

Abstract
Aptamers recognize their targets with extraordinary affinity and specificity. The aptamer-based therapeutic, Macugen, is derived from a modified 2′fluoro pyrimidine RNA inhibitor to vascular endothelial growth factor (VEGF) and is now being used to treat the wet form of age-related macular degeneration. This VEGF165 aptamer binds specifically to the VEGF165 isoform, a dimeric protein with a receptor-binding domain and a heparin-binding domain (HBD). To understand the molecular recognition between VEGF and this aptamer, binding experiments were used to show that the HBD contributes the majority of binding energy in the VEGF165–aptamer complex. A tissue culture-based competition assay demonstrated that the HBD effectively competes with VEGF165 for aptamer binding in vivo. Comparison of NMR spectra revealed that structural features of the smaller HBD–aptamer complex are present in the full-length VEGF164–aptamer complex. These data show that the HBD provides the binding site for the aptamer and is the primary determinant for the affinity and specificity in the VEGF165–aptamer complex.
Keywords: age-related macular degeneration, Macugen, RNA, NMR
Vascular endothelial growth factor (VEGF) is an essential growth factor and is the key angiogenic factor for development, proliferation, and maintenance of a healthy vasculature. VEGF is also involved in pathologic angiogenesis, such as age-related macular degeneration (AMD), ischemic heart disease, tumor growth, and metastasis (1, 2). One isoform, VEGF165, is the major inducer of abnormal blood vessel growth and leakage in the wet form of AMD, a vascular disease of the eye that leads to rapid vision loss and blindness in millions worldwide (1, 3, 4). Inhibiting VEGF165 in the eye is therefore a promising approach for controlling angiogenesis in AMD (5).
VEGF165 consists of two domains, a receptor-binding domain that is present in all four isoforms of VEGF and a heparin-binding domain (HBD) that distinguishes VEGF165 from the other soluble isoforms (1). The 3D structures of each domain have previously been determined (6, 7). The receptor-binding domain interacts with cell-surface tyrosine kinase receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR, Flk-1), triggering the signaling cascade that leads to angiogenesis (1, 2). Antibody fragments that target the receptor-binding domain and therefore inhibit receptor binding are currently used in cancer treatment and are also in clinical trials for wet AMD (1). These antibodies inhibit all isoforms of VEGF. In contrast, a nucleic acid-based inhibitor, Macugen, currently used in treatment of wet AMD (5) is an isoform-specific inhibitor of VEGF165 that does not recognize VEGF121, which lacks the HBD (8).
Aptamers are nucleic acids that are generated by the SELEX (systematic evolution of ligands by exponential enrichment) in vitro evolution technology (9, 10). The VEGF165 aptamer, also called NX1838 or pegaptanib sodium, is a potent inhibitor of angiogenesis and was used to develop the drug Macugen (5, 11). This aptamer is a modified RNA derived from a 2′fluoro pyrimidine aptamer and also contains 2′O-methyl purine modifications to enhance stability against endonucleases (Fig. 1A). For effective treatment of AMD in humans, the aptamer is further modified with a 5′ polyethyleneglycol moiety and a 3′ dT attached via a 3′-3′ linkage to confer favorable pharmacokinetic properties and for protection against exonucleases (8).
Fig. 1.

The HBD competes with VEGF165 for aptamer binding in vivo. (A) Sequence and secondary structure of the VEGF aptamer. The subscripts m and f indicate the 2′-OMethyl- and 2′-fluoro-modified residues, respectively, and A represents unmodified ribo-A. The arrow points to the photo-crosslinking site at U14. (B) In vivo competition binding assay for the HBD and VEGF165. Relative mRNA expression levels of TF in HUVEC in culture are normalized to untreated cells. Increasing amounts of HBD (2.3, 4.7, 9.4, 18.8, 37.5, 75, 150, and 300 nM) were added to HUVEC treated with VEGF165 and aptamer. The data show that addition of HBD can effectively reverse the inhibition of VEGF165-induced TF expression by the aptamer. Controls include VEGF165- or VEGF121-induced cells, untreated cells, or addition of DTT-inactivated HBD.
This aptamer binds to VEGF165 with extremely high affinity (Kd = 50 pM), and animal studies and clinical data have demonstrated the efficacy of the aptamer in preventing blood vessel growth and arresting the progression of wet AMD (8, 11–14). However, the detailed molecular mechanism by which this aptamer achieves isoform-specific inhibition of VEGF165 is not well understood. An efficient photo-crosslink has been obtained between the HBD and the aptamer, suggesting this aptamer recognizes VEGF165 by targeting the HBD (8). The focus of the studies here is to characterize the role of the HBD in isoform-specific recognition of VEGF165 by the aptamer. NMR spectroscopy was used to compare the isolated HBD–aptamer complex and the full-length VEGF164–aptamer complex. The results lead to valuable insights into the molecular mechanism of inhibition of this aptamer therapeutic.
Materials and Methods
Sample Preparation. Pegylated and nonpegylated aptamers (Fig. 1 A) were obtained from Transgenomics (Omaha, NE) and exchanged into NMR buffer (10 mM Tris-d11, pH 7.0/100 mM NaCl/0.55 mM CaCl2/0.05 mM EDTA/90% H2O/10% D2O) by repeated concentration in Centricon YM-3 centrifugal filter devices (Millipore), resulting in a 1-mM NMR sample.
NMR quantities of 15N-labeled HBD (VEGF111–165) were produced from a Pichia pastoris expression system (Invitrogen). Multiple copies of the expression cassette were stably inserted into the genome of strain KH71, and a methanol-inducible AOX1 promoter controlled expression of secreted HBD. Cells were grown in minimal media with glycerol supplemented with histidine, according to the supplier's recommendations. The cells were transferred to methanol containing minimal media (30–40 OD600/ml) with 10 g/liter 15N ammonium sulfate, and protein induction was maintained for 32 h at 30°C. The media were collected, and the HBD was purified on a 5-ml heparin Sepharose column (HS Hitrap, Amersham Pharmacia) in 10 mM Tris, pH 7.5, with a salt gradient between 0.1 and 1.5 M NaCl. The peak containing the folded HBD was identified by NMR spectroscopy and exchanged into NMR buffer. Yields of ≈7 mg/liter were obtained as determined with the BCA protein assay (Pierce).
The HBD–aptamer complex was formed either by adding 15N-labeled HBD to the aptamer or adding the aptamer to the HBD. 1D imino proton spectra of the aptamer and 2D 1H-15N heteronuclear single quantum correlation (HSQC) spectra of the protein were used to monitor complex formation. The final sample concentration was 0.5 mM, and a small excess (<15%) of aptamer was added to samples used for resonance assignment of the HBD in the HBD–aptamer complex.
The mouse protein VEGF164 was used for the NMR studies on full-length VEGF. This protein is identical to human VEGF165 except that it is missing the C-terminal Arg residue. The aptamer binds with equal affinity to the full-length human (VEGF165) and mouse proteins. NMR quantities of deuterated 15N-labeled VEGF164 were produced from the P. pastoris expression system (Invitrogen) as described above for the HBD, except that cells were first conditioned to 25% D2O in glycerol containing minimal media before transferring the cells to expression media in 80% D2O (with methanol and 10 g/liter 15N-labeled ammonium sulfate). VEGF164 was purified as described above for the HBD and exchanged into NMR buffer. The VEGF164–aptamer complex was formed by adding VEGF164 and aptamer at a 1:1 ratio to a final concentration of 0.1 mM in NMR buffer. 1D imino proton and 2D 1H-15N transverse relaxation optimized spectroscopy (TROSY)-HSQC spectra were used to monitor complex formation (15).
EMSA. EMSAs were used to determine the affinity of the aptamer for the HBD. Increasing amounts of HBD were incubated with 500 pM 32P-labeled aptamer in NMR buffer at 25°C for 30 min. Free and HBD-bound aptamers were resolved on a native 12% polyacrylamide gel at 4°C in 0.5 mM CaCl2, 45 mM Tris·HCl, and 35 mM boric acid, pH 8.3. The data were fit to a two-state binding curve [fraction aptamer bound = [HBD]/(Kd + [HBD])].
In Vivo Competition Assay. A VEGF-induced tissue factor (TF) expression assay using human umbilical vein endothelial cells (HUVEC) (16) was used to test whether the HBD can compete with full-length VEGF165 for aptamer binding in vivo. HUVEC were cultured in M200 media supplemented with 1× low supplement growth serum (Cascade Biologics, Portland, OR) and antibiotics at 37°C. Four hours before the experiment, HUVEC were serum-starved in M200 media with 1% FBS and antibiotics. All treatments afterward were performed in these media. HUVEC were incubated with VEGF165 (328 pM), VEGF165 plus 1.5 nM pegylated aptamer, or VEGF165 plus 1.5 nM pegylated aptamer and increasing concentrations of HBD (2.3–300 nM) for 1 h. HUVEC were also incubated with DTT-inactivated HBD, VEGF121, or media alone (untreated cells) as control experiments. Total RNA was isolated by RNeasy Mini RNA Isolation Kits (Qiagen, Valencia, CA). TF mRNA expression levels were measured by real-time RT-PCR (TaqMan) analysis using primers specific for human TF mRNA (Applied Biosystems). The TF mRNA levels were normalized to the untreated sample.
NMR Spectroscopy. NMR experiments were carried out on Varian Inova 500-, 600-, or 800-MHz spectrometers, equipped with triple resonance z axis pulsed-field gradient probes. The 600-MHz spectrometer used a cryogenic probe. The TROSY-HSQC experiments of the free VEGF164 and VEFG164–aptamer complex were performed at 800 MHz. The 31P 1D data were acquired with a broadband probe at 500 MHz. 1D data were processed with the program felix (Accelrys, San Diego). 2D and 3D data were processed with the program nmrpipe (17) and analyzed by the program sparky (18). External 2-2-dimethyl-2-silapentane-5-sulfonate was used for the 1H and 15N reference, and external sodium phosphate was used as the 31P reference.
Resonance Assignments. 1H, 15N backbone resonance assignments of the HBD and the HBD–aptamer complex were obtained from 3D 1H-15N NOESY-HSQC (150- and 200-ms mixing times) and 1H-15N HSQC-total correlation spectroscopy (80-ms mixing time) experiments at 10°C. Limited resonance assignments of free aptamer and the aptamer in the HBD–aptamer complex were obtained by using homonuclear NOESY and 15N-ω1-filtered NOESY experiments in 90% H2O/10% D2O.
The average chemical shift differences of the amide proton and nitrogen resonances between the free HBD and HBD in the complex were calculated by using Eq. 1 (19),
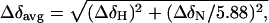 |
[1] |
where ΔδH and ΔδN are the chemical shift differences of the amide proton and nitrogen resonances between free HBD and HBD in the complex.
Results
The VEGF Aptamer Forms a Specific Complex with the HBD of VEGF165. EMSAs were used to probe the affinity for the aptamer to the HBD (Fig. 6, which is published as supporting information on the PNAS web site). A Kd of 12 ± 8 nM was obtained (ΔG° = 10.8 kcal/mol). Because the aptamer binds to VEGF165 with 50 pM affinity (ΔG° = 14.0 kcal/mol), the bulk of the binding energy arises from interactions with the HBD. No binding to the HBD was observed in the absence of Ca2+. Previous studies showed that calcium ions are also required for aptamer binding to full-length VEGF165 where no binding was observed with Mg2+ (8).
2D NOESY spectra were used to assign the imino proton resonances and confirm the secondary structure of the aptamer shown in Fig. 1 A. Fig. 2A shows the titration of aptamer with HBD, where the imino proton spectrum changes until a 1:1 complex is formed and no free aptamer is observed (the U10 resonance at 11.74 ppm is indicative of free aptamer). Further addition of HBD resulted in no change in the imino proton spectrum (data not shown). The formation of the 1:1 HBD–aptamer complex was also monitored by 2D 1H-15N HSQC spectra. Both 1D imino proton and 2D HSQC spectra exhibited slow exchange of the NMR resonances, as expected for a tight binding complex.
Fig. 2.

Characteristic NMR fingerprint of aptamer in complex with the HBD and VEGF164. (A) Imino proton spectra of the aptamer at 10°C upon titration with HBD. The molar ratios are shown on the left. (B) Comparison of the imino proton spectra of the aptamer at 40°C. (Top) The free aptamer. (Middle) The HBD–aptamer complex. (Bottom) The VEGF164–aptamer complex. (C) Comparison of the 31P NMR spectra of the aptamer. (Top) The free aptamer at 25°C. (Middle) The HBD–aptamer complex at 25°C. (Bottom) The VEGF164–aptamer complex at 45°C.
The HBD Competes In Vivo with Full-Length VEGF165 for Aptamer Binding. A VEGF-induced TF expression assay in HUVEC was used to demonstrate the direct binding of the aptamer to the HBD in vivo (16). HUVEC up-regulate the expression of TF in response to VEGF165, and this up-regulation can be suppressed by the aptamer (Fig. 1B). Upon addition of HBD, the suppression of TF expression by the aptamer is reversed, because the HBD competes with full-length VEGF165 for aptamer binding. HBD reverses the inhibition of VEGF165 by the aptamer with an EC50 in the low nM range (Fig. 1B). Competition of HBD and VEGF165 for aptamer binding was observed only with native HBD, and no effect was observed upon addition of 300 nM DTT-inactivated HBD. The results on the unfolded HBD demonstrate that the competition between the HBD and VEGF165 for aptamer binding is not simply caused by favorable electrostatic interactions between the positively charged HBD and the aptamer. Furthermore, VEGF121-induced TF expression in HUVEC was not affected by the addition of aptamer and/or HBD, confirming the specificity of the aptamer for VEGF165. These results demonstrate that HBD can effectively compete with full-length VEGF165 for aptamer binding in vivo.
Comparing the Aptamer Conformation in the HBD–Aptamer Complex and the VEGF164–Aptamer Complex. Fig. 2B shows the imino proton spectra of free aptamer, the aptamer bound to the HBD, and the aptamer bound to VEGF164. The imino proton spectra of the two complexes are very similar, demonstrating that the secondary structure of the aptamer is maintained between the HBD– and the VEGF164–aptamer complexes. The two additional resonances in the VEGF164–aptamer complex were tentatively assigned to the imino proton of U20 (11.61 ppm) in the internal loop of the aptamer and to the amide proton of C57 (11.96 ppm), based on previously published assignments of the receptor-binding domain of VEGF (20).
31P chemical shifts are a sensitive probe of unusual backbone conformations of oligonucleotides and are especially valuable for studying protein–nucleic acid interactions (21, 22). Fig. 2C compares the 31P spectra of the free aptamer with the aptamer bound in either the HBD or the VEGF164 complex. There is a striking change in the 31P spectrum of the aptamer when bound to HBD, with resonances dispersed over 7 ppm. This 31P fingerprint is therefore diagnostic of the HBD–aptamer interaction. Ignoring the differences in line widths resulting from the size difference between the two complexes (63 versus 15.6 kDa), the 31P spectra of the two complexes are essentially the same. These data demonstrate that the backbone conformation of the aptamer and the interactions with the phosphates are conserved between the HBD–aptamer and the VEGF164–aptamer complexes.
The HBD–Aptamer Complex Produces a Characteristic Amide Backbone NMR Fingerprint. Fig. 3 shows a superposition of the 1H-15N HSQC spectra of free HBD and the HBD bound to the aptamer. Amide backbone resonance assignments of the free HBD (at pH 7.0) were obtained from 1H-15N NOESY-HSQC spectra, starting from the published resonance assignments at pH 5.5 (6). Resonance assignments for the HBD–aptamer complex were made by 1H-15N heteronuclear NMR experiments (1H-15N NOESY-HSQC, 1H-15N HSQC-total correlation spectroscopy). In the 1H-15N HSQC spectrum of the HBD–aptamer complex, 48 of the 50 expected amide backbone crosspeaks were assigned, and the two N-terminal resonances were not observed (Fig. 3). The secondary structure of the HBD in the complex with the aptamer was assessed from sequential and medium-range NH-NH and NH-Hα NOE patterns in the 1H-15N NOESY-HSQC spectra (Fig. 7, which is published as supporting information on the PNAS web site). The β1-β2-α-β3-β4 secondary structure of the free HBD (6) is basically unchanged in the complex, allowing the use of chemical shift mapping to probe the aptamer binding site.
Fig. 3.

Superposition of 1H-15N HSQC spectra at 10°C of free HBD (blue) and aptamer-bound HBD (red). Backbone resonance assignments are indicated in blue (free HBD), red (HBD–aptamer complex), and black (free and bound HBD). Boxes illustrate the NMR fingerprint created by residues S121, T142, and T157 in the HBD–aptamer complex.
The weighted average 1H/15N backbone chemical shift changes were determined for each residue by using Eq. 1. These chemical shift changes are clustered in three regions in the amino acid sequence (Fig. 4A). The residues S121, T142, and T157 represent the largest backbone chemical shift perturbations in each affected region. Comparison of the 1H-15N HSQC spectra of free HBD and aptamer-bound HBD (Fig. 3) reveals that these three residues form a well resolved 1H/15N NMR fingerprint that is diagnostic of the HBD–aptamer complex.
Fig. 4.

HBD chemical shift changes upon aptamer binding. (A) The weighted average 1H/15N backbone amide chemical shift changes (Δδavg) of the HBD upon binding to the aptamer. Three regions with large chemical shift changes are labeled as I, II, and III. The residues with the largest chemical shift changes in each region are boxed (S121, T142, and T157). The positions of the secondary structure elements are shown for the free HBD (6). (B) Secondary structural model for the previously determined NMR structure of the free HBD (6). Colors used to illustrate 1H/15N backbone chemical shift changes (Δδavg) upon aptamer binding are: red, >0.5 ppm; magenta, 0.3–0.5 ppm; yellow, 0.1–0.3 ppm; gray, <0.1 ppm. The three regions with large chemical shift changes are labeled I, II, and III.
Comparing the HBD Conformation in the HBD–Aptamer Complex and the VEGF164–Aptamer Complex. The NMR fingerprint created by the amide backbone resonances of the HBD–aptamer complex was used as a structural probe for the VEGF164–aptamer complex. 1H-15N HSQC spectra of the 45-kDa VEGF164 homodimer and the 63-kDa VEGF164–aptamer complex were obtained by the application of TROSY sequences at 800 MHz combined with partial deuteration of VEGF164 and higher temperatures (35°C) (15). The presence of only one set of resonances for the protein and aptamer in these spectra confirm that the 2:2 VEGF164–aptamer complex forms a symmetric dimer in solution.
Fig. 5 shows a superimposition of TROSY-HSQC spectra on the HBD–aptamer and VEGF164–aptamer complexes. The triad of residues S121, T142, and T157 displays the same crosspeak pattern in both the HBD– and VEGF164–aptamer complexes. The crosspeak for S121 is shifted away from other crosspeaks and has a similar position in both complexes. Fig. 5B shows that crosspeaks for residues T142 and T157 in the VEGF164–aptamer complex overlap exactly with those in the HBD–aptamer complex. Additional evidence for these assignments is the absence of crosspeaks at these positions in the spectrum of free VEGF164 (boxes in Fig. 5C), demonstrating that these crosspeaks do not arise from the receptor-binding domain. Further analysis showed that almost all of the 1H/15N crosspeaks in HBD–aptamer complex overlap with crosspeaks in the full-length complex with only two exceptions, where changes for these two residues arise from the presence of the receptor binding domain (data not shown).
Fig. 5.

Protein NMR fingerprint of HBD–aptamer and VEGF164–aptamer complexes. (A) Superposition of the 1H-15N TROSY-HSQC spectra of the HBD–aptamer complex (red) and VEGF164–aptamer complex (blue) acquired at 35°C at 800 MHz. The characteristic signature created by S121, T142, and T157 is emphasized (boxes). (B) Expansion of the spectral region containing the T142 and T157 crosspeaks in the aptamer-bound spectra of the HBD (red) and VEGF164 (blue). The circles indicate the position of these crosspeaks in the free HBD and free VEGF164. (C) The same spectral region as B showing the 1H-15N TROSY-HSQC spectra of free VEGF164. The T142 and T157 amide crosspeaks are circled; the boxes indicate the positions of these crosspeaks in the VEGF164–aptamer complex.
Discussion
The VEGF Aptamer Recognizes the HBD in the VEGF165–Aptamer Complex. The studies here are designed to probe the intermolecular interactions between the aptamer and the HBD in VEGF165. Previous studies have shown that the aptamer does not recognize the shorter VEGF121 isoform, which lacks the C-terminal HBD of VEGF165 (8). In addition, the negatively charged aptamer is predicted to target the positively charged HBD (calculated pI of 11.6), and a unique photo-crosslink has been observed between C137 in the HBD of VEGF165 and a 5-iodo U14-modified aptamer (8). We have shown that most of the binding energy for the aptamer is provided by the HBD, with a ΔΔG° between the HBD– and VEGF165–aptamer complexes of ≈3 kcal/mol or ≈23% of the binding energy. Furthermore, results from the in vivo VEGF-induced TF expression assay demonstrated that the aptamer suppresses VEGF165-induced TF expression with an EC50 that is equivalent to the Kd measured in vitro (Figs. 1B and 6).
The Local Structure of the HBD–Aptamer Complex is Conserved in the Full-Length VEGF164–Aptamer Complex. NMR spectroscopy was used to probe the conformations of both the aptamer and the HBD in the HBD–aptamer and the VEGF165–aptamer complexes. Imino proton spectra provide information on nucleic acid secondary structure, and the spectra in Fig. 2B show there is no significant change in secondary structure of the aptamer in these two complexes. The observation of the U20 imino proton resonance (Fig. 2B Bottom) results from slower hydrogen exchange of this imino proton in the VEGF164–aptamer complex compared with the HBD–aptamer complex. This difference in hydrogen exchange kinetics, combined with the observed difference in aptamer binding energies between the HBD and VEGF164, suggest that the receptor binding domain is either providing additional interactions with the aptamer or indirectly stabilizing new or existing interactions of the aptamer with the HBD.
The Ca2+ requirement for the formation of the VEGF165–aptamer complex is specific to this aptamer family and is not observed in other high-affinity aptamers to VEGF165 (8, 23–25). The results here show that Ca2+ is also required for the HBD–aptamer complex formation. However, no Ca2+ dependence was observed in the NMR spectra of the free aptamer or free HBD (data not shown), and the same small change in melting temperature of the aptamer (3°C) was observed upon addition of either Ca2+ or Mg2+ (8) Thus it appears the Ca2+ binding site is formed only in the aptamer–protein complex, and this Ca2+ binding site may contribute to the isoform-specific recognition of the aptamer.
31P chemical shifts are sensitive to the local environment of phosphates in nucleic acids (21). The HMG box protein SRY bound to its DNA promoter sequence is one of the few protein–nucleic acid systems that have been characterized by 31P NMR (22). This complex shows a 31P chemical shift dispersion of 2.8 ppm with a number of resonances shifted away from standard DNA duplex 31P chemical shifts. These nonstandard 31P chemical shifts can arise from a variety of factors, including salt bridges between Arg or Lys residues and the phosphate groups, intermolecular or intramolecular hydrogen bonding, or metal coordination (21, 22, 26). A large percentage (10 of 28) of the 31P resonances in the HBD–aptamer complex have nonstandard chemical shifts (Fig. 2C Middle) consistent with the presence of multiple side-chain–aptamer or metal–aptamer interactions. Furthermore, except for the differences in linewidth, the 31P fingerprint of the VEGF164–aptamer complex (Fig. 2C Bottom) is essentially unchanged from the HBD–aptamer complex. These data provide strong evidence that the aptamer engages in similar specific intermolecular interactions in both the HBD- and full-length VEGF164 complexes.
The conformation of the HBD in the complex was also monitored by analysis of the diagnostic 1H/15N chemical shifts of S121, T142, and T157 produced upon aptamer binding (Figs. 3 and 4). Fig. 5A shows that this NMR signature is indeed observed in the spectrum of the VEGF164–aptamer complex. These data are consistent with the HBD adopting a similar structure in the HBD– aptamer and VEGF164–aptamer complexes.
Recognition of the HBD in the VEGF165–Aptamer Complex. The binding and NMR data show that the HBD–aptamer complex represents an excellent model system for understanding the interactions in the larger VEGF165–aptamer complex. The 1H/15N chemical shift changes induced by aptamer binding can be used to map the recognition surface on the HBD. Fig. 4B maps the chemical shifts changes on the secondary structure of the free HBD (6). The residues with large chemical shift changes (including the NMR fingerprint S121, T142, and T157) are located in the two central loops (regions I and III in Fig. 4A) and before the α helix (region II). S121 in region I displays the largest chemical shift change upon aptamer binding. The mechanism for formation of a photo-crosslink between 5-iodo-U14 and C137 requires π stacking of the base and the disulfide bond (8, 27). Thus the photo-crosslink to C137, which forms a disulfide bond with C120, is consistent with the aptamer being close to S121. The observed chemical shift changes suggest that the aptamer interacts with the two central loops in the HBD (regions I and III in Fig. 4A). Therefore the chemical shift changes in region II could arise from either the large aptamer wrapping around the HBD to interact with region II or a local conformational change in the hinge region II that brings the two central loops together to form a contiguous binding site for the aptamer.
Most of the structurally characterized aptamers bind their ligands by an induced fit-type mechanism with the aptamer being well ordered in the complex but only partially structured in the free form (28, 29). This type of induced fit requires an entropic cost for ordering the aptamer, which will reduce the overall binding affinity. Thus, higher affinity could be obtained by a mechanism where two rigid bodies form a complex with no additional ordering of the ligand or the aptamer. In our system, the HBD forms a relatively rigid secondary structure stabilized by four disulfide bonds. In addition, the secondary structure of the aptamer is thermodynamically stabilized by the 2′fluoro modifications (8), which greatly enhance helix formation because of their strong preference for the 3′endo sugar pucker (30). Therefore, this VEGF aptamer may be more structured in the free state than most other RNA-based aptamers. Because the stable secondary structure provides a preformed scaffold, only limited folding of the internal loop may be required for VEGF164 binding.
Biological Implications for VEGF165 Inhibition by the Aptamer. The VEGF aptamer is a potent inhibitor of VEGF165-mediated angiogenesis and effectively blocks the interactions with the cell surface tyrosine kinase receptors VEGFR-1 and VEGFR-2 (8). All isoforms of VEGF, including VEGF121, which lacks the HBD, activate VEGFR-1 and VEGFR-2 (1). Thus, it might initially seem unlikely that an aptamer that interacts with the HBD would block receptor binding. However, the results here clearly show that the aptamer interacts predominantly with the HBD of full-length VEGF165. One possibility is that the aptamer inhibits VEGF165 through a steric interference mechanism, where the bulky aptamer prevents interaction of the receptor-binding domain with the cell-surface receptors. Several natural antiangiogenic proteins have been previously identified that support this hypothesis. The metalloprotease ADAMTS1/METH1 (31), heparin affinity regulatory peptide HARP (32), and connective tissue growth factor CTGF (33) have been shown to specifically target the VEGF165 isoform by binding to the HBD (1). Thus this nucleic acid aptamer appears to be mimicking a natural protein-associated interference mechanism for inhibiting the angiogenic activity of VEGF165.
Another possible role of the HBD is to increase the local concentration of VEGF165 at the cell surface by interacting with heparan sulfate proteoglycans (1). Having VEGF165 bound to the cell surface greatly enhances the probability of receptor binding by restricting diffusion of VEGF165 to two dimensions. Thus the therapeutic activity of this aptamer may arise by capturing soluble VEGF165, therefore preventing interactions with heparan on cell surface proteoglycans.
VEGF165-induced signaling through VEGFR-2 is further enhanced by the coreceptor neuropilin 1 (34, 35). Association of VEGFR-2 and neuropilin 1 leads to tyrosine kinase activity of VEGFR-2 at ≈10-fold lower concentrations of VEGF165 than of VEGF121 (35). Neuropilin 1 recognizes the HBD of VEGF165 (34). Thus an important function of the VEGF aptamer may be to block the interaction between neuropilin 1 and VEGF165, thereby suppressing VEGF165-induced pathology of the vasculature.
Conclusions
The VEGF aptamer inhibits angiogenesis by specifically recognizing the HBD of VEGF165. The data presented here demonstrate that the HBD is the primary target of the aptamer and that interactions with the HBD provide most of the binding energy in the VEGF165–aptamer complex. The aptamer not only binds to the HBD in vitro, but also effectively competes with VEGF165 for aptamer binding in vivo. Comparison of NMR spectra of the HBD– and VEGF164–aptamer complexes demonstrates that the same molecular interactions are present in both complexes. Therefore, the HBD–aptamer complex is an important model system for understanding the molecular recognition in the full-length VEGF165–aptamer complex. We propose that both the HBD and VEGF165 use the same mechanism of recognition for binding to the aptamer and that interactions with the HBD lead to the isoform-specific inhibition of VEGF165.
Supplementary Material
Acknowledgments
We thank Drs. R. Shoemaker and A. Fowler for help with running the NMR spectrometers and Drs. D. Wuttke and L. Gold for valuable comments on the manuscript. This work was supported in part by National Institutes of Health Grant AI33098 and the W. M. Keck Foundation initiative in RNA science at the University of Colorado. The NMR instrumentation was purchased with partial support from National Institutes of Health Grants RR11969 and RR16649, National Science Foundation Grants 9602941 and 0230966, and the W. M. Keck Foundation.
Author contributions: J.-H.L., A.P., and F.J. designed research; J.-H.L., M.D.C., A.D.E., D.K., Y.-S.N., and F.J. performed research; A.D.E., D.K., Y.-S.N., D.T.S., and F.J. contributed new reagents/analytic tools; J.-H.L., M.D.C., D.K., Y.-S.N., D.T.S., A.P., and F.J. analyzed data; and J.-H.L., A.P., and F.J. wrote the paper.
Conflict of interest statement: A.D.E., D.K., Y.-S.N., and D.T.S. all are employees of Eyetech Pharmaceuticals.
Abbreviations: AMD, age-related macular degeneration; HBD, heparin-binding domain; HSQC, heteronuclear single quantum correlation; TROSY, transverse relaxation optimized spectroscopy; TF, tissue factor; HUVEC, human umbilical vein endothelial cells.
References
- 1.Ferrara, N., Gerber, H. P. & LeCouter, J. (2003) Nat. Med. 9, 669–676. [DOI] [PubMed] [Google Scholar]
- 2.Tammela, T., Enholm, B., Alitola, K. & Paavonen, K. (2005) Cardiovasc. Res. 65, 550–553. [DOI] [PubMed] [Google Scholar]
- 3.Witmer, A. N., Vrensen, G., Van Noorden, C. J. F. & Schlingemann, R. O. (2003) Prog. Retin. Eye Res. 22, 1–29. [DOI] [PubMed] [Google Scholar]
- 4.Ishida, S., Usui, T., Yamashiro, K., Kaji, Y., Amano, S., Ogura, Y., Hida, T., Oguchi, Y., Ambati, J., Miller, J. W., et al. (2003) J. Exp. Med. 198, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng, E. & Adamis, A. (2005) Can. J. Ophthalmol. 40, 352–368. [DOI] [PubMed] [Google Scholar]
- 6.Stauffer, M. E., Skelton, N. J. & Fairbrother, W. J. (2002) J. Biomol. NMR 23, 57–61. [DOI] [PubMed] [Google Scholar]
- 7.Wiesmann, C., Fuh, G., Christinger, H. W., Eigenbrot, C., Wells, J. A. & deVos, A. M. (1997) Cell 91, 695–704. [DOI] [PubMed] [Google Scholar]
- 8.Ruckman, J., Green, L. S., Beeson, J., Waugh, S., Gillette, W. L., Henninger, D. D., Claesson-Welsh, L. & Janjic, N. (1998) J. Biol. Chem. 273, 20556–20567. [DOI] [PubMed] [Google Scholar]
- 9.Tuerk, C. & Gold, L. (1990) Science 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 10.Nimjee, S., Rusconi, C. & Sullenger, B. (2005) Annu. Rev. Med. 56, 555–583. [DOI] [PubMed] [Google Scholar]
- 11.Bell, C., Lynam, E., Landfair, D. J., Janjic, N. & Wiles, M. (1999) In Vitro Cell Dev. Biol. Anim. 35, 533–542. [DOI] [PubMed] [Google Scholar]
- 12.Gragoudas, E., Adamis, A., Cunningham, E. J., Feinsod, M. & Guyer, D. (2004) N. Engl. J. Med. 351, 2805–2816. [DOI] [PubMed] [Google Scholar]
- 13.Schachat, A. P. (2005) Ophthalmology 112, 531–532. [DOI] [PubMed] [Google Scholar]
- 14.Fine, S., Martin, D. & Kirkpatrick, P. (2005) Nat. Rev. Drug Discov. 4, 187–188. [DOI] [PubMed] [Google Scholar]
- 15.Pervushin, K., Riek, R., Wider, G. & Wüthrich, K. (1997) Proc. Natl. Acad. Sci. USA 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucerna, M., Mechtcheriakova, D., Kadl, A., Schabbauer, G., Schäfer, R., Gruber, F., Koshelnick, Y., Müller, H.-D., Issbrücker, K., Clauss, M., et al. (2003) J. Biol. Chem. 278, 11433–11440. [DOI] [PubMed] [Google Scholar]
- 17.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 18.Goddard, T. D. & Kneller, D. G. (2003) sparky (University of California, San Francisco).
- 19.Farmer, B. T., Constantine, K., Goldfarb, V., Friedrichs, M., Wittekind, M., Yanchunas, J. J., Robertson, J. & Mueller, L. (1996) Nat. Struct. Biol. 3, 995–997. [DOI] [PubMed] [Google Scholar]
- 20.Pan, B. & Fairbrother, W. J. (2002) J. Biomol. NMR 22, 189–190. [DOI] [PubMed] [Google Scholar]
- 21.Gorenstein, D. G. (1984) Phosphorous-31 NMR: Principles and Applications (Academic, Orlando, FL).
- 22.Castagne, C., Murphy, E. C., Gronenborn, A. M. & Delepierre, M. (2000) Eur. J. Biochem. 267, 1223–1229. [DOI] [PubMed] [Google Scholar]
- 23.Jellinek, D., Green, L. S., Bell, C. & Janjic, N. (1994) Biochemistry 33, 10450–10456. [DOI] [PubMed] [Google Scholar]
- 24.Green, L. S., Jellinek, D., Bell, C., Beebe, L. A., Feistner, B. D., Gill, S. C., Jucker, F. M. & Janjic, N. (1995) Chem. Biol. 2, 683–695. [DOI] [PubMed] [Google Scholar]
- 25.Burmeister, P., Lewis, S., Silva, R., Preiss, J., Horwitz, L., Pendergrast, P., McCauley, T., Kurz, J., Epstein, D., Wilson, C. & Keefe, A. (2005) Chem. Biol. 12, 25–33. [DOI] [PubMed] [Google Scholar]
- 26.Gueron, M. & Shulman, R. G. (1975) Proc. Natl. Acad. Sci. USA 72, 3482–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golden, M. C., Collins, B. D., Willis, M. C. & Koch, T. H. (2000) J. Biotechnol. 81, 167–178. [DOI] [PubMed] [Google Scholar]
- 28.Williamson, J. R. (2000) Nat. Struct. Biol. 7, 834–837. [DOI] [PubMed] [Google Scholar]
- 29.Hermann, T. & Patel, D. J. (2000) Science 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 30.Cummins, L., Owens, S., Risen, L., Lesnik, E., Freier, S., McGee, D., Guinosso, C. & Cook, P. (1995) Nucleic Acids Res. 23, 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luque, A., Carpizo, D. R. & Iruela-Arispe, M. L. (2003) J. Biol. Chem. 278, 23656–23665. [DOI] [PubMed] [Google Scholar]
- 32.Heroult, M., Bernard-Pierrot, I., Delbe, J., Hamma-Kourbali, Y., Katsoris, P., Varritault, D., Papadimitriou, E., Plouet, J. & Courty, J. (2004) Oncogene 23, 1745–1753. [DOI] [PubMed] [Google Scholar]
- 33.Inoki, I., Shiomi, T., Hashimoto, G., Enomoto, H., Nakamura, H., Makino, K., Ikeda, E., Takata, S., Kobayashi, K. & Okada, Y. (2002) FASEB J. 16, 219–221. [DOI] [PubMed] [Google Scholar]
- 34.Soker, S., Takashima, S., Miao, H. Q., Neufeld, G. & Klagstrun, M. (1998) Cell 92, 735–745. [DOI] [PubMed] [Google Scholar]
- 35.Whitaker, G. B., Limberg, B. & Rosenbaum, J. S. (2001) J. Biol. Chem. 276, 25520–25531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.