

Abstract
The death-domain kinase RIP (receptor-interacting protein) is an important effector of tumour necrosis factor (TNF) signalling and is essential for TNF-induced nuclear factor-κB activation. However, the function of RIP in the TNF-induced activation of mitogen-activated protein kinases (MAPKs) has not been fully investigated. In this report, using Rip null (Rip−/−) mouse fibroblast cells, we investigated whether RIP is required for TNF-induced activation of the MAPKs extracellular-signal-related kinase (ERK), p38 and c-Jun amino-terminal kinase (JNK). We found that TNF-induced activation of ERK, p38 and JNK is decreased in Rip−/− cells. The activation of these kinases by interleukin-1 is normal in Rip−/− cells. More importantly, we showed that the kinase activity of RIP is needed for ERK activation.
Introduction
The pro-inflammatory cytokine tumour necrosis factor (TNF) has an important function in diverse cellular events, such as septic shock, induction of other cytokines, cell proliferation, differentiation and apoptosis (Tartaglia & Goeddel, 1992; Tracey & Cerami, 1993; Vandenabeele et al., 1995). The molecular mechanisms that regulate TNF-mediated responses have been studied intensively in recent years (Nagata & Golstein, 1995; Liu & Han, 2001). For tumour necrosis factor receptor 1 (TNFR1)-mediated signalling, several molecules, including TNFR-associated factor 2 (TRAF2) and the death-domain kinase RIP (receptor-interacting protein), have been identified as important effectors (Rothe et al., 1995; Stanger et al., 1995; Hsu et al., 1996a, b; Liu et al., 1996). TRAF2 is a member of the TRAF protein family (Rothe et al., 1995). Whereas the TRAF domain of TRAF2 is responsible for its recruitment to the TNFR complex, its ring- and zinc-finger domains are crucial for transducing the TNF signal to downstream molecules (Baud et al., 1999). RIP is a death-domain kinase, and its death domain is required for its recruitment to the TNFR signalling complex (Stanger et al., 1995; Hsu et al., 1996b; Ting et al., 1996). However, the function of RIP kinase activity in TNF signalling is not fully understood (Hsu et al., 1996b; Ting et al., 1996). Several lines of evidence indicate that both RIP and TRAF2 are involved in the TNF-induced activation of the transcription factor nuclear factor-κB (NF-κB; Rothe et al., 1995; Stanger et al., 1995; Hsu et al., 1996a, b; Liu et al., 1996; Yeh et al., 1997; Kelliher et al., 1998; Devin et al., 2000). Although neither RIP or TRAF2 are essential for TNF-induced apoptosis, a recent study suggested that RIP is essential for TNF-induced necrotic cell death (Holler et al., 2000).
Mitogen-activated protein kinases (MAPKs) are one group of signal-transducing enzymes that have important functions in mediating responses to various extracellular stimuli (Karin, 1998; Davis, 1999). Activation of MAPKs is one of the many cellular responses to TNF (Liu & Han, 2001). Three subgroups of MAPKs have been identified: extracellularsignal-regulated kinases (ERKs), c-Jun amino-terminal kinases (JNKs; also known as stress-activated protein kinases (SAPKs)) and p38 MAPKs (Karin, 1998; Davis, 1999). The activation of different types of MAPKs by TNF is cell-typespecific (Karin, 1998; Davis, 1999; Liu & Han, 2001). Although the TNF-induced activation of MAPKs has been studied intensively in the past few years, the molecular mechanisms that regulate the activation of these MAPKs by TNF are still not fully understood (Karin, 1998; Davis, 1999; Liu & Han, 2001). It has been shown that TRAF2 is essential for TNF-induced JNK activation (Liu et al., 1996; Reinhard et al., 1997; Yeh et al., 1997), and overexpression of TRAF2 leads to the activation of ERK and p38 (Liu & Han, 2001). However, the role of RIP in the TNF-induced activation of ERK and p38 is not clear, although it has been suggested that RIP is not required for TNF-induced JNK activation (Kelliher et al., 1998).
In this study, we investigated whether RIP is involved in p38 and ERK activation by TNF by using Rip null (Rip−/−) mouse fibroblasts. We also re-evaluated the role of RIP in TNF-induced JNK activation. In wild-type mouse embryonic fibroblast (MEF) cells, all three types of MAPK are activated potently by TNF. In Rip−/− cells, we found that RIP is required for TNF-induced activation of all three types of MAPK. Ectopic expression of RIP in Rip−/− cells partially restored the activation of JNK, ERK and p38 in response to TNF. Moreover, our results also indicated that the kinase activity of RIP is only required for the activation of ERK, but not of JNK and p38. To clarify the involvement of TRAF2 in TNF-induced ERK and p38 activation, Traf2 null (Traf2−/−) MEFs were also studied.
Results
RIP is required for TNFR1-mediated MAPK activation
To determine whether RIP is involved in TNF-induced p38, ERK and JNK activation, we measured the activation of these MAPKs in Rip−/− mouse fibroblasts, using wild-type fibroblasts as a control. In these cells, mouse TNF-α binds to both TNFR1 and TNFR2, whereas human TNF-α binds only to TNFR1 (Lewis et al., 1991). As TNFR1 and TNFR2 elicit some overlapping responses, we used human TNF-α to study TNFR1-induced MAPK activation. We detected p38 and ERK activation using anti-phospho-p38 and anti-phospho-ERK antibodies, and measured JNK1 activation using an in vitro kinase assay with glutathione-S-transferase (GST)–c-Jun as a substrate. As shown in Fig. 1A, all three types of MAPK were activated by TNF in wild-type fibroblasts. By contrast, in Rip−/− cells, the activation of these three types of MAPK was decreased to ∼40% for p38, ∼70% for ERK and ∼60% for JNK (Fig. 1A). The protein levels of p38, ERK, and JNK in Rip−/− cells were similar to those in wild-type cells (Fig. 1A). These results suggested that RIP might be required for the TNF-induced activation of p38, ERK and JNK. However, as the result of analysing TNF-induced JNK activation in Rip−/− cells was contradictory to a previous report (Kelliher et al., 1998), we also measured TNF-induced JNK activation by western blotting using an anti-phospho-JNK antibody. As shown in Fig. 1B, the activation of JNK by TNF was detected in wild-type fibroblasts but not in Rip−/− cells. This result further confirmed that RIP is also required for TNF-induced JNK activation. To investigate whether TRAF2 is required for p38 and ERK activation by TNF, we analysed p38 and ERK phosphorylation after TNF treatment in Traf2−/− cells. As shown in Fig. 1A, the phosphorylation of p38 and ERK was decreased in Traf2−/− cells, indicating that TRAF2 also functions in the TNF-induced activation of p38 and ERK. Consistent with a previous study (Yeh et al., 1997), JNK activation by TNF in Traf2−/− cells was also decreased. More importantly, as shown in Fig. 1C, the activation of p38, ERK and JNK in response to treatment with interleukin-1 (IL-1) was normal in Rip−/− and Traf2−/− cells (that is, it was similar to that in wild-type cells), suggesting that the pathways that mediate the activation of these MAPKs are intact in the Rip and Traf2 knockout cell-lines. These results also suggest that both RIP and TRAF2 are essential for the TNF-induced activation of p38, ERK and JNK.
Figure 1.
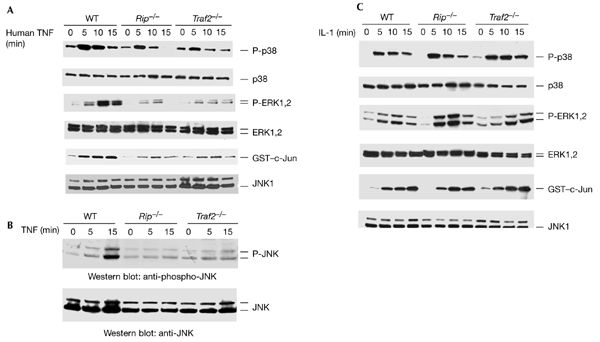
TNFR1-mediated p38, ERK and JNK activation require both TRAF2 and RIP. (A) Human-TNF-induced p38, ERK and JNK activation in wild-type, Rip−/− and Traf2−/− fibroblasts. Mouse fibroblasts were treated with human TNF (40 ng ml−1) for various durations or were left untreated as a control. Cell extracts were used for western blotting and in in vitro kinase assays to measure the activation of p38, ERK and JNK. (B) JNK activation in wild-type, Rip−/−, and Traf2−/− cells was measured using an anti-phospho-JNK antibody. (C) Interleukin-1 (IL-1)-induced p38, ERK and JNK activation. Wild-type, Rip−/− and Traf2−/− fibroblasts were either treated with IL-1 (4 ng ml−1) for various durations or were left untreated as a control. The activation of p38, ERK and JNK was measured as described in (A). These experiments were repeated three times. ERK, extracellular-signal-related kinase; GST, glutathione-S-transferase; hTNF, human TNF; JNK, c-Jun amino-terminal kinase; P-ERK, phospho-ERK; P-JNK, phospho-JNK; P-p38; phospho-p38; RIP, receptor-interacting protein; TNF, tumour necrosis factor; TNFR1, TNF receptor 1; TRAF2, TNFR-associated factor 2.
To rule out the possibility that Rip−/− cells have defects in the TNFR1-mediated MAPK activation pathway, we tested whether the TNF-induced activation of MAPKs could be reconstituted in these cells. We ectopically expressed MYC-tagged RIP in Rip−/− cells and treated the transfected cells with TNF. The activation of p38, ERK and JNK was measured by detecting their phosphorylation or kinase activities, as shown in Fig. 1. As shown in Fig. 2A, RIP expression restored p38, ERK and JNK activation in response to TNF treatment in Rip−/− cells. The expression level of MYC–RIP is shown in the bottom panel of Fig. 2A. Because only a certain percentage of cells were transfected with MYC–RIP, TNF-induced activation of MAPKs was only partially restored in the Rip−/− cells. When haemagglutinin (HA)-tagged p38, ERK and JNK were co-transfected with MYC–RIP, the reconstitution efficiency was much higher (Fig. 3A,C). To test whether the expression of TRAF2 in Traf2−/− cells also restores the activation of these three MAPKs in response to TNF, we used a previously established Traf2−/− cell line (Traf2−/− TRAF2) in which FLAG–TRAF2 is stably expressed. As shown in Fig. 2B, the activation of p38, ERK and JNK by TNF was restored in Traf2−/− TRAF2 cells. These results further suggest that the failure of p38, ERK and JNK activation in response to TNF treatment in Rip−/− and Traf2−/− cells is due to the absence of RIP and TRAF2, respectively.
Figure 2.
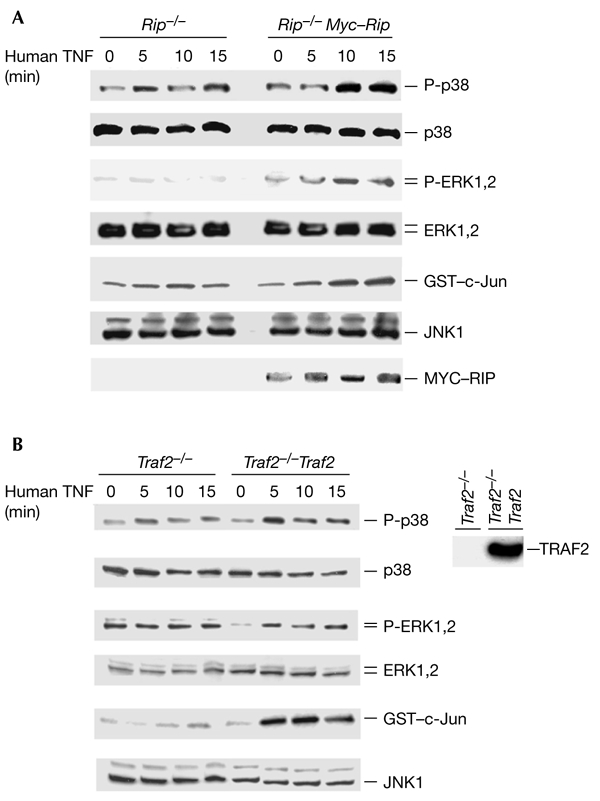
TNF-induced activation of p38, ERK and JNK are restored in Rip−/− or Traf2−/− cells by ectopically expressing MYC–RIP or FLAG–TRAF2. (A) Reconstitution of TNF-induced p38, ERK and JNK activation in Rip−/− fibroblasts. Wild-type or Rip−/− cells were transfected with 2 µg of either the MYC–RIP expression plasmid or an empty vector, in 100-mm dishes. Twenty-four hours after transfection, the transfected cells were treated with 40 ng ml−1 of human TNF for various durations or were left untreated as a control. The amounts of protein in cell extracts were quantified by performing protein assays and were then either immunoprecipitated with anti-JNK1 antibody to enable a kinase assay to be performed, or were resolved by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) followed by western blotting with anti-JNK1, anti-phospho-p38, anti-p38, anti-phospho-ERK, anti-ERK or anti-MYC antibodies. (B) Reconstitution of TNF-induced p38, ERK and JNK activation in Traf2−/− fibroblasts. Traf2−/− cells and Flag–Traf2 stably transfected cells were treated with human TNF-α (40 ng ml−1) for various durations or were left untreated as a control. The amounts of protein in cell extracts were quantified by performing protein assays and were then resolved by SDS–PAGE for western blotting with anti-JNK, anti-phospho-p38, anti-p38, anti-phospho-ERK, anti-ERK and anti-MYC antibodies. To measure JNK activity, the cell extracts were used in in vitro kinase assays. These experiments were repeated three times. ERK, extracellular-signal-related kinase; GST, glutathione-S-transferase; JNK, c-Jun amino-terminal kinase; P-ERK, phospho-ERK; P-p38, phospho-p38; RIP, receptor-interacting protein; TNF, tumour necrosis factor; Traf2, TNFR-associated factor 2.
Figure 3.
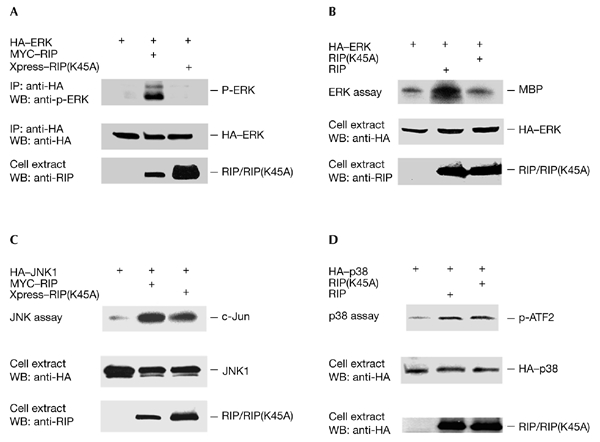
The RIP kinase domain is necessary for activating ERK. (A) Rip−/− cells were co-transfected in 60-mm dishes with 1 µg of the haemagglutinin (HA)–ERK1 expression plasmid and 0.5 µg of either the MYC–RIP expression plasmid, the Xpress–RIP(K45A) expression plasmid or an empty vector. Cells were collected 24 h after transfection. The HA–ERK1 content was quantified by western blotting (WB) using an anti-HA antibody, cell extracts were immunoprecipitated (IP) using an anti-HA antibody, and the immunoprecipitates were resolved by SDS–polyacrylamide gel electrophoresis for western blotting using anti-phospho-ERK1. The expression levels of MYC–RIP or Xpress–RIP(K45A) were detected using an anti-RIP antibody. (B) Experiments similar to those described in (A) were performed to measure ERK activity using an in vitro kinase assay with MBP as the substrate. In these experiments, Xpress-tagged RIP and RIP(K45A) were used, and these were detected using an anti-Xpress antibody. (C) Rip−/− cells were transfected in 60-mm dishes with 1 µg of the HA–JNK1 expression plasmid and 0.5 µg of either the MYC–RIP expression plasmid, the Xpress–RIP(A45K) expression plasmid or an empty vector. Twenty-four hours after transfection, cells were collected and the HA–JNK1 content was quantified by western blotting using an anti-HA antibody. Cell extracts were then immunoprecipitated with an anti-HA antibody for use in a kinase assay. The expression levels of RIP and RIP(K45A) were detected using an anti-RIP antibody. (D) Similar experiments to those described in (C) were performed using an HA–p38 plasmid, and HA–p38 activity was measured by performing an in vitro kinase assay with glutathione-S-transferase–ATF2 as the substrate. These experiments were repeated three times. ERK, extracellular-signal-related kinase; JNK, c-Jun amino-terminal kinase; RIP, receptor-interacting protein.
The kinase activity of RIP is required for ERK activation
Although RIP has been shown to have an important function in TNF signalling, its kinase activity has been found to be dispensable in several studies (Hsu et al., 1996b; Liu et al., 1996; Ting et al., 1996). However, it has been reported recently that the kinase activity of RIP is necessary for TNF-induced necrotic cell death (Holler et al., 2000). In this study, we tested whether the kinase activity of RIP is required for the activation of ERK. As ectopic expression of RIP can lead to the activation of ERK (data not shown; but see Fig. 3A,B), wild-type RIP and a kinase-dead form, RIP(K45A), were co-expressed with HA–ERK1 in Rip−/− cells, and HA-ERK1 was then immunoprecipitated to determine its activity, as described in Fig. 1. As shown in Fig. 3A (top panel), whereas wild-type RIP induced the activation of ERK, the kinase-dead RIP protein did not. The amount of immunoprecipitated HA–ERK1 is also shown in Fig. 3A (middle panel), as are the expression levels of RIP and RIP(K45A) (bottom panel). To confirm that the kinase activity of RIP is indeed essential for ERK activation, we performed similar experiments to those shown in Fig. 3A, except that ERK activity was analysed in an in vitro kinase assay using MBP as the substrate. Consistent with the results shown in Fig. 3A, whereas overexpression of wild-type RIP activated ERK, overexpression of the kinase-dead RIP(K45A) form did not increase the activity of ERK (Fig. 3B). When a similar experiment was performed with HA–JNK1, consistent with a previous report (Liu et al., 1996), the kinase activity of RIP did not have a role in JNK activation (Fig. 3C). In a similar way, the function of the kinase activity of RIP in p38 activation was tested using HA–p38. As shown in Fig. 3D, the kinase activity of RIP was not required for the activation of p38. Therefore, these results suggest that the kinase activity of RIP is only essential for ERK activation.
Discussion
TNF is a pleiotropic pro-inflammatory cytokine that has a crucial function in many physiological and pathological processes. Many cellular responses, including the activation of NF-κB and MAPKs, are elicited in response to TNF (Tartaglia & Goeddel, 1992; Tracey & Cerami, 1993; Vandenabeele et al., 1995; Liu & Han, 2001). It is thought that all cellular responses to TNF are mediated by the effector molecules of TNF receptors. In this study, we evaluated the role of two TNF effector proteins, RIP and TRAF2, in the TNF-induced activation of the MAPKs, p38, ERK and JNK in mouse fibroblast cells. We found that both RIP and TRAF2 are required for the TNF-induced activation of p38, ERK and JNK. Although previous studies have suggested that TRAF2 is essential for TNF-induced JNK activation, the function of these two effectors in TNF-induced p38 and ERK activation remains unclear. Our findings indicate that, as for NF-κB activation, the activation of p38, ERK and JNK is also mediated by both RIP and TRAF2.
The death-domain kinase RIP has been found to have a crucial function in TNF-induced NF-κB activation, but not in JNK activation (Kelliher et al., 1998). In addition, it was recently reported that RIP is essential for TNF-induced necrotic cell death (Holler et al., 2000). Our study suggests that RIP is required for TNF-induced activation of all three types of MAPK. However, our finding that RIP is required for TNF-induced JNK activation is contradictory to a previous study (Kelliher et al., 1998). Because the same Rip−/− cells were used in both studies, the cause for the discrepancy is unclear. Interestingly, a recent study using mice in which Rip2 (a homologue of Rip) is knocked out suggested that RIP2 is required for TOLL-like-receptor (TLR)-induced NF-κB activation and for the activation of all three types of MAPK (Kobayashi et al., 2002). Our study suggests that RIP, like RIP2 in TLR signalling, mediates most TNF-induced pathways, but not the apoptotic pathway. However, it is still unclear as to how the signal is transduced from RIP to its downstream targets. For example, it has been proposed that MAP3Ks such as MEKK1 and ASK1 may be the immediate targets of RIP and TRAF2, but studies with Map3k-null cells have suggested otherwise (Xia et al., 2000; Yujiri et al., 2000; Tobiume et al., 2001). We also analysed the role of two of the MAP3Ks, MEKK1 and MEKK3, in the TNF-induced activation of p38, ERK and JNKs using MEKK1- or MEKK3-null cells. We found that deletion of either MEKK1 or MEKK3 had no effect on the TNF-induced activation of p38, ERK and JNK (data not shown). One possibility is that there is redundancy among these MAP3Ks in the TNF-induced activation of MAPKs. Therefore, double or even triple Map3k knock-out mice will be necessary to further address this issue. To test whether Raf is involved in TNF-induced ERK activation, we performed co-immunoprecipitation experiments with anti-TNFR1 and examined whether Raf is present in the signalling complex. We found that neither Raf-A nor Raf-B were present in the TNFR1 complex after TNF treatment (data not shown). Therefore, it is still not clear what are the immediate downstream targets of RIP and TRAF2. Nevertheless, because the kinase domain of RIP is only required for activating ERK, but not JNK and p38, it is likely that RIP interacts with different downstream targets to activate different MAPKs.
Methods
Reagents and plasmids.
The anti-RIP antibody was purchased from Transduction Laboratories. Anti-TRAF2, anti-HA and anti-Xpress antibodies were purchased from Santa Cruz. Anti-MYC and anti-JNK1 antibodies were purchased from Pharmingen. Anti-ERK, anti-p38, anti-phospho-ERK, anti-phospho-p38 and anti-phospho-JNK were from NEB. Human and mouse TNF-α and IL-1 were purchased from R&D Systems. The mammalian expression plasmids that were used to express RIP, TRAF2, HA–JNK1, HA–p38 and HA–ERK1 have been described previously (Liu et al., 1996; Baud et al., 1999; Devin et al., 2000).
Cell culture and transfection.
Wild-type mouse fibroblast cells were cultured in DMEM supplemented with 10% FBS (or 10% FCS), 2 mM glutamine, 100 units ml−1 penicillin and 100 μg ml−1 streptomycin. Rip−/− and Traf2−/− cells were cultured in the same medium, except that 0.3 mg ml−1 G418 was included. Transfections were performed with Lipofectamine PLUS reagent (Invitrogen), following the instructions provided by the manufacturer.
Western blot analysis.
For western blotting, cells were treated with human TNF-α, as indicated in the figure legends, and were then collected in M2 lysis buffer (20 mM Tris, pH 7, 0.5% NP40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 0.5 mM PMSF, 20 mM β-glycerol phosphate, 1 mM sodium vanadate, 1 μg ml−1 leupeptin, 1 μg ml−1 aprotinin, 1 μg ml−1 pepstatin and 10 mM pNpp). Fifty micrograms of the cell lysates were separated using 4–20% or 10% SDS–polyacrylamide gels, and western blotting was performed with the appropriate antibodies. The proteins were visualized by enhanced chemiluminescence (ECL; Amersham) in accordance with the manufacturer's instructions.
Kinase assays.
Cells were collected in M2 lysis buffer as described in Devin et al. (2000). JNK1, HA–JNK1, HA–p38 and HA–ERK1 were immunoprecipitated with anti-JNK1 (Pharmingen) and anti-HA antibodies, respectively. In vitro kinase assays were performed as described previously, using GST–c-Jun peptides (residues 1–79) and MBP as exogenous substrates for measuring JNK and ERK activities, respectively (Liu et al., 1996; Devin, et al., 2000). The HA–p38 kinase assay was performed using GST–ATF2 as the substrate (Cell Signaling Technology).
Acknowledgments
We thank W.-C. Yeh and T.W. Mak for Traf2−/− fibroblasts, and M. Kelliher for Rip−/− fibroblasts. We are grateful to J. Woo for assistance with manuscript preparation.
References
- Baud V., Liu Z.G., Bennett B., Suzuki N., Xia Y. & Karin M. (1999) Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev., 13, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.J. (1999) Signal transduction by the c-Jun N-terminal kinase. Biochem. Soc. Symp., 64, 1–12. [DOI] [PubMed] [Google Scholar]
- Devin A., Cook A., Lin Y., Rodriguez Y., Kelliher M. & Liu Z.G. (2000) The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity, 12, 419–429. [DOI] [PubMed] [Google Scholar]
- Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J.L., Schneider P., Seed B. & Tschopp J. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature Immunol., 1, 489–495. [DOI] [PubMed] [Google Scholar]
- Hsu H., Shu H.B., Pan M.G. & Goeddel D.V. (1996a) TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell, 84, 299–308. [DOI] [PubMed] [Google Scholar]
- Hsu H., Huang J., Shu H.B., Baichwal V. & Goeddel D.V. (1996b) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity, 4, 387–396. [DOI] [PubMed] [Google Scholar]
- Karin M. (1998) Mitogen-activated protein kinase cascades as regulators of stress responses. Ann. NY Acad. Sci., 851, 139–146. [DOI] [PubMed] [Google Scholar]
- Kelliher M.A, Grimm S., Ishida Y., Kuo F., Stanger B.Z. & Leder P. (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity, 8, 297–303. [DOI] [PubMed] [Google Scholar]
- Kobayashi K., Inohara N., Hernandez L.D., Galan J.E., Nunez G., Janeway C.A., Medzhitov R. & Flavell R.A. (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature, 416, 194–199. [DOI] [PubMed] [Google Scholar]
- Lewis M., Tartaglia L.A., Lee A., Bennett G.L., Rice G.C., Wong G.H., Chen E.Y. & Goeddel D.V. (1991) Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl Acad. Sci. USA, 88, 2830–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.G. & Han J. (2001) Cellular responses to tumor necrosis factor. Curr. Issues Mol. Biol. 3, 79–90. [PubMed] [Google Scholar]
- Liu Z.G, Hsu H., Goeddel D.V. & Karin M. (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell, 87, 565–576. [DOI] [PubMed] [Google Scholar]
- Nagata S. & Golstein P. (1995) The Fas death factor. Science, 267, 1449–1456. [DOI] [PubMed] [Google Scholar]
- Reinhard C., Shamoon B., Shyamala V. & Williams L.T. (1997) Tumor necrosis factor-α-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J., 16, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M., Sarma V., Dixit V.M. & Goeddel D.V. (1995) TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science, 269, 1424–1427. [DOI] [PubMed] [Google Scholar]
- Stanger B.Z., Leder P., Lee T.H., Kim E. & Seed B. (1995) RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell, 81, 513–523. [DOI] [PubMed] [Google Scholar]
- Tartaglia L.A. & Goeddel D.V. (1992) Two TNF receptors. Immunol. Today, 13, 151–153. [DOI] [PubMed] [Google Scholar]
- Ting A.T., Pimentel-Muinos F.X. & Seed B. (1996) RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J., 15, 6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Tobiume K., Matsuzawa A., Takahashi T., Nishitoh H., Morita K., Takeda K., Minowa O., Miyazono K., Noda T. & Ichijo H. (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep., 2, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey K.J. & Cerami A. (1993) Tumor necrosis factor, other cytokines and disease. Annu. Rev. Cell Biol., 9, 317–343. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P., Declercg W., Beyaert R. & Fiers W. (1995) Two tumour necrosis factor receptors: structure and function. Trends Cell. Biol., 5, 392–399. [DOI] [PubMed] [Google Scholar]
- Xia Y., Makris C., Su B., Li E., Yang J., Nemerow G.R. & Karin M. (2000) MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl Acad. Sci. USA, 97, 5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh W.C. et al. (1997) Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity, 7, 715–725. [DOI] [PubMed] [Google Scholar]
- Yujiri T. et al. (2000) MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-κB activation. Proc. Natl Acad. Sci. USA, 97, 7272–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]