

Abstract
The recent discovery of an alternate pathway for indirectly charging tRNACys has stimulated a re-examination of the evolutionary history of Cys-tRNACys formation. In the first step of the pathway, O-phosphoseryl-tRNA synthetase charges tRNACys with O-phosphoserine (Sep), a precursor of the cognate amino acid. In the following step, Sep-tRNA:Cys-tRNA synthase (SepCysS) converts Sep to Cys in a tRNA-dependent reaction. The existence of such a pathway raises several evolutionary questions, including whether the indirect pathway is a recent evolutionary invention, as might be implied from its localization to the Euryarchaea, or, as evidence presented here indicates, whether this pathway is more ancient, perhaps already in existence at the time of the last universal common ancestral state. A comparative phylogenetic approach is used, combining evolutionary information from protein sequences and structures, that takes both the signature of horizontal gene transfer and the recurrence of the full canonical phylogenetic pattern into account, to document the complete evolutionary history of cysteine coding and understand the nature of this process in the last universal common ancestral state. Resulting from the historical study of tRNACys aminoacylation and the integrative perspective of sequence, structure, and function are 3D models of O-phosphoseryl-tRNA synthetase and SepCysS, which provide experimentally testable predictions regarding the identity and function of key active-site residues in these proteins. The model of SepCysS is used to suggest a sulfhydrylation reaction mechanism, which is predicted to occur at the interface of a SepCysS dimer.
Keywords: aminoacyl-tRNA synthetase, cysteine, methanogenic archaea, O-phosphoserine, structure and sequence-based phylogeny
In 1996, the first complete genomic sequence of an archaeon (Methanococcous jannaschii) revealed the surprising absence of four of the standard aminoacyl-tRNA synthetases (aaRSs) (1). The existence of two protein families of aaRSs, unrelated in both sequence and structure, suggests that nature has independently arrived at two solutions for matching tRNAs with their cognate amino acids (2). The aaRSs were originally thought to obey the so-called class rule, stating that a particular amino acid is ligated to its cognate tRNA by a member of only one of the two classes. The discovery of a class I LysRS broke the class rule and explained the absence of the typical class II LysRS in Methanococcous jannaschii and other methanogenic archaea (3). It was also quickly recognized that in Methanococcous jannaschii tRNAAsn and tRNAGln are aminoacylated with their cognate amino acids by an indirect mechanism, whereby a nondiscriminating aaRS first mischarges the tRNA with the corresponding diacid precursor, which is subsequently converted to the cognate species by a second amidotransferase enzyme (4–6). The indirect aminoacylation mechanisms are a critical intersection between the metabolic and information processing subsystems of the cell.
Although three of Methanococcous jannaschii's missing synthetases were found within 2 years of the publication of its genome sequence, the nature of Cys-tRNACys formation in Methanococcous jannaschii, and two other methanogenic archaea, Methanopyrus kandleri and Methanothermobacter thermoutotrophicus, remained unknown for more than a decade (7, 8). Our recent bioinformatic study first pointed to the presence of a putative class II CysRS in Methanococcous jannaschii (gene MJ1660) and related euryarchaea. MJ1660 and its orthologs form a monophyletic gene family that is obviously unrelated to the typical class I CysRS (9). The story was soon confirmed and elaborated by the elegant biochemical and genetic work of Söll and colleagues (10), who showed that the suspected class II CysRS, now properly renamed SepRS (O-phosphoseryl-RS), actually loaded O-phosphoserine (Sep), a precursor of cysteine, onto tRNACys. The noncognate Sep-tRNACys is then converted to the cognate pairing by the enzyme Sep-tRNA:Cys-tRNA synthase (SepCysS) (MJ1678). The coincidence of a class rule violation and an unexpected indirect mechanism are part of the reason why the mechanism Cys-tRNACys formation in Methanococcous jannaschii remained mysterious for so long. The fact that the class II aaRS involved in this pathway was (and is still) consistently misannotated as a PheRS α-chain in all of the sequence databases did not help matters and underscores the fundamental problems of gene annotation based on sequence similarity alone.
As the idea began to emerge that some of the missing synthetases could be accounted for by indirect mechanisms, Olsen and Woese (11) were prompted to call for a “rethinking of our concept of tRNA charging, its evolution, and even the evolutionary relationship between translation and intermediary metabolism.” Herein, sequence and structure data are cooperatively used to document the major evolutionary events in the history of Cys-tRNACys formation and begin the process of rethinking the above issues. Specifically, protein structures are used to establish accurate alignments of distantly related proteins, whereas sequences define phylogenetic patterns and help to elucidate the determinants of molecular recognition in structural models of the enzyme–substrate complexes of SepRS and SepCysS.
Methods
Sequences were obtained from the Integrated Microbrial Genomes database (12), Swiss-Prot (13), the National Center for Biotechnology Information, and ref. 14. Protein structures were extracted from the Protein Data Bank (15) and the Structural Classification of Proteins/ASTRAL database, version 1.67 (16, 17). Multiple structure alignments were performed by using stamp (18) as implemented in vmd version 1.83 (19), and clustal (20) was used for multiple sequence alignment. Phylogenetic analysis, as in refs. 21 and 22, involved a combination of maximum parsimony using paup4b10 (23) and maximum likelihood with phyml (24) and protml (25). modeller 6.2 (26) was used for building homology models. Relaxed models of the enzyme–substrate complexes for SepRS and SepCysS resulted from equilibration in the molecular dynamics package namd (27) with the charmm force field (28).
Detailed methods can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Results and Discussion
The phylogenetic trees shown in Fig. 1 are a mapping of the known evolutionary history of Cys-tRNACys formation, which is revealed from the comparative analysis of three homologous groups of proteins. Fig. 1a includes the euryarchaeal SepRSs and their closest relatives in the class II aaRS family, the α- and β-subunits of PheRS. The second group (Fig. 1b) contains the bacterial-type class I CysRS and its closest class I aaRS relatives, GluRS and GlnRS. Fig. 1c includes the SepCysSs and the cysteine desulfurases. Individual sequence phylogenies of PheRS, GluRS, GlnRS, and CysRS have been examined previously in detail (21), and the ancient relationships between members of the class I and, separately, among the class II aaRSs (whose structures are known) have been derived by using structural phylogeny (2, 9, 29). Here, we focus on orthologous and paralogous relationships, using a combination of protein structures to obtain accurate alignments of distant homologs and additional sequence data to explore the phylogenetic patterns in detail, especially by expanding the euryarchaeal portion of the trees for direct comparison with the SepRS and SepCysS phylogenies.
Fig. 1.

Phylogenetic trees are shown for homologous groups including: SepRS, α- and β-chains of PheRS (rooted by AlaRS, see Fig. 4, which is published as supporting information on the PNAS web site) (a), GluRS and CysRS (rooted by LysRS, see Fig. 5, which is published as supporting information on the PNAS web site) (b), and SepCysS and the cysteine desulfurases (rooted by alanine-glyoxylate aminotransferase, Protein Data Bank ID code 1h0c) (c). Organism names are color-coded as Bacteria (red), Archaea (blue), and Eucarya (green). Local bootstrap probabilities, with <90% support, are shown to the right of each branching. Protein Data Bank codes for crystallographic structures used in the structural alignment are in parentheses. Purple circles represent the base of the UPT, bifurcations before these points occured during the time of LUCAS. Alternate views of the trees are available in Figs. 4–7, which are published as supporting information on the PNAS web site.
The full canonical phylogenetic pattern (21, 30) emerges three times in Fig. 1 and has four defining characteristics: (i) general congruence with the rRNA phylogeny, including (ii) a deep division (observed in long branches and corresponding sequence signatures) separating the bacterial from the archaeal genre of the molecule, such that the sequence differences within the archaeal-eukaryotic group or within the bacterial clade are far less than those between the groups, (iii) the presence of a distinct eukaryotic version of the molecule that is of the archaeal genre, and (iv) an ancestral base positioned between the bacterial domain and the archaeal-eukaryotic group. The evolutionary period associated with bifurcations in the trees that occurred before this base represents the evolution of the last universal common ancestral state (LUCAS) itself (sensu ref. 31), and the subsequent period involves the divergence of the main organismal lineages, radiation of the major taxonomic divisions, and speciation. Throughout Fig. 1, where a node can be defined that corresponds to the base of the universal phylogenetic tree (UPT), i.e., the rRNA tree, it is labeled (purple circles).
The Evolution of PheRS from Sequence and Structure. Based on sequence comparisons among the class II aaRSs, the catalytic domain of SepRS is most similar to its counterpart in the α-subunit of PheRS (α-PheRS). The PheRSs conform to the rRNA phylogeny at the highest taxonomic ranks and in subordinate groupings as well (Fig. 1a). The eukaryotes are confidently grouped together and are clearly of the archaeal type. The euryarchaeal sequences conform to accepted archaeal taxonomy (30, 32, 33), distinguished by a specific relationship between Methanococcoides burtonii and the genus Methanosarcina, in addition to well supported clusters of the Methanomicrobiales and Methanococcales. The clear sisterhood between the Methanomicrobiales and Archaeoglobus was previously established with rRNA (34, 35), ribosomal protein, and transcription factor phylogenies (22, 33). A recent analysis of environmental rRNA sequences from a group of anaerobic methane-oxidizing archaea (ANME-1) (31) supports the inclusion of ANME-1 in this group. In agreement with the work cited above, Methanopyrus kandleri and Methanothermobacter thermoautotrophicus are deeply branching euryarchaea, neither showing well supported kinship with other euryarchaeal groups. With minor exceptions, the same archaeal phylogenetic pattern recurs throughout Fig. 1, as it is also evident among the β-PheRSs and the SepRSs (Fig. 1a), the GluRSs in Fig. 1b, and the SepCysSs in Fig. 1c.
The comparative analysis of 3D protein structure uncovers evolutionary events that predate the emergence of the main organismal lineages and provides support for how PheRS evolved into its unique tetrameric form. Most class II aaRSs are homodimers in nature with the catalytic domain, responsible for the aminoacylation reaction, being the only component shared, and thus defining this protein family. PheRS is an (αβ)2 tetramer with the catalytic domain residing in α-PheRS and a homologous catalytic-like domain in the β-subunit (β-PheRS), which is not capable of catalyzing aminoacylation (37). Lacking the evolutionary constraint to preserve enzymatic function, β-PheRS has incurred more evolutionary change than the α-chain. This evolutionary process left an average of 15% sequence identity between the α- and β-PheRSs, necessitating the use of a structural superposition of these paralogs to establish an accurate alignment. The phylogenetic patterns of the α- and β-PheRSs are largely congruent, but the elevated evolutionary tempo in the β-subunits has led to some difficulty in the phylogenetic reconstruction. There is a large separation between the bacterial and archaeal versions of β-PheRS, yet the tree is not reliably rooted between the Bacteria and Archaea.
Although the β-PheRSs show 15% average sequence identity (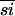 ) with either the SepRSs or the α-PheRSs, at 23%
) with either the SepRSs or the α-PheRSs, at 23%  , the SepRSs and α-PheRSs are more similar to each other than either are to the β-PheRSs. The similarity data might lead one to assume that α-PheRS and SepRS descended from the more recent common ancestor. Because the β-PheRSs, at 27%
, the SepRSs and α-PheRSs are more similar to each other than either are to the β-PheRSs. The similarity data might lead one to assume that α-PheRS and SepRS descended from the more recent common ancestor. Because the β-PheRSs, at 27% 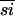 within the group, evolved at a faster rate than the α-PheRSs (40%
within the group, evolved at a faster rate than the α-PheRSs (40% 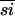 ) or the SepRSs (69%
) or the SepRSs (69% 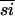 ), a tree topology that gives a monophyletic cluster of the PheRS subunits to the exclusion of the SepRSs is suggested, albeit with low support (local bootstrap probabilities 43%). SepRS may have diverged from PheRS before generation of the β-subunit, but, certainly, the SepRSs arise as a bona fide outgroup to the α-PheRSs (Fig. 4). Examination of the quaternary structure of PheRS shows that α- and β-PheRS are in the same relative orientation as the two catalytic domains of a typical class II aaRS homodimer. The association between the α- and β-subunits appears to be a molecular fossil of an α2 dimeric or α4 tetrameric [similar to the related AlaRS (38)] ancestral state for the PheRSs.
), a tree topology that gives a monophyletic cluster of the PheRS subunits to the exclusion of the SepRSs is suggested, albeit with low support (local bootstrap probabilities 43%). SepRS may have diverged from PheRS before generation of the β-subunit, but, certainly, the SepRSs arise as a bona fide outgroup to the α-PheRSs (Fig. 4). Examination of the quaternary structure of PheRS shows that α- and β-PheRS are in the same relative orientation as the two catalytic domains of a typical class II aaRS homodimer. The association between the α- and β-subunits appears to be a molecular fossil of an α2 dimeric or α4 tetrameric [similar to the related AlaRS (38)] ancestral state for the PheRSs.
Phylogenetic Pattern in the SepRSs. Major features of established euryarchaeal taxonomy are preserved in the phylogeny of the SepRSs (Fig. 1a). The SepRS gene is completely absent from the euryarchaeal orders Thermococcales, Halobacteriales, and Thermoplasmatales, and pseudogene searches of all of the completely sequenced microbial genomes failed to reveal any additional SepRS or SepCysS genes or gene remnants. A feature that is exclusively common to the organisms that encode SepRS/SepCysS in their genomes is the presence of the methanogenesis genes, either for generating or oxidizing methane, as in the case of the ANME-1 group (36) and Archaeoglobus, which contains five of the typical seven enzymes in this pathway (39, 40). Partial loss of the methanogenesis genes in Archaeoglobus, the least derived of the nonmethanogenic euryarchaeal phenotypes, and complete loss of these genes in the same orders that lack SepRS/SepCysS, leads to the suggestion of a tentative evolutionary linkage between SepRS/SepCysS and the methanogenesis genes. In Methanococcus maripaludis (10), and possibly in some other organisms (Table 1, which is published as supporting information on the PNAS web site), SepRS/SepCysS is the sole route for cysteine biosynthesis. Based on the completely sequenced genomes, only in Methanosarcina barkeri, Methanosarcina acetivorans, and Methanospirillum hungatei, which all have the class I CysRS and the bacterial pathway for cysteine biosynthesis (Fig. 8, which is published as supporting information on the PNAS web site), does the SepRS/SepCysS system appear to be functionally redundant. Interestingly, Methanosarcina mazei lost one of the two genes in the bacterial pathway, which has been conserved in the other members of its lineage, instead preferring to retain SepRS/SepCysS for cysteine biosynthesis (Table 1).
Although the SepRSs are in general agreement with established euryarchaeal taxonomy and a recent phylogeny of the methanogenesis genes (37), it is striking that the Methanomicrobiaceae appear as a deep branch. This placement may be the result of either an increased evolutionary tempo, which could have “erased” sequence signatures that may have suggested their expected recent common ancestry with the Methanosarcinaceae, or, perhaps, the Methanomicrobiaceae received their SepRS from an as yet unknown, deeply branching euryarchaeal order. Enough of the established euryarchaeal phylogenetic pattern remains that it is fair to conclude that SepRS is at least as old as the euryarchaeal lineage itself.
The Evolutionary Relationship Between PheRS and SepRS. Our analysis further demonstrates that SepRS and the associated indirect aminoacylation pathway for tRNACys are truly ancient, already present at the time of the LUCAS. If SepRS had recently diverged from PheRS, one would expect a phylogenetic pattern such as that observed for AsnRS or GlnRS. All known examples of GlnRS are specifically related to the eukaryotic version of GluRS (Fig. 1b and ref. 21), and the AsnRSs were derived from the archaeal version of AspRS. Both are clear cases of a recent divergence, occurring well after the base of the UPT, from duplication of a specific aaRS. Were this the case for SepRS, one might see a specific relationship between, say, the archaeal α-PheRSs and the SepRSs. Instead, the data presented in Fig. 1a show that SepRS diverged from PheRS well before the base of the UPT. Note the bifurcation between the SepRS and PheRS occurred before the UPT base points (Fig. 1a, purple circles) defined for both the α- and β-PheRSs.
Evolutionary Conservation and Modeling Reveal Molecular Recognition in SepRS. In the known SepRSs, there are 123 invariant residues, constituting ≈22% of the molecule. By mapping the evolutionary conservation onto a tertiary structure model, a reasonable prediction of the residues responsible for substrate recognition in SepRS can be made. Structural homology among the class II aaRSs indicates that the backbone structure of the aaRSs is highly conserved in the vicinity of the active site (39), so the closely related α-PheRS serves as an ideal structural template for generating a homology-based model of SepRS.
The presence of a K[MRK]SK motif that is highly conserved among the SepRSs has recently been reported (9). Because the aminoacylation reaction involves the binding of ATP, the aaRSs use conserved, positively charged side chains to stabilize the ATP in the active-site pocket. In the class I aaRSs, ATP interacts favorably with the KMSK motif, whereas, for the class II aaRSs, two completely conserved arginines play an analogous role. The SepRSs contain both the class II arginines and a KMSK-like motif. In Methanococcous jannaschii, our alignment places the KKSK motif in a short “recognition loop” that is structurally highly conserved (Fig. 9, which is published as supporting information on the PNAS web site). Although not all of the class II aaRSs use this loop for substrate recognition, PheRS, the closest relative of SepRS, uses it to position two phenylalanines and one valine in direct contact with the side chain of the substrate. In SepRS, according to the alignment and homology model, the serine (S283*) and lysine (K284*) in this loop, as well as an additional arginine (R141*), are predicted to directly recognize the phosphate moiety of Sep. (see Fig. 2).
Fig. 2.

The sequence of SepRS aligned to a multiple structural alignment of class II aaRS catalytic domains, cocrystallized with their cognate aminoacyladenylate substrate or substrate analog. A phylogeny based the tertiary structure similarity metric QH shows the evolutionary relationships between these aaRSs (2, 29). Adenylate recognition residues (blue), residues contributing nonsequence specific backbone interactions (green), residues that recognize the side chain of the cognate amino acid (magenta), and residues contacting the carbonyl and α-amide of the cognate amino acid (brown) are within 4 Å of the substrate. A view of the modeled, equilibrated (after 1-ns molecular dynamics simulation) active site of SepRS is shown with the O-phosphoseryl-adenylate (ball and stick) and key substrate recognition residues (stick).
Class I CysRS and GluRS Phylogeny. Among the archaea in the GluRS tree (Fig. 1b), the crenarchaeal examples are specifically related to each other, but the tree does not show a clear divide between the Euryarchaea and the Crenarchaea. Much of the typical euryarchaeal pattern is evident, but notice the placement of Methanospirillum hungatei with respect to the other euryarchaea, which, much like the SepRSs of the Methanomicrobiaceae, is not grouping among the Methanosarcinaceae–Archaeoglobus cluster. Even with such idiosyncrasies, the presence of the full canonical pattern indicates that the root of the GluRS tree (Fig. 1b, purple circle) is equivalent to the base of the UPT.
In the CysRS tree (Fig. 1b), although the archaeal and eukaryotic examples do not conform to rRNA phylogeny, there is a well supported grouping of the genus Methanosarcina, indicating that the CysRS gene was horizontally transferred, perhaps from the Spirochete lineage, to the Methanosarcina before the speciation events that produced the three members of this genus. That the bacterial CysRSs conform in large measure to the rRNA-based bacterial taxonomy (7, 21), and that the eukaryotic and archaeal examples do not, is a clear signal that the class I CysRS is of bacterial origin. The divergence between CysRS and GluRS occurred before the time symbolized by the base of the UPT (Fig. 1b, purple circle). It is puzzling that, while the class I CysRS was extant at the time of LUCAS, it was only originally used and conserved among the bacterial lineages. Only later, after the emergence of the major bacterial phyla was CysRS horizontally transferred, in multiple independent events, to members of the archaeal and eukaryotic domains.
Evolutionary Relationship of SepCysS to the Cysteine Desulfurases. SepCysS, which converts Sep-tRNACys to Cys-tRNACys, is most closely related to pyridoxal 5′-phosphate (PLP)-dependent cysteine desulfurase. Patterns of sequence conservation between SepCysS and the cysteine desulfurases not only suggest descent from a common ancestral gene, but also a homologous reaction mechanism. The cysteine desulfurases are found in two distantly homologous groups that can only be accurately related to one another with a structural alignment. Group I includes the NiFS and IscS genes, and group II is composed of cysteine sulfinate desulfinases. Both groups are monophyletic (Fig. 1c), and each display some of the bacterial groupings observed in the rRNA tree.
In group I, the mitochondrial proteins are specifically related to the Rickettsiales, a parasitic group of α-Proteobacteria. There are also two separate clades of the α-, β-, and γ-Proteobacteria, indicating a paralogous relationship between the two major phyletic clusters, NiFS and IscS, within group I. Similarly, among the group II desulfurases, the rRNA-based bacterial groupings are clearly distinguishable, and the phylogenetic relationships between the Euryarchaea follow established taxonomy (Fig. 7). In general, the cysteine desulfurases fail to display the canonical phylogenetic pattern, as a deep split between the archaeal and bacterial genres is not evident, so it is not possible to choose a node in these trees that is equivalent to the base of the UPT.
The SepCysS proteins form a monophyletic group with respect to the cysteine desulfurases, indicating a divergence in function. SepRS and SepCysS have a common evolutionary history, the latter displaying additional gene duplications. Archaeoglobus fulgidus and Methanococcoides burtonii have two SepCysS genes. As in established archaeal taxonomy, A. fulgidus 1 (gene AF0028) is closely related to the Methanosarcinaceae, yet A. fulgidus 2 (AF0181) branches as deeply as Methanopyrus kandleri. Similarly, the position of the Methanococcoides burtonii 1 is as expected, yet Methanococcoides burtonii 2 is specifically related to the Methanospirillum hungatei protein. Their relationship is supported by sequence signature analysis, a unique N-terminal extension, and genomic context (see Supporting Text). The unexpected Methanospirillum hungatei–Methanococcoides burtonii 2 cluster is deeply branching with respect to the other euryarchaeal sequences. Although Methanococcoides burtonii 2 is the result of horizontal gene transfer, the reason for the global placement of Methanospirillum hungatei, as discussed above for SepRS, is uncertain. The partial sequence of SepCysS from Methanogenium frigidum, not shown in Fig. 1c, is specifically related to the Methanospirillum hungatei SepCysS, with 63% sequence identity over the aligned fragment.
SepCysS Modeling and Active-Site Identification. The cysteine desulfurases have the PLP-dependent transferase fold, and, by removal of a thiol group, convert cysteine to alanine. Structures of cysteine desulfurases were used to model the structure and active-site configuration of SepCysS. A free substrate cysteine in the group I desulfurases is the sulfur donor for a variety of biomolecules, such as Fe–S clusters and modified nucleotides (42). In analogy with the desulfurase reaction mechanism (43), formation of a persulfide is potentially important for the sulfhydrylation reaction catalyzed by SepCysS. Consistent with experimental observations (10), i.e., that MJ1678 required Na2S and PLP to convert Sep-tRNACys to Cys-tRNACys, the SepCysS reaction mechanism is likely very similar to that of the related PLP-dependent enzymes, in which PLP forms an internal aldimine bond with a conserved lysine (K̇234) (see Fig. 3). In the model, the residues interacting with PLP in the cysteine desulfurases are also well conserved in the SepCysSs, and a region of positive electrostatic potential was found in MJ1678 to correspond to the putative binding site of the tRNA acceptor stem (Fig. 10, which is published as supporting information on the PNAS web site).
Fig. 3.

Alignment of modeled structure of SepCysS (MJ1678) with templates and active-site architecture (after 2-ns molecular dynamics equilibration). The regions surrounding the substrate and cofactor are shown in the alignment. Important active-site residues, within 4 Å of the substrate and cofactor, are color-coded according to four interaction types: contact with the Sep side chain (magenta), contact with the α-amide and carboxyl groups of amino acids (green), contact with PLP (blue), and the two cysteines, putatively involved in catalysis (brown). Sequence signatures, residues with >70% conservation in an evolutionary profile (9) for each homologous group, are underlined or boxed.
Residues predicted to interact with Sep are completely conserved in the SepCysSs, whereas most are not conserved in the cysteine desulfurases. In the equilibrated model, which suggests that the active sites in the SepCysS homodimer are formed at the dimerization interface, K265* from chain B and R42* and R362 from chain A directly contact the phosphate group of Sep. SepCysS also has two invariant cysteine residues (see brown in Fig. 3), which, from chain B, are in somewhat close proximity to the Sep and PLP bound to chain A. Either of these residues may play a role in persulfide formation, in analogy to C325 in an NiFS-like protein (Protein Data Bank ID code 1ecx) (43). Although the natural sulfur donor is unknown, analogy with other cysteine biosynthetic pathways suggest a simple sulfide or, perhaps, a persulfide, thiosulfate, or thiocarboxylate donated by another sulfur carrier protein (44–46). In the final steps of catalysis, the Sep moiety from Sep-tRNACys will form an external aldimine bond with PLP before the thiol transfer from one of the nearby Cys residues to form Cys-tRNACys. The highlighted residues in Fig. 2 for SepRS and Fig. 3 for SepCysS are suggested for mutational analysis.
Conclusion
While it is commonly stated that the genetic code is enforced by the aaRSs, this is not always the case (47). The indirect pathway for charging tRNACys, as well as indirect pathways for asparagine and glutamine, indicate that some aaRSs have evolved to mischarge a tRNA, and this “mistake” is only later corrected by an additional enzyme. A relevant question is whether the indirect mechanisms predate the direct mechanisms, is one more primitive than the other? The phylogenetic distribution of the indirect pathways for charging tRNAAsn and tRNAGln and the late emergence and narrow phylogenetic range of the direct pathways, catalyzed by AsnRS and GlnRS, suggest that in both cases, the indirect pathway is the primordial route (21, 47). In the case of cysteine, the direct and indirect pathways are of equally ancient origin.
The SepCysS tree displays evidence of horizontal gene transfer events that, nevertheless, do not obscure its shared evolutionary path with SepRS. In large part, this history is congruent with established euryarchaeal taxonomy, indicating that SepRS and SepCysS were in existence at the time of origin of the euryarchaeal lineage. The CysRS phylogeny, on the other hand, is in good agreement with bacterial taxonomy, and its archaeal and eukaryotic examples are most likely the result of several independent horizontal gene transfer events. By reconciling this and other horizontal gene transfer events with the recurrence of the full canonical phylogenetic pattern, we conclude that the origin of both the bacterial system for cysteine coding, CysRS, and the archaeal version, SepRS/SepCysS, predate the base of the UPT. Although the genetic code established during the evolution of LUCAS was universal, the mechanism for cysteine coding was not. If both systems developed contemporaneously, why was one sequestered to the archaeal lineages, whereas the other was vertically inherited only among the Bacteria? These appear to be signature genes (48) or, sensu Darwin (49), essential characters, which are defining components of each lineage. The fact that lineage-defining genes could be present during the time of LUCAS suggests that the organismal lineages themselves were becoming defined in this era.
The phylogenetic distributions of SepRS/SepCysS and the methanogenesis genes are identical. Despite the fact that many of these organisms have received the bacterial mechanisms for cysteine coding and biosynthesis, these horizontally transferred genes have not successfully displaced SepRS/SepCysS. In one striking case, Methanosarcina mazei appears to have lost a key gene in the bacterial cysteine biosynthetic pathway, preserving its native archaeal route. Also, a recent bioinformatic study found that, as compared with other microbes, Archaeoglobus and the methanogens have a preponderance of proteins containing iron-sulfur cluster motifs of the form CX2CX2CX3C (50). These data all suggest an unresolved link between primary energy production, sulfur metabolism, and cysteine coding in these organisms that demands further investigation. To obtain a clearer picture of the phylogenetic distribution of SepRS/SepCysS and the methanogenesis genes, directed environmental sequencing efforts are called for, and genetic experiments in Methanosarcina acetivorans, e.g., ref. 51, which contains both the archaeal and bacterial routes for cysteine coding and biosynthesis, will help in understanding why this organism has retained the archaeal pathway and shed light on the reason for the apparent functional redundancy. Directed by hypotheses generated from the evolutionary data, such work will lead to a deeper understanding of these archaea and the connection between information processing and metabolism in cellular evolution.
Acknowledgments
We thank James M. Briggs (University of Houston, Houston) for providing the force-field parameters for PLP. The work was supported by National Science Foundation Grant MCB04-46227 and Department of Energy Grant DE-FG02-05ER64144. P.O. was supported by a postdoctoral fellowship from the Institute for Genomic Biology at the University of Illinois at Urbana–Champaign.
Author contributions: P.O., A.S., C.R.W., and Z.A.L.-S. designed research; P.O. and A.S. performed research; P.O., A.S., C.R.W., and Z.A.L.-S. analyzed data; and P.O., A.S., C.R.W., and Z.A.L.-S. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: RS, tRNA synthetase; aaRS, aminoacyl-tRNA synthetase; Sep, O-phosphoserine; SepCysS, Sep-tRNA:Cys-tRNA synthase; SepRS, O-phosphoseryl-RS; LUCAS, last universal common ancestral state; PLP, pyridoxal 5′-phosphate; UPT, universal phylogenetic tree.
References
- 1.Bult, C. J., White, O., Olsen, G. J., Zhou, L., Fleischmann, R. D., Sutton, G. G., Blake, J. A., FitzGerald, L. M., Clayton, R. A., Gocayne, J. D., et al. (1996) Science 273, 1058–1073. [DOI] [PubMed] [Google Scholar]
- 2.O'Donoghue, P. & Luthey-Schulten, Z. (2003) Microbiol. Mol. Biol. Rev. 67, 550–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibba, M., Morgan, S., Curnow, A. W., Pridmore, D. R., Vothknecht, U. C., Gardner, W., Lin, W., Woese, C. R. & Söll, D. (1997) Science 278, 1119–1122. [DOI] [PubMed] [Google Scholar]
- 4.Curnow, A. W., Hong, K., Yuan, R., Kim, S., Martins, O., Winkler, W., Henkin, T. M. & Söll, D. (1997) Proc. Natl. Acad. Sci. USA 94, 11819–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curnow, A., Tumbula, D., Pelaschier, J., Min, B. & Söll, D. (1998) Proc. Natl. Acad. Sci. USA 95, 12838–12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tumbula, D., Vothknecht, U., Kim, H., Ibba, M., Min, B., Li, T., Pelaschier, J., Stathopoulos, C., Becker, H. & Söll, D. (1999) Genetics 152, 1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, T., Graham, D. E., Stathopoulos, C., Haney, P. J., Kim, H., Vothknecht, U., Kitabatake, M., Hong, K., Eggertsson, G., Curnow, A. W., et al. (1999) FEBS Lett. 462, 302–306. [DOI] [PubMed] [Google Scholar]
- 8.Ruan, B., Nakano, H., Tanaka, M., Mills, J. A., DeVito, J. A., Min, B., Low, K. B., Battista, J. R. & Söll, D. (2004) J. Bacteriol. 186, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sethi, A., O'Donoghue, P. & Luthey-Schulten, Z. (2005) Proc. Natl. Acad. Sci. USA 102, 4045–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sauerwald, A., Zhu, W., Major, T. A., Roy, H., Palioura, S., Jahn, D., Whitman, W., Yates, J. R., III, Ibba, M. & Söll, D. (2005) Science 307, 1969–1972. [DOI] [PubMed] [Google Scholar]
- 11.Olsen, G. J. & Woese, C. R. (1996) Trends Genet. 12, 377–379. [DOI] [PubMed] [Google Scholar]
- 12.Markowitz, V., Korzeniewski, F., Palaniappan, K., Szeto, E., Werner, G., Padki, A., Zhao, X., Dubchak, I., Hugenholtz, P., Anderson, I., et al. (2006) Nucleic Acids Res., in press. [DOI] [PMC free article] [PubMed]
- 13.Bairoch, A., Apweiler, R., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M., et al. (2005) Nucleic Acids Res. 33, D154–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saunders, N. F. W., Thomas, T., Curmi, P. M. G., Mattick, J. S., Kuczek, E., Slade, R., Davis, J., Franzmann, P. D., Boone, D., Rusterholtz, K., et al. (2003) Genet. Res. 13, 1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H. N., Shindyalov, I. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andreeva, A., Howorth, D., Brenner, S. E., Hubbard, T. J. P., Chothia, C. & Murzin, A. G. (2004) Nucleic Acids Res. 32, D226–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandonia, J. M., Hon, G., Walker, N. S., Conte, L. L., Koehl, P., Levitt, M. & Brenner, S. E. (2004) Nucleic Acids Res. 32, D189–D192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell, R. B. & Barton, G. B. (1992) Proteins Struct. Funct. Genet. 14, 309–323. [DOI] [PubMed] [Google Scholar]
- 19.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 20.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woese, C. R., Olsen, G., Ibba, M. & Söll, D. (2000) Microbiol. Mol. Biol. Rev. 64, 202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brochier, C., Forterre, P. & Gribaldo, S. (2004) Genome Biol. 5, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swofford, D. (2003) paup*: Phylogenetic Analysis Using Parsimony (* and Other Methods) (Sinauer, Sunderland, MA), Version 4.
- 24.Guindon, S. & Gascuel, O. (2003) Syst. Biol. 52, 696–704. [DOI] [PubMed] [Google Scholar]
- 25.Adachi, J. & Hasegawa, M. (1996) Comput. Sci. Monogr. 28, 1–150. [Google Scholar]
- 26.Marti-Renom, M. A., Stuart, A., Fiser, A., Sanchez, F., Melo, F. & Sali, A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 291–325. [DOI] [PubMed] [Google Scholar]
- 27.Kale, L., Skeel, R., Bhandarkar, M., Brunner, R. A., Gursoy, N. K., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Phys. 151, 283–312. [Google Scholar]
- 28.MacKerell, A. D., Bashford, D., Bellott, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 29.O'Donoghue, P. & Luthey-Schulten, Z. (2005) J. Mol. Biol. 346, 875–894. [DOI] [PubMed] [Google Scholar]
- 30.Woese, C. R. (1987) Microbiol. Rev. 51, 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woese, C. R. (2002) Proc. Natl. Acad. Sci. USA 99, 8742–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schleper, C., Jurgens, G. & Jonuscheit, M. (2005) Nat. Rev. Microbiol. 3, 479–488. [DOI] [PubMed] [Google Scholar]
- 33.Brochier, C., Forterre, P. & Gribaldo, S. (2005) BMC Evol. Biol. 5, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woese, C. R., Achenbach, L., Rouviere, P. & Mandelco, L. (1991) Syst. Appl. Microbiol. 14, 364–371. [DOI] [PubMed] [Google Scholar]
- 35.Rouviere, P., Mandelco, L., Winker, S. & Woese, C. R. (1992) Syst. Appl. Microbiol. 15, 363–371. [DOI] [PubMed] [Google Scholar]
- 36.Hallam, S. J., Putnam, N., Preston, C. M., Detter, J. C., Rokhsar, D., Richardson, P. M. & DeLong, E. F. (2004) Science 305, 1457–1462. [DOI] [PubMed] [Google Scholar]
- 37.Mosyak, L., Reshetnikova, L., Goldgur, Y., Delarue, M. & Safro, M. G. (1995) Nat. Struct. Biol. 2, 537–547. [DOI] [PubMed] [Google Scholar]
- 38.Putney, S. D., Sauer, R. T. & Schimmel, P. R. (1981) J. Biol. Chem. 256, 198–204. [PubMed] [Google Scholar]
- 39.Klenk, H. P., Clayton, R. A., Tomb, J. F., White, O., Nelson, K. E., Ketchum, K. A., Dodson, R. J., Gwinn, M., Hickey, E. K., Peterson, J. D., et al. (1997) Nature 390, 364–370. [DOI] [PubMed] [Google Scholar]
- 40.Moran, J. J., House, C. H., Freeman, K. H. & Ferry, J. G. (2005) Archaea 1, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bapteste, E., Brochier, C. & Boucher, Y. (2005) Archaea 1, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mihara, H. & Esaki, N. (2002) Appl. Microbiol. Biotechnol. 60, 12–23. [DOI] [PubMed] [Google Scholar]
- 43.Kaiser, J. T., Clausen, T., Bourenkow, G. P., Bartunik, H. D., Steinbacher, S. & Huber, R. (2000) J. Mol. Biol. 297, 451–464. [DOI] [PubMed] [Google Scholar]
- 44.Kredich, N. M. (1996) in Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, eds. Neidhardt, F. C., Curtiss, R., III, Gross, C. A., Ingraham, J. L., Lin, E. C. C., Low, K. B., Magasanik, B., Reznikoff, W., Riley, M., Schaechter, M. & Umbarger, H. (Am. Soc. Microbiol., Washington, DC), 2nd Ed., pp. 514–527.
- 45.White, R. H. (2003) Biochim. Biophys. Acta 1624, 46–53. [DOI] [PubMed] [Google Scholar]
- 46.Burns, K. E., Baumgart, S., Dorrestein, P. C., Zhai, H., McLafferty, F. W. & Begley, T. P. (2005) J. Am. Chem. Soc. 127, 11602–11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ibba, M., Becker, H. D., Stathopoulos, C., Tumbula, D. L. & Söll, D. (2000) Trends Biochem. Sci. 25, 311–316. [DOI] [PubMed] [Google Scholar]
- 48.Graham, D. E., Overbeek, R., Olsen, G. J. & Woese, C. R. (2000) Proc. Natl. Acad. Sci. USA 97, 3304–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Darwin, C. (1859) The Origin of the Species by Means of Natural Selection or the Preservation of Favored Races in the Struggle for Life (John Murray, London).
- 50.Major, T. A., Burd, H. & Whitman, W. B. (2004) FEMS Microbiol. Lett. 239, 117–123. [DOI] [PubMed] [Google Scholar]
- 51.Rother, M., Boccazzi, P., Bose, A., Pritchett, M. A. & Metcalf, W. W. (2005) J. Bacteriol. 187, 5552–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]