prove valuable in other tissue engineering applications.
Methods
Cells and tissues.
Human aortic SMCs that were isolated from a two-year-old male were propagated in SMGM-2 containing 10–20% fetal bovine serum, proline, glycine, ascorbic acid, all at 50 μg ml−1 and alanine at 20 μg ml−1 (BioWhittaker). These cells were infected at early passage with amphotropic retroviruses that were derived from pBABEhygro or from a pBABEhygro derivative encoding hTERT cDNA (Meyerson et al., 1997) tagged with the FLAG epitope. Cells were then selected using 60 μg ml−1 hygromycin, as previously described (Armbruster et al., 2001). HUVECs (BioWhittaker) were cultured in EGM-2 (in the absence of hydrocortisone and ascorbic acid; BioWhittaker) supplemented with 10% FBS and 150 μg ml−1 heparin (GIBCO-BRL).
Telomerase activity, hTERT expression, telomere analysis and cytogenetic analysis.
1.0-μg samples of lysates that were isolated from SMCs were assayed for telomerase activity as described previously (Kim & Wu, 1997). As controls, duplicate lysates were heat inactivated for 2 min at 85 °C. RT–PCR assays to detect hTERT and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were carried out as described previously (Armbruster et al., 2001), with the exception that endogenous hTERT mRNA was detected using primers 5′-CGTGCCACTCCTGGGGTCAC-3′ and 5′-CCTGGGACGTAGAGCCCGGC-3′. TRFs were visualized by resolving 1 μg of genomic DNA digested with HinfI and RsaI on 0.5% agarose gels, which were hybridized with a 32P-labelled (CCCTAA)3 probe, followed by three washes with 15× sodium chloride sodium citrate, as described previously (Counter et al., 1992). G-banded metaphase chromosomes from late-passage (107 PDs) hTERTsMC were prepared and analysed by standard cytogenetic procedures.
Blood-vessel culture and endothelialization.
Blood-vessel bioreactors, pulsatile-flow systems and PGA meshes were prepared as described previously (Niklason et al., 1999, 2001) and seeded with either uninfected SMCs (11 × 106 cells; PD 20) or SMCs infected with hTERT-encoding retrovirus (8 × 106 cells; PD 32; after 9 PDs with selection). Bioreactors were filled with hygromycin-free culture medium, and meshes were pulsed internally using silicone tubing (165 beats per min, 1% radial distension) with humidification and 10% CO2 at 37 °C. Vessels were endothelialized with 3.6 × 106 HUVECs at passage 2 by removing the inner silicone tubing, injecting the HUVECs into the vessel lumen, and allowing the cells to adhere for 16 h with periodic rotation.
Mechanical testing and calculations.
Vessel mechanics were quantified using a circumferential-stretch test, similar to that described previously (Donovan et al., 1990; Seliktar et al., 2000). Two wires were inserted through the lumen of a vessel segment, to which increasing tension was applied. The distending vessel was imaged using a digital video camera with a 10-μm pixel size (XL1; Canon), and the external radius was measured. The wall area from each crosssectional image was used to calculate the average wall thickness, the circumferential wall stress, and the theoretical rupture-strength, as previously described (Pagani et al., 1979).
Western-blot analysis.
For pRb blots, duplicate cell cultures were seeded at the same densities, collected and solubilized after three days (subconfluent) or seven days (confluent; Jiang et al., 1999; Morales et al., 1999). For p53 blots, duplicate subconfluent cultures were either left untreated or were subjected to 6 Gy of irradiation for 1.2 min, collected 4 h later, and solubilized (Morales et al., 1999). For p16 blots, cell cultures were collected and solubilized. Blots using lysates from cells and engineered arteries were performed as described in Higgins et al. (2002), using primary antibodies to β-actin, calponin, tropoelastin (Sigma), SM-MHC (Biomedical Technologies), hyper-phosphorylated pRb (Cell Signaling Technology), p53, c-Myc (Wang et al., 2000; Santa Cruz) and p16 (PharMingen).
Immunostaining.
Vessel samples were fixed in formalin, dehydrated, and embedded in paraffin. 5-μm sections were deparaffinized and immunostained for the presence of DNA-strand breaks (Apoptag Apoptosis Detection Kit; Intergen), PCNA (Niklason et al., 1999), or vWF (DAKO), in accordance with the manufacturers' instructions. A Student's t-test was performed to compare the number of TUNEL-positive or PCNA-positive cells in the control and hTERTsMC vessels.
Cell density analysis.
As described in Niklason et al. (1999), vessel samples were weighed and lyophilized to obtain wet and dry weights, respectively. The samples were incubated in papain (1 ml; 0.7 μg ml−1) at 60 °C overnight. 2 ml of Hoechst dye (Polysciences) was added to each of the samples, which were then analysed in a spectrofluorimeter (excitation at 365 nm, emission at 458 nm), calibrated using calf thymus DNA standards. A Student's t-test was performed to compare cell densities between control and hTERTsMC vessels.
Collagen analysis.
Papain-digested samples were incubated in 6 N HCl at 115 °C for 18 h, neutralized, reacted with p-dimethylaminobenzaldehyde and chloramine-T, and quantified at a wavelength of 555 nm. A 1:10 w/w ratio of hydroxyproline and collagen was used to calculate the collagen content of the vessels. A Student's t-test was performed to compare collagen contents between control and hTERT-SMC vessels.
Acknowledgments
We thank R. Nelson and J. Flowers for technical assistance and T. Veldman for cytogenetic analysis of SMCs. This work was supported by grants from the American Federation for Aging Research to L.E.N. and from the National Cancer Institute (grant number CA82481) to C.M.C. C.M.C. is a Leukemia and Lymphoma Society Scholar.
References
- Allsopp R.C., Vaziri H., Patterson C., Goldstein S., Younglai E.V., Futcher A.B., Greider C.W. & Harley C.B. (1992) Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl Acad. Sci. USA, 89, 10114–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster B.N., Banik S.S.R., Guo C., Smith A.C. & Counter C.M. (2001) N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vitro. Mol. Cell. Biol., 21, 7775–7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierman E.L. (1978) The effect of donor age on the in vitro life span of cultured human arterial smooth-muscle cells. In Vitro, 14, 951–955. [DOI] [PubMed] [Google Scholar]
- Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S. & Wright W.E. (1998) Extension of lifespan by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bonin L.R., Madden K., Shera K., Ihle J., Matthews C., Aziz S., Perez-Reyes N., McDougall J.K. & Conroy S.C. (1999) Generation and characterization of human smooth muscle cell lines derived from atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol, 19, 575–587. [DOI] [PubMed] [Google Scholar]
- Caro C.G., Pedley T.J., Schroter R.C. & Seed W.A. (1978) The Mechanics of the Circulation. Oxford Univ. Press, Oxford, UK. [Google Scholar]
- Chang E. & Harley C.B. (1995) Telomere length and replicative aging in human vascular tissues. Proc. Natl Acad. Sci. USA, 92, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K. & Mitchell J.R. (2002) Telomerase in the human organism. Oncogene, 21, 564–579. [DOI] [PubMed] [Google Scholar]
- Counter C.M., Avilon A.A., LeFeuvre C.E., Stewart N.G., Greider C.W., Harley C.B. & Bacchetti S. (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan D.L., Schmidt S.P., Townshend S.P., Njus G.O. & Sharp W.V. (1990) Material and structural characterization of human saphenous vein. J. Vasc. Surg., 12, 531–537. [PubMed] [Google Scholar]
- Drayton S. & Peters G. (2002) Immortalisation and transformation revisited. Curr. Opin. Genet. Dev., 12, 98–104. [DOI] [PubMed] [Google Scholar]
- Elenbaas B., Spirio L., Koerner F., Fleming M., Zimonjic D., Donaher J., Popescu N., Hahn W. & Weinberg R. (2001) Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev., 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W. & Weinberg R.A. (1999) Creation of human tumour cells with defined genetic elements. Nature, 400, 464–468. [DOI] [PubMed] [Google Scholar]
- Harley C.B. (2002) Telomerase is not an oncogene. Oncogene, 21, 494–502. [DOI] [PubMed] [Google Scholar]
- Harley C.B. et al. (1994) Telomerase, cell immortality, and cancer. Cold Spring Harb. Symp. Quant. Biol., 59, 307–315. [DOI] [PubMed] [Google Scholar]
- Higgins S.P., Solan A. & Niklason L.E. Effects of polyglycolic acid on porcine smooth muscle cell growth and differentiation. J. Biomed. Mater. Res. (in the press). [DOI] [PubMed] [Google Scholar]
- Jiang X.-R. et al. (1999) Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nature Genet., 21, 111–114. [DOI] [PubMed] [Google Scholar]
- Kempczinski R.F. (2000) in Vascular Surgery (ed. Rutherford, R.), 527–532. W.B. Saunders, Denver, Colorado, USA. [Google Scholar]
- Kim N.W. & Wu F. (1997) Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res., 25, 2595–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozik A., Bradbury E.M. & Zalensky A. (1998) Increased telomere size in sperm cells of mammals with long terminal (TTAGGG)n arrays. Mol. Reprod. Dev., 51, 98–104. [DOI] [PubMed] [Google Scholar]
- L'Heureux N., Paquet S., Labbe R., Germain L. & Auger F.A. (1998) A completely biological tissue-engineered human blood vessel. FASEB J., 12, 47–56. [DOI] [PubMed] [Google Scholar]
- Langer R. & Vacanti J.P. (1993) Tissue engineering. Science, 260, 920–926. [DOI] [PubMed] [Google Scholar]
- Lefkowitz R.J. & Willerson J.T. (2001) Prospects for cardiovascular research. JAMA, 285, 581–587. [DOI] [PubMed] [Google Scholar]
- Lundberg A.S. & Weinberg R.A. (1999) Control of the cell cycle and apoptosis. Eur. J. Cancer, 35, 531–539. [PubMed] [Google Scholar]
- Lundberg A.S. et al. (2002) Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene, 21, 4577–4586. [DOI] [PubMed] [Google Scholar]
- Meyerson M. et al. (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- Morales C.P., Holt S.E., Ouellette M., Kaur K.J., Yan Y., Wilson K.S., White M.A., Wright W.E. & Shay J.W. (1999) Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nature Genet., 21, 115–118. [DOI] [PubMed] [Google Scholar]
- Nakamura T.M. & Cech T.R. (1998) Reversing time: origin of telomerase. Cell, 92, 587–590. [DOI] [PubMed] [Google Scholar]
- Niklason L.E. (1999) Replacement arteries made to order. Science, 286, 1493–1494. [DOI] [PubMed] [Google Scholar]
- Niklason L.E., Gao J., Abbott W.M., Hirschi K., Houser S., Marini R. & Langer R. (1999a) Functional arteries grown in vitro. Science, 284, 489–493. [DOI] [PubMed] [Google Scholar]
- Niklason L.E. et al. (2001) Morphologic and mechanical characteristics of bovine engineered arteries. J. Vasc. Surg., 33, 628–638. [DOI] [PubMed] [Google Scholar]
- Owens G.K. (1995) Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev., 75, 487–517. [DOI] [PubMed] [Google Scholar]
- Pagani M., Mirsky I., Baig H., Manders W.T., Kerkhoff P. & Vatner S.F. (1979) Effects of age on aortic pressure–diameter and elastic stiffness–stress relationships in unanesthetized sheep. Circ. Res., 44, 420–429. [DOI] [PubMed] [Google Scholar]
- Rich J., Guo C., McLendon R., Bigner D., Wang X. & Counter C. (2001) A genetically tractable model of human glioma formation. Cancer Res., 61, 3556–3560. [PubMed] [Google Scholar]
- Sedivy J.M. (1998) Can ends justify the means?: telomeres and the mechanisms of replicative senescence and immortalization in mammalian cells. Proc. Natl Acad. Sci. USA, 95, 9078–9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliktar D., Blcak R.A., Vito R.P. & Nerem R.M. (2000) Dynamic mechanical conditioning of collagen-gel blood vessel constructs induces remodeling in vitro. Ann. Biomed. Eng., 28, 351–362. [DOI] [PubMed] [Google Scholar]
- Shay J.W. & Bacchetti S. (1997) A survey of telomerase activity in human cancer. Eur. J. Cancer, 33, 787–791. [DOI] [PubMed] [Google Scholar]
- Vacanti J.P. & Langer R. (1999) Tissue engineering: the design and fabrication of living replacement devices for surgical reconstruction and transplantation. Lancet, 354 (suppl. 1), SI32–SI34. [DOI] [PubMed] [Google Scholar]
- Vaziri H. & Benchimol S. (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol., 8, 279–282. [DOI] [PubMed] [Google Scholar]
- Wang J., Hannon G.J. & Beach D.H. (2000) Risky immortalization by telomerase. Nature, 405, 755–756. [DOI] [PubMed] [Google Scholar]
- Williams S.K. (2000) Tissue engineered vascular grafts: from bench to clinical use. FASEB J., 14, A305. [Google Scholar]
- Yang J., Chang E., Cherry A.M., Bangs C.D., Oei Y., Bodnar A., Bronstein A., Chiu C-P. & Herron G.S. (1999) Human endothelial cell life extension by telomerase expression. J. Biol. Chem., 274, 26141–26148. [DOI] [PubMed] [Google Scholar]
- Yang J., Nagavarapu U., Relloma K., Sjaastad M.D., Moss W.C., Passaniti A. & Herron G.S. (2001) Telomerized human microvasculature is functional in vivo. Nature Biotechnol., 19, 219–224. [DOI] [PubMed] [Google Scholar]