

Abstract
In utero exposure to Δ9-tetrahydrocannabinol (Δ9-THC), the active component from marijuana, induces cognitive deficits enduring into adulthood. Although changes in synaptic structure and plasticity may underlie Δ9-THC-induced cognitive impairments, the neuronal basis of Δ9-THC-related developmental deficits remains unknown. Using a Boyden chamber assay, we show that agonist stimulation of the CB1 cannabinoid receptor (CB1R) on cholecystokinin-expressing interneurons induces chemotaxis that is additive with brain-derived neurotrophic factor (BDNF)-induced interneuron migration. We find that Src kinase-dependent TrkB receptor transactivation mediates endocannabinoid (eCB)-induced chemotaxis in the absence of BDNF. Simultaneously, eCBs suppress the BDNF-dependent morphogenesis of interneurons, and this suppression is abolished by Src kinase inhibition in vitro. Because sustained prenatal Δ9-THC stimulation of CB1Rs selectively increases the density of cholecystokinin-expressing interneurons in the hippocampus in vivo, we conclude that prenatal CB1R activity governs proper interneuron placement and integration during corticogenesis. Moreover, eCBs use TrkB receptor-dependent signaling pathways to regulate subtype-selective interneuron migration and specification.
Keywords: corticogenesis, drug abuse, neurotrophin
Endogenous cannabinoids, such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG), modulate synaptic plasticity by the retrograde control of neurotransmitter release (1). Accordingly, a compartmentalization of endocannabinoid (eCB) synthesis and action has been demonstrated in adult brain (1–3). eCBs are predominantly synthesized in dendritic compartments and signal through presynaptic Gi/o protein-coupled CB1 cannabinoid receptors (CB1Rs) (4–6). The adult phenotype of cannabinoid systems is achieved through a series of developmentally regulated events culminating in high CB1R expression on cholecystokinin (CCK)-containing GABAergic interneurons (CB1R+ cells) in the hippocampus and neocortex (3, 5, 6).
Recent studies have demonstrated the existence of functional CB1Rs in developing cortical neurons (7). A coincidence of eCB synthesis and release and CB1R activation within axonal growth cones was postulated to provide an eCB-driven reinforcement loop that regulates axonal growth and guidance (8). Although the functional significance of CB1Rs during assembly of cortical neuronal circuitries is unknown, their developmental impact is illustrated by long-lasting cognitive, motor, and social disturbances in offspring exposed prenatally to cannabis (9, 10).
The majority of cortical interneurons are derived from extracortical precursor pools and undergo long-distance migration before inhabiting specific cortical layers (11, 12). Interneuron specification is in part governed by epigenetic cues within the neocortex, including brain-derived neurotrophic factor (BDNF) (12–14). Considering that pyramidal cells in the juvenile neocortex harbor the capacity of eCB synthesis and release (15), we hypothesized that eCBs can act as target-derived factors regulating interneuron migration and morphogenesis during embryonic development.
By sorting CB1R+ interneurons, we show that AEA acts as chemoattractant and regulates interneuron migration by TrkB receptor transactivation. However, prolonged AEA exposure antagonizes the BDNF-induced differentiation of cortical interneurons. We also demonstrate that neuronal differentiation is associated with the simultaneous recruitment of CB1Rs and TrkB receptors to axon terminal segments of developing CB1R+ interneurons. Coincident targeting of these receptors establishes a spatially segregated signaling platform where eCBs induce the assembly of CB1R/TrkB complexes. These findings provide evidence on the physiological role of eCBs during cortical development. Increased density of hippocampal CCK+ interneurons after prenatal Δ9-tetrahydrocannabinol (Δ9-THC) treatment reveals that overactivation of CB1Rs on developing neurons can affect their postnatal positioning and suggests that interfering with eCB signaling during embryogenesis can prevent the proper patterning of cortical neuronal networks.
Materials and Methods
Sorting of Interneurons. Immunomagnetic cell isolation with rabbit antibodies recognizing the N terminus of the rat CB1R (4) and the Kv3.1b subunit was performed on embryonic days (E)19/20 as previously described in ref. 14. Interneurons sorted from fetal rat cortices were maintained in DMEM/F12 containing 2% B27 or used in Boyden chamber assays. Isolated interneurons were exposed to 50–100 ng/ml BDNF and 25–100 nM AEA (Tocris Cookson, Bristol, U.K.) with or without 0.1 μM Src kinase inhibitor PP2, 25 μM phosphatidylinositol 3-kinase inhibitor LY 294002, 10 μM phospholipase C-γ inhibitor ET-18-OCH3, and 10 μM mitogen-activated protein kinase kinase inhibitor PD 98059 (all inhibitors from Cell Signaling Technology, Beverly, MA).
PCR Analysis. RT-PCRs were performed as described in ref. 14 with the primers described in refs. 14 and 16. Real-time quantitative PCR was carried out according to Castelo-Branco et al. (17). Total RNA not incubated with reverse transcriptase was used as negative control. BDNF primers were 5′-TTTCTGCTGGAGGAATACAAAAA-3′ (forward) and 5′-ATGGGATTACACTTGGTCTCGTA-3′ (reverse, 270 bp). Expression levels were normalized to ribosomal 18S rRNA housekeeping standard obtained for every sample in parallel assays in two independent experiments.
Electrophysiology. Electrical signals in whole-cell configuration were recorded as described in ref. 14. CB1R+ neurons on coverslips were washed in DMEM before measurements. Input resistance plots were constructed after incrementing somatic current injections. The GABAergic nature of spontaneous inhibitory postsynaptic currents (sIPSCs) was determined by stepwise depolarization of the recorded cell (-70 to +20 mV) with 20 mM Cl- in the pipette solution. The GABAA receptor antagonist(+)-bicuculline (Tocris Cookson) was applied at 1 μM in the extracellular solution for 5 min, and sIPSC recordings were performed at -70 mV holding potential.
Immunocytochemistry and Morphometry. Immunolabeling of free-floating sections of adult Sprague–Dawley rats and cell cultures was performed as described in ref. 14 with the following primary antibodies: rabbit anti-CB1R (C-terminal, 1:500), goat anti-CB1R (C-terminal, 1:500), rabbit anti-fatty-acid amide hydrolase (1:1,000), mouse anti-nonphosphorylated neurofilament (1:1,000; SMI-32, Affinity Research Products, Exeter, U.K.), mouse anti-GAD65/67 (GAD, glutamic acid decarboxylase; 1:1,000; Stressgen Biotechnologies, Victoria, Canada), mouse anti-β-III-tubulin (1:500, Promega), rabbit anti-vesicular GABA transporter (1:500; Synaptic Systems, Goettingen, Germany), and mouse anti-TrkB receptor (1:500, BD Biosciences). Immunoreactivities were visualized with carbocyanine 2- and 3-conjugated secondary antibodies (1:200 in 2% BSA; Jackson ImmunoResearch). F-actin was revealed by Alexa Fluor 546-tagged phalloidin (Molecular Probes). After staining with 0.5% 1,1′-dioctadecyl-3,3,3′,3′-tetra-methylindocarbocyanine perchlorate (Molecular Probes) for 20 min, images were captured with a laser-scanning microscope (LSM510, Zeiss). Neurite growth was studied at 6 days in vitro (DIV) with neuronj.
Chemotaxis Assay. BDNF and AEA (×106 diluted stock in ethanol) were added to the lower wells of Boyden chambers, whereas 0.1 μM K252a (Calbiochem), 200 nM N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251, Tocris Cookson), and 0.1 μM PP2 were applied in the upper chambers of a 96-well microplate (ChemoTx System, NeuroProbe, Gaithersburg, MD). CB1R+ interneurons (5,000 cells per well) were added to the upper chamber and allowed to migrate through polycarbonate filters (8-μm pore size) for 16 h at 37°C. Cells that accumulated in the bottom chambers were fixed, stained with cresyl-violet, and counted on the entire floor surface.
Cell Transfection. nnr5 PC12-TrkB cells stably transfected with a TrkB cDNA (18) were transiently cotransfected with a pcDNA3 plasmid containing the full-length rat CB1R sequence fused with the hemagglutinin epitope and a 5′ preprolactin signal sequence. Forty-eight hours later, the cells were serum-starved overnight. Stimulation with BDNF, AEA, and 2-AG (both from ×106 diluted stock in ethanol) was performed for the indicated periods. AM251 was applied for 60 min before exposure to AEA or 2-AG.
Immunoprecipitation and Western Blotting. Protein samples were analyzed as described in ref. 14. Western blotting of BDNF from culture media was performed by using rabbit anti-human BDNF antibody (1:1,000, Promega) according to established protocols (19). Immunoprecipitation was performed on interneurons at 3 DIV (20). Primary antibodies were mouse anti-TrkB receptor (1:1,000, BD Biosciences), rabbit anti-CB1R (C-terminal 15 amino acid residues, 1:1,000), mouse anti-hemagglutinin (1:1,000; Covance, Richmond, CA), mouse anti-phosphotyrosine (1:1,000; PY-99, Santa Cruz Biotechnology), phospho-Src kinase (1:1,000; Tyr-416, Cell Signaling Technology), and rabbit anti-Src kinases (1:2,000; c-Src, Santa Cruz Biotechnology). Blots were scanned in a Storm 840 fluorimager and quantified with imagej 1.32j. Changes in phosphorylation states were expressed as folds of the untreated control value.
Δ9-THC Administration and in Situ Hybridization. Long–Evans female rats (Charles River Breeding Laboratories) were implanted with a permanent catheter secured to the right jugular vein and administered a daily i.v. injection of Δ9-THC (0.15 mg/kg) or vehicle from gestational day 5 to postnatal day 2. One hour after the last treatment, pups were anaesthetized and their brains were dissected. Sense and antisense probes were synthesized by in vitro transcription of pGEM-T vectors (Promega) carrying inserts of the rat CCK (position 314–606) and preproenkephalin (position 388–435) cDNAs in the presence of [35S]uridine 5′-[α-thio]triphosphate (NEN). In situ hybridization was performed as described in ref. 21. Cell counts and densitometry were performed on the entire hippocampal surface in serial sections (at coordinates -2.80 to -4.3 mm relative to bregma). After determining the surface area, cell numbers and hybridization signal density were expressed as millimeters squared and disintegrations per minute per milligram of tissue, respectively.
Statistics. Data in pharmacological experiments were evaluated by using ANOVA with Bonferroni's post hoc test. Group comparisons were performed with Student's t test. A P value of <0.05 was considered as statistically significant. Data were expressed as means ± SEM.
Results
CB1Rs Identify a Subset of GABAergic Interneurons. High CB1R expression on CCK+ interneurons throughout development and adulthood (Fig. 1A) (3–6) prompted us to postulate that stimulation of CB1Rs on partially specified interneurons by eCBs is instrumental for their proper positioning and morphogenesis. To determine whether eCBs affect interneuron migration and differentiation, we generated subset-specific interneuron cultures from E19/20 rat cortices by immunomagnetic sorting for the CB1R (13, 16). Isolated neurons (72,000 ± 16,000 cells per embryo) exhibited bipolar or multipolar somatodendritic morphologies (22) and were immunoreactive for the GABA-synthesizing enzyme GAD (>96%), vesicular GABA transporter, β-III-tubulin, fatty-acid amide hydrolase, and CB1Rs (Fig. 1 B–F′; see also Supporting Materials and Methods and Table 1, which are published as supporting information on the PNAS web site). CB1Rs were localized to axon varicosities (Fig. 1 E and F) and growth cones (Fig. 1F′) in immature interneurons and were predominant on axon terminals of mature cells (data not shown). RT-PCR confirmed the expression of CB1Rs, CCK, somatostatin, and neuropeptide Y mRNA transcripts by isolated neurons (Fig. 1G), corroborating the neurochemical identity of developing CB1R+ interneurons (3) (Fig. 6C, which is published as supporting information on the PNAS web site).
Fig. 1.

AEA induces the migration of CB1R-expressing interneurons. (A) In the adult neocortex, CB1Rs are localized to axon terminals of GABAergic basket cells providing perisomatic innervation (arrows) to pyramidal cells (Pyr). (B) Isolated CB1R+ interneurons exhibit multipolar morphology. (C–F′)CB1Rs and fatty-acid amide hydrolase (FAAH) accumulate in axon varicosities and growth cones. (Scale bars: A, 15 μm; B and D, 30 μm; C, 7 μm; E, F, and F′, 3 μm.) (G) Isolated cells express interneuron markers, including GAD65, CCK, somatostatin (SOM), neuropeptide Y (NPY), calretinin (CR), and CB1R but not CB2R mRNA transcripts. (H) Isolated neurons show regular (Upper) or irregular (Lower) discharge patterns at 8 DIV. (I) sIPSCs reveal a reversal potential of approximately -40 mV, characteristic of GABA (n = 12), and slow time courses τ = 63.3 ± 16.5 ms at 6 DIV. (J) BDNF (F(3,28) = 3.471; P = 0.035) and AEA (F(3,28) = 4.948; P = 0.010) induced interneurons migration and AM251 blocked the AEA-induced chemotaxis. BDNF or AEA presented in the upper and lower chambers did not induce cell migration. (K) BDNF (F(5,69) = 50.835; P < 0.001) but not AEA (P > 0.4) induced the migration of PV+ interneurons, which did not express CB1Rs as shown by Western blotting (Inset). Results of two to five independent experiments were expressed as percentages of the basal migration rate in the presence of vehicle. ○, P < 0.05 vs. control; *, P < 0.05 vs. 100 ng/ml BDNF, +, P < 0.05 vs. 100 nM AEA. CB, calbindin D28K; F-A, F-actin; tubulin, β-III-tubulin, VIP, vasoactive intestinal polypeptide.
CCK+ interneurons exhibit a predominant regular-spiking phenotype in vivo (5, 22). Whole-cell recordings of cultured CB1R+ cells revealed irregular or regular discharge patterns (Fig. 1H)with frequencies ranging from 22 to 54 Hz. Synapse formation paralleled the development of neuronal excitability (Fig. 6 A and B). The reversal potential of sIPSCs was equivalent to that calculated for GABA (-48 mV, n = 12) (Fig. 1I) and verified by the GABAA receptor antagonist(+)-bicuculline (100 nM)-induced block of sIP-SCs (n = 5) (data not shown). Collectively, our data show that selectively sorted CB1R+ interneurons acquire morphological and electrophysiological properties of CCK+ GABAergic interneurons.
eCBs Are Chemoattractants. The layer-specific patterning of the developing cortex is achieved by the active migration of pyramidal cells and interneurons (12). Hence, we asked whether eCBs can induce interneuron migration. The attractive properties of AEA were analyzed in a Boyden chamber assay. We observed maximal ligand-directed migration toward a 100 nM AEA concentration (Fig. 1J) equivalent to its calculated EC50 value in microglia migration assays (16). Treatment with the specific CB1R antagonist AM251 blocked AEA-induced chemotaxis, confirming the involvement of CB1R activation. CB1R dependency of this AEA-induced migratory response was substantiated by showing that parvalbumin (PV)-expressing interneurons lacking CB1Rs (14) do not migrate toward AEA gradients (Fig. 1K).
Next, we tested whether BDNF, whose receptor, TrkB, is expressed on interneurons during cortical positioning (12, 13), induces interneuron migration (23). BDNF significantly increased the migration of CB1R+ and PV-expressing interneurons at 50–100 ng/ml concentrations (Fig. 1 J and K). Notably, AEA modulated the BDNF-induced migration of CB1R+ interneurons in an additive fashion (Fig. 1J). Neither AEA nor BDNF induced chemokinesis upon disrupting their gradients by adding these compounds to both wells of Boyden chambers (Fig. 1J). Our data thus demonstrate that eCBs act as chemoattractants and cooperate with BDNF signaling to control interneuron migration.
eCBs Inhibit Interneuron Differentiation. BDNF controls interneuron differentiation by triggering somatic growth, dendrite development, and synapse establishment (13, 14). In contrast, eCBs were shown to inhibit synapse formation (24). Hence, we studied whether eCBs affect interneuron development and interact with neurotrophin signaling by culturing CB1R+ interneurons in the presence of AEA, BDNF, or both for 6 DIV. BDNF induced significant neurite branching and elongation (Fig. 2 A and C). In contrast, AEA inhibited neurite extension and also attenuated the BDNF-induced growth response when applied simultaneously (Fig. 2 A, A′, and C). The AEA-induced inhibition of neurite growth depended on CB1R activation because AM251 significantly attenuated these effects. Reduced hyperpolarization of interneuron membranes after AEA and combined treatments lent further support to the AEA-induced inhibition of interneuron differentiation (Fig. 2B). Because AEA alone or in combination with BDNF did not affect synapse differentiation (Fig. 7, which is published as supporting information on the PNAS web site), we conclude that eCBs act as potent inhibitors of dendrite arborization in developing interneurons.
Fig. 2.

AEA and BDNF coregulate neurite development of CB1R+ interneurons. (A and A′) Examples of cellular morphologies. (Scale bar: 40 μm.) (B) Resting membrane potentials at 6 DIV (n = 7 cells per condition). (C) BDNF (100 ng/ml) induces neurite extension and branching. In contrast, AEA (100 nM) inhibits neurite formation (n > 20 cells per condition) and suppresses the BDNF-induced neurite growth. AM251 antagonizes the effects of AEA. Significant BDNF–AEA interaction was noted in total length: F(3,47) = 6.696, P = 0.011. All treatments lasted for 6 DIV. Experiments were performed in triplicate. ○, P < 0.05; ○○, P < 0.01 vs. control; *, P < 0.05; **, P < 0.01 vs. 100 ng/ml BDNF; +, P < 0.05 vs. 100 nM AEA; #, P < 0.05 vs. combination.
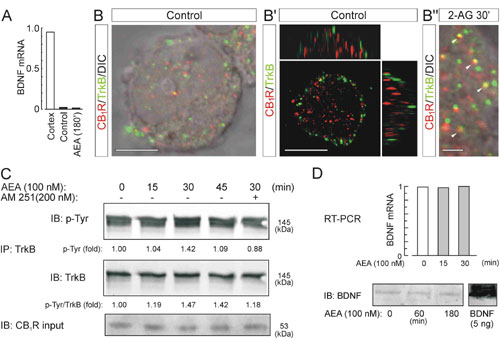
eCBs Activate TrkB Receptors. Identification of integrated signaling responses by the formation of Trk/G protein-coupled receptor signaling complexes (25), and their affinity for G protein-coupled receptor-mediated receptor transactivation (26, 27) suggest that CB1Rs could regulate TrkB activity. Therefore, we assessed the spatial distribution of CB1Rs and TrkB receptors in interneurons. CB1Rs and TrkB receptors colocalized on axon terminal segments of developing interneurons, providing a platform for signaling interactions (Fig. 3A). Treatment of CB1R+ interneurons with AEA induced a transient increase (at 15–30 min) in the phosphorylation of membrane-targeted, fully glycosylated TrkB receptors (Fig. 3B). In contrast, BDNF induced direct TrkB phosphorylation as early as 5 min.
Fig. 3.

AEA activates TrkB receptors and induces the formation of CB1R/TrkB heterocomplexes. (A) TrkB and CB1Rs are codistributed along varicose axon segments of CB1R+ interneurons (arrows). a, axon segment; V, varicosity. (Scale bar: 10 μm.) (B) Time course of AEA activation of TrkB receptors on interneurons. BDNF was used as positive control. After immunoprecipitation, TrkB receptors phosphorylation was detected by immunoblotting (IB) with PY99 anti-phosphotyrosine (p-Tyr) antibodies. (C) AEA induces the formation of CB1R/TrkB heterocomplexes, as revealed by coimmunoprecipitation (IP) of the two receptors. Receptor complex formation was confirmed by detection of the hemagglutinin tag of transfected CB1Rs. (D) 2-AG also induces TrkB receptor transactivation at 30 min, which is blocked by AM251. (E) At 30 min, AEA activates Src family kinases and is blocked by PP2. PP2 also abolishes tyrosine phosphorylation of the TrkB receptor. Data were expressed as folds of the corresponding unstimulated samples. Each panel is an example from two to seven independent experiments.
To overcome the sparseness of CB1R+ interneurons, we transiently transfected nnr5 PC12-TrkB cells (18) with the full-length rat CB1R (PC12TrkB/CB1R cells). CB1R and TrkB receptors were found on the plasma and intracellular membranes of PC12TrkB/CB1R cells, and their close proximity suggested their simultaneous targeting to specific membrane compartments (Fig. 8B, which is published as supporting information on the PNAS web site). The time course of AEA-induced TrkB receptor activation in PC12TrkB/CB1R cells was comparable with that observed in primary cells, with its maximum after 30 min of stimulation (Fig. 8C). Application of the CB1R antagonist AM251 eliminated TrkB activation, confirming that this mechanism was mediated by CB1Rs. The spatial association of the two receptor types upon CB1R activation together with the eCB-induced transactivation of Trk receptors presents a possibility for heterocomplex formation by CB1R and TrkB receptors.
Real-time quantitative PCR and Western analyses on both cortical cultures and PC12TrkB/CB1R cells demonstrated that AEA-induced TrkB transactivation was not associated with increased BDNF expression or release from preexisting intracellular pools (Fig. 8 A and D). Therefore, it is unlikely that transactivation of TrkB receptors involves BDNF.
Assembly of CB1R/TrkB Complexes. The formation of CB1R/TrkB complexes was tested after AEA stimulation of PC12TrkB/CB1R cells by immunoprecipitation of TrkB receptors. Immunoblot detection revealed CB1Rs in immunoprecipitated fractions after AEA exposure that was indicative of the assembly of CB1R/TrkB receptor complexes (Fig. 3C). TrkB transactivation and CB1R/TrkB receptor complex formation were also induced by 2-AG and inhibited by AM251 (Fig. 3D).
Previous studies have highlighted a role for Src family kinases in TrkA and epidermal growth factor receptor transactivation (27–29). Here, AEA-activated Src family kinases, which was reversed by the selective Src family kinase inhibitor PP2 (Fig. 3E). Significantly, PP2-induced inhibition of Src family kinases abolished the AEA-induced activation of TrkB receptors (Fig. 3E). Thus, our data suggest that CB1R-mediated TrkB receptor association and transactivation is a mechanism triggered by multiple eCBs, and the regulation of TrkB receptor activity is mediated through Src tyrosine kinases.
Physiological Significance of CB1R/TrkB Signaling Interactions. We addressed whether eCB-induced TrkB receptor transactivation is a requirement for interneuron migration by inhibiting the Trk family of receptor tyrosine kinases. K252a (30), applied in the upper wells of the Boyden chamber assay, effectively blocked AEA-induced interneuron migration, supporting our concept that the CB1Rs uses the TrkB receptor signaling system to couple to a migratory response (Fig. 4A).
Fig. 4.

The significance of CB1R/TrkB signaling interactions during interneuron development. (A) Interneuron migration induced by 100 nM AEA is abolished by 100 nM K252a. ○, P < 0.05 vs. control; **, P < 0.01 vs. AEA. (B) Morphogenic effects of AEA (100 nM) in combination with site-specific inhibitors of TrkB receptor signaling at 6 DIV. F(5,69) = 9.124, P < 0.0001 (branching); F(5,69) = 5.190, P < 0.0001 (total length); F(5,69) = 4.359, P = 0.002 (average length). *, P < 0.05. **, P < 0.01 vs. AEA; ○, P < 0.05; ○○, P < 0.01 vs. LY294002; #, P < 0.05 vs. ET-18-OCH3.(C and C′) Sequential treatment with 100 ng/ml BDNF (0–3 DIV) and 100 nM AEA plus 0.1 μM PP2 (4–6 DIV) mimicked the BDNF-induced growth response over 6 DIV. Horizontal dashed lines indicate the effects of 100 ng/ml BDNF continuously applied for 6 DIV. F(2,31) = 5.023, P = 0.013 (dendrite branching). ○, P < 0.05 vs. control; *, P < 0.05 vs. BDNF. (Scale bar: 10 μm.)
Next, we tested whether modulation of TrkB signaling affects the AEA-induced inhibition of interneuron differentiation by exposure to AEA in combination with particular inhibitors for 6 DIV (Fig. 4B). Blockade of Src family kinases and mitogen-activated protein kinase by PP2 and PD98059, respectively, significantly enhanced neurite extension. In contrast, inhibition of phosphatidylinositol 3-kinase by LY 294002 was detrimental for neurite growth. Antagonism of phospholipase C-γ activity (ET-18-OCH3) did not affect neurite development. To address whether prolonged inhibition of CB1R/TrkB receptor signaling interactions by abating Src kinase activity could revert the AEA-induced suppression of neurite growth, we exposed BDNF-primed interneurons to AEA with or without PP2. Sequential BDNF and AEA/PP2 treatments triggered neurite formation, mimicking the effects of continuous BDNF treatment (Fig. 4 C and C′). Collectively, our data show that eCBs suppress neurite development of cortical interneurons by preferentially affecting Src family kinase-mediated signaling pathways.
Δ9-THC Increases Interneuron Density In the Hippocampus. To provide a proof of concept for the putative role of CB1R signaling during CNS development, we tested whether prenatal Δ9-THC treatment affects the packing density of CCK-expressing interneurons in the early postnatal hippocampus. We detected CCK expression by in situ hybridization after Δ9-THC treatment that mimicked the effects and pharmacokinetic profile of marijuana smoking in humans (31). Δ9-THC significantly increased the density of CCK+ cells and putative interneurons in the strata radiatum, lacunosum-moleculare of the CA1–CA3 subfields (Fig. 5A and B), and the hilar region. Unchanged preproenkephalin mRNA transcript levels, known to reveal CB1R-negative interneurons (3), suggest that Δ9-THC effects predominate on hippocampal interneuron subsets that constitutively express CB1Rs. We concluded that prenatal Δ9-THC exposure can hamper interneuron positioning during corticogenesis.
Fig. 5.

Prenatal Δ9-THC treatment increases the packing density of CCK-expressing interneurons in the postnatal hippocampus. (A) CCK in situ hybridization reveals increased densities of putative interneurons (arrows) across the CA3 subfield. Dashed square indicates magnification location in B. (Scale bar: 250 μm.) g, stratum granulosum; pyr, stratum pyramidale; rad, stratum radiatum. (B) Cresyl-violet counterstaining verified that the in situ hybridization signal was derived from single neurons (asterisk). (Scale bar: 5 μm.) (C) Δ9-THC treatment increases the density of CCK-expressing interneurons without affecting preproenkephalin expression, a marker of vasoactive intestinal polypeptide-containing GABAergic interneurons (Fig. 6C), in the dorsal hippocampus [n = 8 (control), n = 11 (Δ9-THC)].
Discussion
Cortical interneurons are derived from nonoverlapping segments of the ganglionic eminences (11, 12), migrate tangentially, and populate cortical laminae in an inside-out pattern (12). Although an array of attractant and repellent signals, including neuroregulins, Slit1/2, and semaphorins, direct long-distance interneuron migration (12, 32), little is known about intrinsic microenvironmental factors that determine the proper placement and phenotypic differentiation of particular interneuron subtypes.
BDNF, selectively expressed and secreted by pyramidal cells, is a differentiation signal for GABAergic interneurons (12–14). However, it is unlikely that BNDF is the sole determinant of interneuron specification. Given that (i) eCBs are synthesized throughout development (33), (ii) CB1Rs are localized on neuronal growth cones (8), (iii) CB1R expression in the neocortex undergoes a striking restriction during the early postnatal period (3), and (iv) prenatal exposure to marijuana hampers cognitive performance (9, 10), we reasoned that modulation of CB1R signaling could determine interneuron placement and morphology and the assembly and plasticity of neuronal networks.
To define developmental processes regulated by CB1R signaling upon stimulation with exogenous or endogenous agonists, we sorted CB1R+ interneurons from embryonic cortices. We identified eCBs as chemoattractants for CB1R+ but not PV+ interneurons. Significantly, K252a, a selective inhibitor of Trk tyrosine kinases receptors (30), abolished the AEA-induced migratory response. These observations establish a signaling interaction between CB1Rs and TrkB receptors and show that eCBs use the TrkB receptor signaling system to evoke interneuron migration. Considering that BDNF-induced TrkB activation exhibits a rapid time-course and AEA-induced TrkB phosphorylation occurs later, similar to other reported G protein-coupled receptor agonists (26), we concluded that sustained activation of TrkB receptors via temporally distinct signaling mechanisms provides a basis for the additive effects of BDNF and AEA when inducing the chemotaxis of interneurons.
The interaction of CB1R and TrkB signaling appear to be localized to defined membrane compartments (34). This hypothesis is supported by the assembly of CB1R/TrkB receptor complexes. In addition, activation of Src family kinases was required for TrkB receptor activation upon agonist stimulation of CB1Rs. These findings provide a cellular platform for the eCB-induced activation event of TrkB receptors.
Next, we addressed whether eCBs affect the morphological specification of CB1R+ interneurons. AEA potently inhibited neurite extension by a CB1R-mediated mechanism, because AM251 blocked the AEA-induced growth arrest. In contrast, BDNF increased neurite growth and branching (12–14, 35). Notably, AEA eliminated the BDNF-induced differentiation response. To understand signaling mechanisms conferring the effects of AEA on neurotrophin-induced neurite development, we used selective inhibitors of kinases (Src family, phosphatidylinositol 3-kinase, mitogen-activated protein kinase kinase, and phospholipase C-γ) known to mediate developmental effects of TrkB receptor activation. Inhibition of Src and mitogen-activated protein kinase kinases induced significant neurite elongation and branching in the presence of AEA. In contrast, phosphatidylinositol 3-kinase inhibition potentiated the AEA-induced diminution of neurite growth. Phospholipase C-γ kinases did not appear to be involved in the AEA-induced inhibition of neurite development. The opposite effects of AEA and BDNF on neurite extension suggest that temporally and spatially distinct signaling events mediated by CB1Rs and TrkB receptors (36) with a central role for Src kinase-induced downstream signaling (37), rather than receptor transactivation, may provide the underlying mechanism.
Our results expose a role for eCBs during CNS development. Specifically, eCBs provide a mechanism for the cellular control of interneuron migration and differentiation by regulating neurotrophin actions. We propose that the placement of interneurons migrating in the neocortex is determined by the temporal availability of target-derived eCBs and neurotrophins. Once proper positioning is acquired, the extension of the dendritic tree is directed by neurotrophins under the regulatory drive of eCBs. Notably, these pathways appear to be significant to the development of cortical neuronal networks, for which a balance of eCB and neurotrophin signaling seems essential for the establishment of inhibitory components. However, interfering with the delicate balance of eCB signaling even with low concentrations of Δ9-THC during embryogenesis triggered aberrant patterning of CCK/CB1R-expressing interneurons in the early postnatal hippocampus. The notion that Δ9-THC significantly affects this principal subset of perisomatic inhibitory basket cells (38) suggests that an altered inhibitory drive on pyramidal cells triggers discordant excitatory output from hippocampal neuronal circuitries (2, 10).
Supplementary Material
Acknowledgments
nnr5 PC12-TrkB cells were provided by Dr. L. Greene (Columbia University, New York). This work was supported by the Swedish Medical Research Council (P.E., G.P., and T.H.), Hedlunds Stiftelse (P.E.), Hjärnfonden (P.E.), Åke Wiberg Stiftelse (T.H.), Svenska Sällskapet för Medicinsk Forskning (G.S.), and National Institute of Health Grants DA11322, DA00286 (to K.M.), and DA12030 (to Y.L.H.).
Author contributions: G.P., Y.L.H., and T.H. designed research; P.B., M.B.D., X.W., S.S., F.L., K.M.S., G.S., and G.P. performed research; P.E. and K.M. contributed new reagents/analytic tools; P.B., M.B.D., G.P., and T.H. analyzed data; and P.B., Y.L.H., and T.H. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: 2-AG, 2-arachidonoylglycerol; AEA, anandamide; AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; BDNF, brain-derived neurotrophic factor; CB1R, CB1 cannabinoid receptor; CCK, cholecystokinin; DIV, day in vitro; En, embryonic day n; eCB, endocannabinoid; GAD, glutamic acid decarboxylase; sIPSC, spontaneous inhibitory postsynaptic current; Δ9-THC, Δ9-tetrahydrocannabinol; PV, parvalbumin.
References
- 1.Wilson, R. I. & Nicoll, R. A. (2002) Science 296, 678-682. [DOI] [PubMed] [Google Scholar]
- 2.Bernard, C., Milh, M., Morozov, Y. M., Ben Ari, Y., Freund, T. F. & Gozlan, H. (2005) Proc. Natl. Acad. Sci. USA 102, 9388-9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morozov, Y. M. & Freund, T. F. (2003) Eur. J. Neurosci. 18, 1213-1222. [DOI] [PubMed] [Google Scholar]
- 4.Katona, I., Sperlagh, B., Sik, A., Kafalvi, A., Vizi, E. S., Mackie, K. & Freund, T. F. (1999) J. Neurosci. 19, 4544-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freund, T. F., Katona, I. & Piomelli, D. (2003) Physiol. Rev. 83, 1017-1066. [DOI] [PubMed] [Google Scholar]
- 6.Marsicano, G. & Lutz, B. (1999) Eur. J. Neurosci. 11, 4213-4225. [DOI] [PubMed] [Google Scholar]
- 7.Mato, S., Del Olmo, E. & Pazos, A. (2003) Eur. J. Neurosci. 17, 1747-1754. [DOI] [PubMed] [Google Scholar]
- 8.Williams, E. J., Walsh, F. S. & Doherty, P. (2003) J. Cell Biol. 160, 481-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fried, P. A., Watkinson, B. & Gray, R. (2003) Neurotoxicol. Teratol. 25, 427-436. [DOI] [PubMed] [Google Scholar]
- 10.Mereu, G., Fa, M., Ferraro, L., Cagiano, R., Antonelli, T., Tattoli, M., Ghiglieri, V., Tanganelli, S., Gessa, G. L. & Cuomo, V. (2003) Proc. Natl. Acad. Sci. USA 100, 4915-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson, S. A., Eisenstat, D. D., Shi, L. & Rubenstein, J. L. (1997) Science 278, 474-476. [DOI] [PubMed] [Google Scholar]
- 12.Xu, Q., de la Cruz, E. & Anderson, S. A. (2003) Cereb. Cortex 13, 670-676. [DOI] [PubMed] [Google Scholar]
- 13.Marty, S., Berninger, B., Carroll, P. & Thoenen, H. (1996) Neuron 16, 565-570. [DOI] [PubMed] [Google Scholar]
- 14.Berghuis, P., Dobszay, M. B., Sousa, K. M., Schulte, G., Mager, P. P., Hartig, W., Gorcs, T. J., Zilberter, Y., Ernfors, P. & Harkany, T. (2004) Eur. J. Neurosci. 20, 1290-1306. [DOI] [PubMed] [Google Scholar]
- 15.Trettel, J., Fortin, D. A. & Levine, E. S. (2004) J. Physiol. 556, 95-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter, L., Franklin, A., Witting, A., Wade, C., Xie, Y., Kunos, G., Mackie, K. & Stella, N. (2003) J. Neurosci. 23, 1398-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castelo-Branco, G., Wagner, J., Rodriguez, F. J., Kele, J., Sousa, K., Rawal, N., Pasolli, H. A., Fuchs, E., Kitajewski, J. & Arenas, E. (2003) Proc. Natl. Acad. Sci. USA 100, 12747-12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ilag, L. L., Lonnerberg, P., Persson, H. & Ibanez, C. F. (1994) J. Biol. Chem. 269, 19941-19946. [PubMed] [Google Scholar]
- 19.Poulsen, F. R., Jahnsen, H., Blaabjerg, M. & Zimmer, J. (2002) Brain Res. 950, 103-118. [DOI] [PubMed] [Google Scholar]
- 20.Paratcha, G., Ledda, F. & Ibanez, C. F. (2003) Cell 113, 867-879. [DOI] [PubMed] [Google Scholar]
- 21.Wang, X., Dow-Edwards, D., Keller, E. & Hurd, Y. L. (2003) Neuroscience 118, 681-694. [DOI] [PubMed] [Google Scholar]
- 22.Galarreta, M., Erdelyi, F., Szabo, G. & Hestrin, S. (2004) J. Neurosci. 24, 9770-9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behar, T. N., Dugich-Djordjevic, M. M., Li, Y. X., Ma, W., Somogyi, R., Wen, X., Brown, E., Scott, C., McKay, R. D. & Barker, J. L. (1997) Eur. J. Neurosci. 9, 2561-2570. [DOI] [PubMed] [Google Scholar]
- 24.Kim, D. & Thayer, S. A. (2001) J. Neurosci. 21, RC146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pyne, N. J., Waters, C., Moughal, N. A., Sambi, B. S. & Pyne, S. (2003) Biochem. Soc. Trans. 31, 1220-1225. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopal, R., Chen, Z. Y., Lee, F. S. & Chao, M. V. (2004) J. Neurosci. 24, 6650-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daub, H., Weiss, F. U., Wallasch, C. & Ullrich, A. (1996) Nature 379, 557-560. [DOI] [PubMed] [Google Scholar]
- 28.Hart, S., Fischer, O. M. & Ullrich, A. (2004) Cancer Res. 64, 1943-1950. [DOI] [PubMed] [Google Scholar]
- 29.Lee, F. S., Rajagopal, R., Kim, A. H., Chang, P. C. & Chao, M. V. (2002) J. Biol. Chem. 277, 9096-9102. [DOI] [PubMed] [Google Scholar]
- 30.Berg, M. M., Sternberg, D. W., Parada, L. F. & Chao, M. V. (1992) J. Biol. Chem. 267, 13-16. [PubMed] [Google Scholar]
- 31.Grotenhermen, F. (2003) Clin. Pharmacokinet. 42, 327-360. [DOI] [PubMed] [Google Scholar]
- 32.Marin, O., Plump, A. S., Flames, N., Sanchez-Camacho, C., Tessier-Lavigne, M. & Rubenstein, J. L. (2003) Development (Cambridge, U. K.) 130, 1889-1901. [DOI] [PubMed] [Google Scholar]
- 33.Bisogno, T., Howell, F., Williams, G., Minassi, A., Cascio, M. G., Ligresti, A., Matias, I., Schiano-Moriello, A., Paul, P., Williams, E. J., et al. (2003) J. Cell Biol. 163, 463-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bari, M., Battista, N., Fezza, F., Finazzi-Agro, A. & Maccarrone, M. (2005) J. Biol. Chem. 280, 12212-12220. [DOI] [PubMed] [Google Scholar]
- 35.Patapoutian, A. & Reichardt, L. F. (2001) Curr. Opin. Neurobiol. 11, 272-280. [DOI] [PubMed] [Google Scholar]
- 36.Rueda, D., Navarro, B., Martinez-Serrano, A., Guzman, M. & Galve-Roperh, I. (2002) J. Biol. Chem. 277, 46645-46650. [DOI] [PubMed] [Google Scholar]
- 37.He, J. C., Gomes, I., Nguyen, T., Jayaram, G., Ram, P. T., Devi, L. A. & Iyengar, R. (2005) J. Biol. Chem. 280, 33426-33434. [DOI] [PubMed] [Google Scholar]
- 38.Morozov, Y. M. & Freund, T. F. (2003) Neuroscience 120, 923-939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}