

Abstract
Archaea and their viruses are poorly understood when compared with the Eukarya and Bacteria domains of life. We report here the crystal structure of the major capsid protein (MCP) of the Sulfolobus turreted icosahedral virus, an archaeal virus isolated from an acidic hot spring (pH 2–4, 72–92°C) in Yellowstone National Park. The structure is nearly identical to the MCP structures of the eukaryotic Paramecium bursaria Chlorella virus, and the bacteriophage PRD1, and shows a common fold with the mammalian adenovirus. Structural analysis of the capsid architecture, determined by fitting the subunit into the electron cryomicroscopy reconstruction of the virus, identified a number of key interactions that are akin to those observed in adenovirus and PRD1. The similar capsid proteins and capsid architectures strongly suggest that these viral capsids originated and evolved from a common ancestor. Hence, this work provides a previously undescribed example of a viral relationship spanning the three domains of life (Eukarya, Bacteria, and Archaea). The MCP structure also provides insights into the stabilizing forces required for extracellular hyperthermophilic proteins to tolerate high-temperature hot springs.
Keywords: crystallography, evolution, hyperthermophile, electron cryomicroscopy
The Sulfolobus turreted icosahedral virus (STIV) infects Sulfolobus solfataricus, an acidophilic hyperthermophilic organism (grows optimally at pH 2–4 and at >80°C) that is emerging as a model for studying hyperthermophilic archaea and their viruses (1). STIV possesses a 17,663-bp circular dsDNA genome that encodes 36 predicted ORFs (2). The viral particle is composed of a 37-kDa major capsid protein (MCP) and several 25-, 12.5-, and 10-kDa minor capsid proteins (2). The electron cryomicroscopy (cryo-EM) image reconstruction of STIV showed a pseudo T = 31 icosahedral capsid with trimers at the quasi sixfold coordinated positions, turret-like appendages at the vertices, and what appears to be an internal lipid membrane sandwiched between the genome and capsid shell (2). The capsid architecture of STIV is reminiscent of the mammalian adenovirus, bacteriophage PRD1, and Paramecium bursaria Chlorella virus (PBCV-1). The viral capsids of adenovirus, PRD1, and PBCV-1 are believed to have descended from a common ancestor (3). By using sequence alignment and modeling techniques, this lineage was recently extended to include additional large-faceted viruses containing a double-barrel trimeric major coat protein (4).
Here we report the crystal structure of the STIV MCP and show its structural homology and sequence similarity to the MCPs of the adenovirus, PRD1, and PBCV-1. We further analyze the capsid architecture of STIV by docking the MCP crystal structure into the cryo-EM reconstruction and calculating a difference map. Our analysis reveals a number of quaternary interactions similar to those observed in adenovirus and PRD1. The structural and sequence comparison between the MCPs of these viruses, and similarities between their capsid architectures support the idea that their viral capsids share a common ancestor. The MCP (an extracellular protein) structure also suggests how proteins may be able to tolerate the extreme physicochemical habitat of high-temperature hot spring environments.
Materials and Methods
Protein Expression and Purification. MCP of STIV was cloned into the pDEST14 expression vector (Gateway system, Invitrogen) according to the manufacturer's protocols. Selenomethionine (SeMet)-labeled protein was expressed in the Escherichia coli BL21 Star (DE3) One Shot (Invitrogen) strain carrying a Rosetta vector (Novagen) by using the Met biosynthesis inhibition protocol (5). After5hofexpressionat37°C,the cells were harvested and stored at –80°C.
Protein purification was carried out at room temperature by using affinity and ion-exchange chromatography. Briefly, the cell pellet was suspended in 20 mM Hepes (pH 8.2), 50 mM Na2SO4,10mM imidazole (pH 8.0), 5 mM 2-mercaptoethanol (β-Me), and 1 mM PMSF (lysis buffer), and lysed through sonication. Soluble material was recovered by centrifugation (7,000 × g for 30 min at 4°C) and loaded onto a Ni-NTA affinity gravity flow column (Novagen). The protein was purified according to the manufacturer's protocol. Fractions containing the protein were pooled and dialyzed against the lysis buffer to remove the imidazole, adjusted to pH 5.2 with 1 M NaAc (pH 5.2), and loaded onto a HiTrap Q HP Amersham Pharmacia column preequilibrated with 20 mM NaAc (pH 5.2) and 5 mM β-Me. The protein was eluted with a 0–1 M NaCl gradient with 5 mM β-Me. The protein-containing fractions were pooled and dialyzed extensively against 20 mM Hepes (pH 8.2) and 50 mM Na2SO4, concentrated to >20 mg·ml–1, flash-frozen in liquid N2, and stored at –80°C.
Crystallization and Structure Determination. Crystals of the native and Se-Met-labeled protein were obtained from a noncommercially available precipitant synergy (PS) screen (condition 46) (6). Diffraction-quality crystals were obtained using the hanging-drop method by mixing a 1:1 ratio of 20 mg·ml–1 protein to a 16% poly(ethylene glycol) 3350, 10% 2-methyl-2,4-pentanediol, 0.2 M ammonium citrate (pH 4.5), 5% (vol/vol) glycerol, 2 mM DTT, 20 mM N,N-dimethyldecylamine-β-oxide, and 0.1 M Bicine (pH 9.0) solution. Crystals were flash-frozen in liquid nitrogen for data collection at 100 K. X-ray diffraction data for a single-wavelength anomalous diffraction experiment at the Se-Met anomalous peak was collected to 2.0 Å at the Lawrence Berkeley National Laboratory (beamline 12.3.1). Diffraction images were processed with the hkl package (see Table 2) (7).
Table 2. Summary of refinement statistics.
| Refined crystal structure | |
| Resolution range,* Å | 30-2.04 (2.17-2.04) |
| No. of reflections* | 99,205 (10,000) |
| Completeness,* % | 76.4 (50.0) |
| R factor,*† % | 20.7 (26.7) |
| Free R factor,*†‡ % | 24.6 (30.9) |
| rmsd bond length, Å | 0.006 |
| rmsd bond angle, ° | 1.4 |
| Refined pseudo-atomic capsid shell model | |
| Method 1 | |
| R factor,† % | 32.8 |
| Temp. factor, Å2 | 276 |
| Method 2 | |
| R factor,† % | 29.9 |
| Temp. factor, Å2 | 258 |
Values in parentheses refer to the highest-resolution bin.
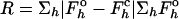 .
.
Calculated from randomly chosen reflections (7%).
The selenium sites (14 of 16) were found by using direct methods with the program shake-and-bake v.2.0 (8). The sites were refined with the program solve (9). Initial phase calculation to 2.6 Å with solve produced low-quality maps that were difficult to interpret. Density modification with the program resolve (9) immensely improved the electron-density map such that the autotrace function of resolve could trace ≈70% of the crystallographic asymmetric unit (ASU). An additional 15% of the ASU was traced with the “baton_build” function of o (10). Refinement of the model and phase extension to 2.0 Å with the program cns (11) allowed the remaining portion of the model to be built in o. Restrained noncrystallographic symmetry was applied for the first few rounds of refinement and removed later near the end of the refinement. The quality of the final model was checked with procheck (12).
Structure Analysis. Buried surface area and available surface area calculations were carried out by using the program cns. Cavity calculations were carried out by using the server CASTp (13).
Pseudoatomic Capsid Model and Difference Map. A pseudoatomic model of the icosahedral ASU was generated by manually docking five of the MCP capsomers into the cryo-EM electron-density maps using the program o. Both enantiomorphs of the cryo-EM reconstruction were used to identify the correct hand of the reconstruction. The trimers were refined with a round of real space refinement by using the “fm_rsr_group” function of o. The placement of the model allowed an educated analysis of the fit. At this point, radial masks were applied to the cryo-EM reconstruction to excluded the viral genome, inner membrane, and turret-like pentamers by using the program mama (14). The remaining maps generously accommodated the model and included surrounding density so as not to bias the fit during rigid-body refinement. Structure factors of the masked electron-density maps were calculated to the resolution of the maps (27 Å) by Fourier transformation using sfall of the ccp4 package (15). Rigid-body refinement of the trimer models was carried out while imposing icosahedral symmetry by using the program x-plor (16). The temperature factor (B) of the refined model was determined by trying values ranging from 50 to 2,000 Å2 and calculating the crystallographic R factor by using x-plor. The B factor giving the lowest R factor was selected. The absolute scale of the cryo-EM maps was calculated by using the refined model of the icosahedral ASU and MCP crystal structure as a reference. The masked maps were scaled by 0.5% increments from 90% to 120% of the initial size using the program mapman (14), structure factors were calculated, and capsomers were manually refitted. Rigid-body and temperature factor refinements were performed for each increment. The R factor, temperature factor, gaps between capsomers, and the number of atoms outside of the map were used as model quality criteria. The van der Waals flag was included during the refinement to reduce steric clash. Particle dimensions were calculated in o.
The structure factors and phases for the pseudoatomic capsid model were calculated with the program x-plor to 27 Å resolution, and a map of the model was calculated by using a grid spacing of 5.2 Å. spider was used for trilinear interpolation of the cryo-EM map to the same grid spacing as the calculated map. The interpolation was crucial to help identify many of the difference map features. The calculated and cryo-EM maps were aligned by using spider (17). The aligned maps were scaled to have the same average and standard deviation in the MCP shell region. A difference map was calculated by using mapman (14). The volume of the individual density difference peaks were calculated with the “floodfill” function of situs (18) by using obtained coordinates of each peak with o. The number of voxels above the MCP threshold were calculated and converted to mass by using the calculated MCP voxel density (see Supporting Text, which is published as supporting information on the PNAS web site).
Results and Discussion
Structure Determination. The MCP with a C-terminal hexahistidine tag was expressed in E. coli and purified to homogeneity by using affinity and ion-exchange chromatography. Crystals of the native and Se-Met-labeled protein were isomorphous. Two single-wavelength anomalous diffraction data sets were collected from a Se-Met MCP crystal at the Se-Met anomalous peak (Table 1). The data were of sufficient quality to identify 14 of the 16 selenium substructures present in the four molecules in the crystallographic ASU. Refinement of the sites and phase calculation produced a low-quality map at 2.6 Å. Subsequent density modification improved the quality of the map and clearly showed the trace of the molecules. More than 70% of the model was autotraced with the program resolve (9). An additional 15% of the model was built manually by using the baton_build feature of o (10) to produce the starting model for phase extension and refinement. The remaining portion of the model was built during refinement. The current model is refined to 2.0 Å, resulting in an R factor of 20.7% and an Rfree of 24.6% (Table 2).
Table 1. Summary of x-ray data.
| Data collection | λ1a | λ1b |
|---|---|---|
| Wavelength, Å | 0.97957 | 0.97957 |
| Max. resolution, Å | 2.6 | 2.0 |
| Measured reflections | 941,689 | 821,050 |
| Space group | C2 | C2 |
| Cell parameters | a = 241.6, b = 82.9, c = 114.8 Å α = γ = 90°, β = 116° | a = 241.6, b = 82.9, c = 114.8 Å α = γ = 90°, β = 116° |
| Res. range,* Å | 30-2.6 (2.69-2.60) | 30-2.0 (2.07-2.00) |
| Unique reflections* | 62,310 (6,008) | 114,006 (7,381) |
| Completeness,* % | 95.5 (90.0) | 83.1 (54.2) |
| Rmerge,*† % | 6.8 (25.6) | 7.2 (29.1) |
| I/σ | 11.8 (2.9) | 16.6 (3.2) |
| Redundancy* | 5.8 (5.1) | 4.7 (3.1) |
| Solvent fraction | 0.58 | 0.58 |
| Substructures (Se) | 14 Se | — |
Values in parentheses refer to the highest-resolution bin.
Rmerge = ΣhΣi\Ihi— 〈Ih 〉/ΣhΣhi.
Structure of the MCP Subunit. There are four molecules in the ASU of the cell, and the electron density for each molecule is continuous from residues Gly-2 to Leu-324. The N-terminal Met is absent in all of the molecules and is probably removed after translation. The 27 residues (325–351) in the C terminus, which includes the hexahistidine tag, show no discernable density and were not modeled. The MCP subunit structure consists of two consecutive barrels (a double barrel), labeled B (residues 2–180) and B′ (residues 181–324) (Fig. 1A). Each barrel can also be described as a β-sandwich composed of eight antiparallel β-strands (labeled B–I for B and B′–I′ for B′). In each barrel, strands B–I–D–G form one of the sheets and strands C–H–E–F form the second sheet. The sheets in each barrel are slightly tilted and have a right-handed twist with respect to one another. There is an extensive hydrophobic core in the interior of each β-sandwich that is primarily responsible for holding the sheets together. There are no hydrogen bonds between strands BC (or B′C′) and FG (or F′G′) leaving the edges of the barrels open. The loops connecting the strands are generally of the same size (five to seven residues), with the exception of the longer loops lde (ld′e′) and lfg (lf′g′). Each barrel is decorated by a number of helices and β-strands. A 10-residue helix (αb) follows the F strand of barrel B (inserted in loop lfg) and is almost orthogonal to that barrel. Helix αb is locked between the two barrels and helps form part of the extensive double-barrel interface. The remaining portion of the interface is formed from interactions between residues in strands F–B′, αb–αb′, and αb–lf′g′. There is ≈2,600 Å2 of buried surface area within the double-barrel interface. An analogous helix (αb′) follows the F′ strand of barrel B′ (inserted in loop lf′g′). The MCP subunit is ≈53 Å wide, 66 Å tall, and 30 Å deep. Barrel B is slightly taller than barrel B′ (≈19 Å) because of a tower formed by loops lde, lfg, and lhi, and strands βa and βc. Strands βa and βc form a small β-sheet.
Fig. 1.

Ribbon representation of viral MCPs. (A) A ribbon representation of the crystal structure of the STIV MCP. Strands are colored in cyan, helices are in yellow, and coils are in gray. (B) Stereoribbons diagram showing the structural homology between the PRD1 P3 (cyan), PBCV-1 vp54 (green), and STIV MCP (yellow). Also shown is the major subunit of cowpea mosaic virus (CpMV; Protein Data Bank ID code 1NY7) in blue for comparison. The CpMV eight-stranded barrel, commonly referred to as a viral jelly roll, is common in capsid proteins present in a wide array of DNA and RNA viruses infecting bacteria, plants, and animals (40–42). The figure was produced with ribbons (43).
The tower is clearly visible in the cryo-EM image reconstruction of the virus and defines a unique, hand-sensitive, orientation of the subunit in the capsid, allowing assignment of the T = 31d enantiomorph of the surface lattice. The cryo-EM-based model also defines the biological trimer, because the subunits are monomeric in the crystal structure (see below). Helix αd extends 12 Å away from the B barrel and separates the two barrels. The C terminus stretches away from the subunit and terminates after Leu-324 because of poorly defined density. However, the density C-terminal to Leu-324 contoured at 0.6σ suggests that the remaining residues continue the helix.
The evolutionary relationship between the adenovirus, PRD1, and PBCV-1 was reported by a number of groups and is rooted in the Linnean approach, where architectural morphology rather than genomics is the determinant (3, 19–21). The relationship among these viruses stems from structural features shared between their capsid architecture and MCPs: hexon, P3, and vp54, respectively. The capsid shells are composed of trimer-forming subunits that are seen as quasi-hexameric capsomers that occupy the predicted quasi sixfold positions in the Caspar–Klug surface lattices. The MCP crystal structure is remarkably similar to the adenovirus, PRD1, and PBCV-1 MCP subunits and shares their structural features (Fig. 1B). The subunits share a double-barrel motif with homologous topology, where there is a major insertion in loops lde (ld′e′) and lfg (lf′g′), with an α-helix in loop lfg of both barrels, and an α-helix that follows the I strand of barrel B to form a barrel–helix–barrel motif (Fig. 1). Superposition of the P3, vp54, and MCP subunits shows that >46% of the residues in each structure overlay on top of one another with an rms deviation of <2.2 Å. A structure-based sequence alignment shows that these proteins share significant sequence homology (33% sequence similarity between the MCP and the hexon, 41% sequence similarity between the MCP and P3, and 38% sequence similarity between the MCP and vp54 using the pam500 scoring matrix) (see Table 3 and Fig. 5, which are published as supporting information on the PNAS web site). Indeed, the major subunit of CpMV, which represents the fusion of two barrel folds (“viral” jelly rolls) found in other icosahedral viruses, shows little structural similarity to the hexon, P3, vp54, and MCP, because the fusion probably occurred in the context of a T = 3 capsid. The similar capsid architecture, homologous MCP structures, and sequence similarity lead us to the conclusion that viral capsid of STIV, adenovirus, PRD1, and PBCV-1 are evolutionarily related.
Structural Stability in the Hot Spring. STIV spends a portion of its life cycle in the acidic high-temperature extracellular environment of the hot springs. Therefore, the capsid must tolerate the high temperature and acidic conditions to maintain infectivity. Experimental data show that the virus is stable in the high-temperature acidic growth media for weeks (data not shown) and that >7 M guanidine hydrochloride is required to completely denature the MCP (data not shown).
The exceptional stability of the capsid can be partly explained through the analysis and comparison of the MCP subunit structure to the hexon, P3, vp54, and the large subunit of CpMV. Packing and cavity volume calculations show that the MCP subunit is a more tightly packed protein with fewer cavities and a smaller total cavity volume than the other subunits (data not shown). For example, the total cavity volume of the P3 subunit, the next tightest packed structure, has more than twice the cavity volume than that calculated for the MCP subunit. Filling cavities inside of tightly packed structure is known to increase the thermal stability of proteins (22). In addition, the Pro content of the MCP (6.7%) is higher than what is observed on average in protein sequences (5.1%) (23). The MCP Pro residues are evenly distributed between the two barrels of the subunit and primarily occur in the loops, where they help form and stabilize a number of the turns. Indeed, a large Pro cluster is localized in the tower section of the first barrel (seven Pros in a sphere of radius 12 Å; data not shown). The tower is predominantly exposed to the extracellular environment (see below); therefore, Pro residues may be a stereochemical adaptation to create a rigid and more stable tower. Pro residues have been suggested to contribute to thermostabilization by reducing flexibility and decreasing the entropy of the unfolded state (24). Additional stability may come from the shorter loops in MCP when compared with the hexon, P3, and vp54 (25). In general, it is difficult to draw a simplified argument as to what structural features underlie the increased thermostability of thermophillic organisms. However, additional factors attributed to increase the thermal stability of proteins, but not found in our analysis, include increased numbers of hydrogen bonds and electrostatic interactions (salt bridges) and increased polar patches on the surface of proteins (26). It may be possible that such differences could not be identified in our comparative analysis because, although not thermophillic, hexon, P3, and vp54 also display unusual stability (19, 20, 27). For example, P3 forms a trimer that requires extensive boiling in SDS to dissociate (28).
MCP Trimer Model. A trimeric STIV capsomer was initially proposed by the cryo-EM image reconstruction of the virus. Moreover, the PRD1 P3 crystal structure could easily be modeled into that reconstruction (2). However, the crystal structure of MCP has four monomer subunits in the ASU with no threefold-related trimers. In contrast to the monomeric MCP, the hexon, P3, and vp54 all form threefold-related trimers in their crystals that are closely similar to the trimers observed in the virus surface lattice. Therefore, it is puzzling why the crystal structure of the MCP is a monomer in the crystal lattice. In an effort to explain the monomeric MCP crystal structure, we generated a pseudotrimer model of the MCP by overlaying three subunits onto the P3 trimer. The resulting model is in good agreement with the molecular envelope of the cryo-EM image reconstruction (see below). The MCP trimer looks like a daisy wheel with six spokes showing pseudohexameric symmetry (Fig. 2A). There are a few atomic clashes and gaps between neighboring monomers of the MCP trimer model that indicate conformational adjustments in the monomer are required for trimer formation. Comparison of the P3 and MCP trimers produces a convincing explanation for the monomeric MCP structure. Both trimers bury little more than 1,140 Å2 of surface area at the edge of each subunit. An extended FG1 loop in the P3 structure contributes an additional 2,220 Å2 of buried surface area to the P3 trimer interface (Fig. 2A); however, this loop is absent in the MCP sequence. The 1,140 Å2 of buried surface area for each MCP trimer interface is below the 1,600 (±400) Å2 found for stable protein–protein interfaces (29). Therefore, we conclude that an equivalent FG1 loop missing in the MCP subunit may be responsible for the monomeric crystal structure. It may also be possible that the presence of viral or host proteins, missing in our recombinant expression system, are required for trimerization or that high temperature may act as an allosteric activator for trimerization. Both hexon and P3 are reported to require chaperonins to ensure the proper formation of a trimer (30, 31). Indeed, crystals of hexon, P3, and vp54 were all produced from protein purified from mature virus particles.
Fig. 2.

Pseudo-atomic model of the STIV icosahedral ASU MCP shell. (A) Ribbon diagram of the STIV MCP trimer (Left) modeled after the P3 trimer (Right) shown down the threefold axis. Each subunit is colored separately. Loop FG1 of P3, colored magenta, is missing in the MCP. (B) Top view of the STIV icosahedral ASU showing the Cα-trace of the refined capsomer model of the MCP (method 1). The peripentonal capsomer is colored in cyan, the facet-edge capsomer is red, the capsomer near the twofold axis is green, the capsomer adjacent to the peripentonal capsomer is yellow, and the capsomer near the icosahedral threefold axis is in magenta. Cryo-EM electron-density mesh is contoured at 1.0σ.(C) Side-view comparison of the two refinement methods in the icosahedral ASU. The yellow model is the Cα-trace of the refined model where each capsomer (trimer) is treated as a rigid body (method 1); the magenta model is the Cα-trace of the refined model where each subunit (monomer) is treated as a rigid body (method 2). Notice the differences between the two models near the peripentonal and edge facet positions. Cryo-EM electron density contoured at 1.0σ. The figure was produced with chimera (44).
Capsid Architecture. A pseudoatomic model of the MCP portion of the STIV shell was generated by manually docking five capsomer (trimer) models into the icosahedral ASU of the cryo-EM map by using the program o (10). The model was improved by rigid body refinement by using x-plor (16) while imposing icosahedral symmetry (see Materials and Methods and Fig. 2B). Two different strategies were used for the refinement. In the first method, each capsomer was treated as a rigid body. In the second method, each subunit (monomer) was treated as a rigid body. The purpose of the second method was to identify differences that may exist between the different capsomers in the icosahedral ASU. The two methods produce slightly different results (Table 2 and Fig. 2C), which include an improved fit of the peripentonal and facet edge subunits when refining each subunit as a rigid body (the second method). The calculated rms deviation between equivalent Cα atoms for the alternately refined models is 4.9 Å for the peripentonal subunits, 2.7 Å for the facet edge subunits, and 1.9 Å for the subunits near the icosahedral threefold axis (negligible at the 27-Å resolution of the cryo-EM map). Therefore, it appears that although the initial capsomer model of the MCP is correct (due to the similarity between the two refined models near the icosahedral threefold axis), the MCP trimers in the icosahedral capsid may not be identical to one another, and the MCP trimer may not be a static oligomer. The disparity of these trimers is supported by the monomer crystal structure and the small calculated surface area buried upon MCP trimer formation.
The average center-to-center distance between neighboring MCP capsomers is 73.8 ± 1.4 Å, virtually identical to the 73.5 ± 0.3 Å distance between the P3 capsomers in the PRD1 virion crystal structure (32). The similarity between the molecular envelope dimensions of the P3 and MCP subunits and the distance between neighboring capsomers in the capsid shells concurrently support the MCP trimer model and pseudoatomic model of the capsid shell. The dimensions of the scaled STIV cryo-EM reconstruction are as follows: vertex top–vertex top, 1,016 Å; vertex base–vertex base, 747 Å; edge–edge, 730 Å; and face–face, 692 Å (Fig. 3A).
Fig. 3.

Capsid architecture of STIV. (A) The scaled cryo-EM image reconstruction of STIV at 27-Å resolution. (B) The refined pseudo-atomic model of the MCP shell. The capsomers interdigitate close to one another to form the icosahedral ASU. The color scheme is the same as in Fig. 2B.(C)Cα trace of the refined icosahedral ASU overlaid onto the difference map of the STIV. The color scheme is the same as in Fig. 2B. The outer viral membrane leaflet closely follows the capsid shell and is colored dark red, the minor capsid proteins (PVI) are blue, Peaks I–V (PI-V) are black, and the vertex appendage is yellow. The C terminus of the MCP crystal structure terminates closely to the large densities protruding from the viral membrane. The proposed C-terminal helices for the MCP subunits are modeled into the difference map to show the interaction with the outer leaflet. These helices were modeled according to the N-terminal helix (first 12 residues) of P3 observed in the PRD1 virus crystal structure. The density bridging the membrane and the capsid shell is strongest near the icosahedral threefold axis and weakest near the edges of the facet. It appears that the flexible C terminus observed in the crystal structure of the MCP may also be flexible in the viral capsid. Difference electron density map is contoured at 100% MCP mass content. Figure was produced with chimera (44).
A difference density map was calculated by subtracting the pseudoatomic model density from the cryo-EM density. Aside from the DNA core, the vertex appendages, and the implicated viral membrane, the difference map revealed a total of six isolated peaks of density near the capsid shell. Peaks I–V (labeled PI to PV in Fig. 3C) are located below the central cavity of each capsomer and are in close proximity to the underlying viral membrane. We speculate that the constituents of these densities may act as a conduit for electrostatic interactions between the negatively charged viral membrane and the positive charged inner surface of the MCP capsomer (data not shown). Similar peaks were observed in PRD1 difference maps (33). The remaining peak (PVI) is a minor capsid protein located beneath the peripentonal capsomer near the base of the vertex (Fig. 3C; see below).
Beneath the MCP shell is a layer of electron density that we assign to the viral lipid membrane (2). A series of density bridges throughout the icosahedral surface facet (with the exception of the peripentonal capsomers and their neighboring subunits) tether this layer to the MCP shell. These bridges are in close proximity to the visible C terminus of the MCP and are large enough to accommodate the 21 residues disordered in the C terminus of the MCP crystal structure (Fig. 3C). The C terminus of MCP is highly basic (PAQVAAIVQQYVARQKRRIKR, calculated pI 12.0), and secondary structure predictions with predator (34), psipred (35), and profsec (B. Rost, personal communication) suggest that it is helical. Therefore, we have modeled the disordered C-terminal residues of the MCP subunit crystal structure as a helix that dips toward and makes contact with the outer leaflet of the lipid membrane (Fig. 3C). Surprisingly, structural superposition of MCP and P3 subunits shows the C and N termini, respectively, of the two proteins to overlay on one another (Fig. 1B). The first three Gln residues in the N terminus of P3 (MA QVQQLTPAQQAA) probably attach to the outer leaflet head groups of the host-derived lipid membrane of PRD1 (32), whereas the C terminus of MCP plays a similar role and attaches to the lipid membrane of STIV (Fig. 3C). The radially averaged peak-to-peak distance between the STIV outer and inner leaflets is ≈38 Å. This distance is within experimental error of the image reconstruction to the reported distance of 25–30 Å for the thickness of the S. solfataricus lipid membrane (36).
Vertex Complex. The vertex complex is a turret or flower-like appendage that extends 135 Å above the outer edge of the MCP shell (Fig. 4). Petal-like attachments branch from the stalk 25 Å above the MCP tower and extend 82 Å laterally. The entire width of the complex is 244 Å. The base of the turret extends 50 Å into the icosahedral capsid from the bottom of the MCP shell to provide structural support for the complex. From above, the assemblage looks like a counterclockwise spinning fan with the tips of the blades pointed clockwise. A subunit from each peripentonal MCP capsomer tightly packs against the vertex complex and carves a groove above its base (Fig. 4). The majority of the interaction between the subunit and the complex occurs above this region, as the pentamer complex interlocks between the two barrels of each subunit. This interaction is analogous to the vertex-capsomer interaction (P3–P30) observed in the PRD1 virus crystal structure (32). The C termini of the peripentonal monomers terminate close to the base of the complex (<2 Å) and are probably in close contact with proteins in this region. The calculated mass of the vertex complex, excluding the cementing protein (PVI), is 637 kDa. There are no single proteins in the predicted ORFs that correlate to a mass of 127 kDa for one of the pentamer subunits; therefore, we conclude that each pentamer subunit is composed of multiple proteins and/or that there may be posttranslational modifications that alter the mass of the complex.
Fig. 4.

Difference density map showing the vertex complex. Surrounding the vertex are the peripentonal capsomers, shown in cyan. The complex measures: a = 244 Å, b = 82 Å, c = 68 Å, d = 92 Å, and e = 205 Å. The viral membrane is colored in red, and the minor capsid proteins at the base of the vertex are blue. Map contoured at 100% MCP mass content. Figure was produced with chimera (44).
Surrounding the base of the complex are density peaks (PVI) that correlate to a 19.8-kDa protein. These peaks are directly beneath the peripentonal MCP capsomers and appear to interact with its C termini. Density can be seen connecting the cementing proteins to the viral membrane (Fig. 4). These proteins may belong to a transmembrane protein similar to P16 in PRD1. Minor capsid proteins surrounding the base of the adenovirus vertices also have been reported (37).
Conclusions
The structural homology shared between the viral capsid proteins and architectures of the mammalian adenovirus, bacteriophage PRD1, and green algae PBCV-1 indicate that their capsids, or capsid proteins, and not the viruses themselves, share a common ancestor because these viruses have genomes of different size and complexity and infect different domains of life. However, a proposal by Bamford (38) suggests that the fundamental structural aspects of the virus, such as the capsid architecture and possibly the genome packaging machinery, are in principle the “self” or “soul” of the virus that have been inherited from a viral ancestor. Whereas attributes such as host recognition and adaptation are more likely acquired from the host through lateral gene transfer, rather than inherited from the ancestor (3). In this view, structural homology can be used to reveal phylogenetic relationships that are otherwise undetectable through genomic comparisons of viruses due to long evolutionary divergence. Consequently, it can be argued that adenovirus, PRD1, and PBCV-1 may share a common ancestor, not just their viral capsids. Here we report the crystal structure of the STIV MCP and show its structural and sequence homology to the adenovirus, PRD1, and PBCV-1 MCPs. We also demonstrate that interactions between the MCP protein shell and other portions of the virus are similar to interactions observed in adenovirus and PRD1. These similarities strongly suggest that these viruses are evolutionarily related and have evolved from a common ancestor. This previously undescribed viral lineage involves all domains of life. It is highly likely that identification and characterization of archaeal viruses will provide additional examples of viral evolutionary relationships that span all the domains of life. Indeed, preliminary characterization of SH1, a spherical halovirus, suggests that this virus too may be related to this lineage (39).
Supplementary Material
Acknowledgments
We thank Ken Frankel for help with the beamline operation and data collection at the SIBLYS beamline (12.3.1) at the Advanced Light Source, supported in part by Department of Energy Grant DE AC03 76SF00098 and National Cancer Institute Grant CA92584. This work was supported by National Institutes of Health Grant R01 GM54076 (to J.E.J.) and National Science Foundation Grant MCB 01322156 (to M.Y.). R.K. was supported by National Institutes of Health Postdoctoral Fellowship F32 AI1065071.
Author contributions: R.K., M.J.Y., and J.E.J. designed research; R.K., L.T., E.T.L., and C.M.L. performed research; R.K. analyzed data; and R.K., M.J.Y., and J.E.J. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ASU, asymmetric unit; cryo-EM, electron cryomicroscopy; MCP, major capsid protein; PBCV-1, Paramecium bursaria Chlorella virus; Se-Met, selenomethionine; STIV, Sulfolobus turreted icosahedral virus.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2BBD).
References
- 1.Pfeifer, F., Palm, P. & Schleifer, K.-H., eds. (1994) Molecular Biology of Archaea (Gustav Fischer, Stuttgart, Germany).
- 2.Rice, G., Tang, L., Stedman, K., Roberto, F., Spuhler, J., Gillitzer, E., Johnson, J. E., Douglas, T. & Young, M. (2004) Proc. Natl. Acad. Sci. USA 101, 7716–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bamford, D. H., Burnett, R. M. & Stuart, D. I. (2002) Theor. Popul. Biol. 61, 461–470. [DOI] [PubMed] [Google Scholar]
- 4.Benson, S. D., Bamford, J. K., Bamford, D. H. & Burnett, R. M. (2004) Mol. Cell 16, 673–685. [DOI] [PubMed] [Google Scholar]
- 5.Van Duyne, G. D., Standaert R. F., Karplus P. A., Schreiver S. L. & Clardy J. (1993) J. Mol. Biol. 229, 105–124. [DOI] [PubMed] [Google Scholar]
- 6.Majeet, S., Ofek, G., Belachew, A., Huang, C.-C. & Kwong, E. (2003) Structure (London) 11, 1061–1070. [DOI] [PubMed] [Google Scholar]
- 7.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 8.Weeks, C. M. & Miller, R. (1999) J. Appl. Crystallogr. 32, 120–124. [Google Scholar]
- 9.Terwillinger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 11.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 12.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- 13.Binkowski, T. A., Naghibzadeh, S. & Liang, J. (2003) Nucleic Acids Res. 31, 3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleywegt, G. J., Zou, J. Y., Kjeldgaard, M. & Jones, T. A. (2000) in Crystallography of Biological Macromolecules, eds. Rossmann, M. G. & Arnold, E. (Kluwer Academics, Dordrecht, The Netherlands), Vol. F, pp. 353–359, 366–367. [Google Scholar]
- 15.Collaborative Computational Project No. 4. (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 16.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S. & Kuszewski, J. (1992) X-PLOR: A System for X-Ray Crystallography and NMR (Yale Univ. Press, New Haven, CT), Version 3.1.
- 17.Frank, J., Radermacher, M., Penczek, P., Zhu, J., Li, Y., Ladjadj, M. & Leith, A. (1996) J. Struct. Biol. 116, 190–199. [DOI] [PubMed] [Google Scholar]
- 18.Wriggers, W., Milligan, R. A. & McCammon, J. A. (1999) J. Struct. Biol. 125, 185–195. [DOI] [PubMed] [Google Scholar]
- 19.Benson, S. D., Bamford, J. K. H., Bamford, D. H. & Burnett, R. (1999) Cell 98, 825–833. [DOI] [PubMed] [Google Scholar]
- 20.Nandhagopal, N., Simpson, A. A., Gurnon, J. R., Yan, X., Baker, T. S., Graves, M. V., Van Etten, J. L. & Rossmann, M. G. (2002) Proc. Natl. Acad. Sci. USA 99, 14758–14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendrix, R. W. (1999) Curr. Biol. 9, R914–R917. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa, K., Nakamura, H., Morikawa, K. & Kanaya, S. (1993) Biochemistry 32, 6171–6178. [PubMed] [Google Scholar]
- 23.McCaldon, P. & Argos, P. (1988) Proteins 4, 99–122. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe, K., Chishiro, K., Kitamura, K. & Suzuki, Y. (1991) J. Biol. Chem. 266, 24287–24294. [PubMed] [Google Scholar]
- 25.Russell, R. J., Ferguson, J. M., Hough, D. W., Danson, M. J. & Taylor, G. L. (1997) Biochemistry 36, 9983–9994. [DOI] [PubMed] [Google Scholar]
- 26.Szilagyi, A. & Zavodszky, P. (2000) Struct. Fold Des. 8, 493–504. [DOI] [PubMed] [Google Scholar]
- 27.Athappilly, F. K., Murali, R., Rux, J. J., Cai, Z. & Burnett, R. M. (1994) J. Mol. Biol. 242, 430–455. [DOI] [PubMed] [Google Scholar]
- 28.Mindich, L., Bamford, D., McGraw, T. & Mackenzie, G. (1982) J. Virol. 44, 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo Conte, L., Chothia, C. & Janin, J. (1999) J. Mol. Biol. 285, 2177–2198. [DOI] [PubMed] [Google Scholar]
- 30.Hanninen, A. L., Bamford, D. H. & Bamford, J. K. (1997) Virology 227, 207–210. [DOI] [PubMed] [Google Scholar]
- 31.Cepko, C. L. & Sharp, P. A. (1982) Cell 31, 407–415. [DOI] [PubMed] [Google Scholar]
- 32.Abrescia, N. G., Cockburn, J. J., Grimes, J. M., Sutton, G. C., Diprose, J. M., Butcher, S. J., Fuller, S. D., San Martin, C., Burnett, R. M., Stuart, D. I., et al. (2004) Nature 432, 68–74. [DOI] [PubMed] [Google Scholar]
- 33.San Martin, C., Huiskonen, J. T., Bamford, J. K. H., Butcher S. J., Fuller, S. D., Bamford, D. H. & Burnett, R. M. (2002) Nat. Struct. Biol. 9, 756–763. [DOI] [PubMed] [Google Scholar]
- 34.Frishman, D. & Argos, P. (1996) Protein Eng. 9, 133–142. [DOI] [PubMed] [Google Scholar]
- 35.Jones, D. T. (1999) J. Mol. Biol. 292, 195–202. [DOI] [PubMed] [Google Scholar]
- 36.Gliozzi, A., Rolandi, R., De Rosa, M. & Gambacorta, A. (1983) J. Membr. Biol. 75, 45–56. [DOI] [PubMed] [Google Scholar]
- 37.Fabry, C. M., Rosa-Calatrava, M., Conway, J. F., Zubieta, C., Cusack, S., Ruigrok, R. W. & Schoehn, G. (2005) EMBO J. 24, 1645–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bamford, D. H. (2003) Res. Microbiol. 154, 231–236. [DOI] [PubMed] [Google Scholar]
- 39.Porter, K., Kukkaro, P., Bamford, J. K., Bath, C., Kivela, H. M., Dyall-Smith, M. L. & Bamford, D. H. (2005) Virology 335, 22–33. [DOI] [PubMed] [Google Scholar]
- 40.Rossmann, M. G. & Johnson, J. E. (1989) Annu. Rev. Biochem. 58, 533–573. [DOI] [PubMed] [Google Scholar]
- 41.Chelvanayagam, G., Heringa, J. & Argos, P. (1992) J. Mol. Biol. 228, 220–242. [DOI] [PubMed] [Google Scholar]
- 42.McKenna, R., Xia, D., Willingmann, P., Ilag, L. L., Krishnaswamy, S., Rossmann, M. G., Olson, N. H., Baker, T. S. & Incardona, N. L. (1992) Nature 355, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carson, M. (1986) J. Mol. Graphics. 5, 103–106. [Google Scholar]
- 44.Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004) J. Comput. Chem. 20, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.