

Abstract
A cancer DNA phenotype, identical to the DNA structure of tumors, has been identified in the prostate glands of certain healthy men over 55 years of age. We now show that the same DNA signature exists in normal tissues adjacent to tumors. This finding implies that the phenotype is maintained in normal prostate cells from its inception through tumor development. The presence of the phenotype in tumors, adjacent normal cells, and in the normal prostate cells of certain older men suggests that it is a potentially critical factor in tumor development and may serve as an early biomarker for cancer risk assessment. Intervention to inhibit the development of the phenotype in healthy men, or to eliminate it once formed, may suppress or even prevent tumor formation.
Keywords: cancer phenotype, DNA structure, cancer biomarkers, cancer prediction and intervention, Fourier transform-infrared spectroscopy
Using established Fourier transform-infrared (FT-IR) statistical models (1-5), we have demonstrated previously a cancer DNA phenotype in the limbs of mice 10 weeks after injecting the carcinogen 3-methylcholanthrene (4). This phenotype was identified in normal tissues 8 weeks before tumor formation. Strikingly, administration of the prodrug cyclophosphamide every 10 days after 3-methylcholanthrene injection delayed tumor formation by about 30% and almost completely suppressed development of the phenotype, thus suggesting that it may also be inhibited by other intervention strategies with less toxic side effects (4).
In a previously reported study (2), a similar cancer DNA phenotype was identified in the prostate glands of certain healthy older men (55-80 years old). Both studies showed differences between the DNA of normal tissues and those tissues with the phenotype characterized by significant alterations in the base and backbone structures. In the prostate, for example, the development of the phenotype appeared to be age-related in that no evidence was found for it in men younger than 36 years of age. A statistical comparison of the FT-IR spectrum of the cancer phenotype with the DNA spectrum of normal prostate tissues of the healthy younger men revealed a broad array of differences in base structures (e.g., N-H and C-O), as well as in vertical base-stacking interactions. Various differences were also found in the phosphodiester-deoxyribose backbone (2). These differences likely arise from constituents in the microenvironment (e.g., reactive oxygen species) capable of inducing conformational changes in the backbone via disruptions in the planar base structure (6).
The previous evidence (2) for a cancer DNA phenotype in certain older men suggests that this structure may respond differently in transcription and replication from the DNA of younger men, thus potentially increasing cancer risk. We now report that the cancer DNA phenotype in the prostate glands of older men is indistinguishable from the phenotype found in histologically normal tissues surrounding tumors. This finding implies that the phenotype is structurally stable, surviving from its first appearance through tumor formation. Furthermore, the presence of the phenotype in the older men and in normal tissues adjacent to tumors suggests that this structure is likely hardwired in progenitor cells (7-9). Evidence for an association between the phenotype and progenitor cells would strengthen the hypothesis that the phenotype is a significant factor in tumor etiology, a biomarker for assessing cancer risk, and a target for early intervention.
Materials and Methods
Tissues. Thirty-nine prostate samples from the peripheral zone were provided by the following donors: Baylor College of Medicine Specialized Program of Research Excellence tissue bank project (Houston) (n = 21) and Washington Pathology Consultants (Seattle) (n = 18). The samples comprised the following groups: (i) tissues from patients, ages 50-75 years, with primary prostate cancer having no evidence of metastasis, consisting of microscopically isolated tumor tissues (n = 9); (ii) matched histologically normal tissues microscopically isolated from the same patients with primary prostate tumors (n = 9); (iii) histologically normal tissues from younger men, ages 16-36 years (n = 9); and (iv) histologically normal tissues from older men, ages 55-80 years (n = 12).
Isolation of High-Purity Tissues. Samples from the Baylor College of Medicine were isolated from fresh prostate tissues via punch biopsy (2). The remaining tissue samples were treated as follows: frozen tumor tissues were inked with different colors to denote posterior or anterior and right or left aspects. Two similarly inked, matching glass microscope slides of stained sections from the adjacent slices of the tumor were also obtained. Tumor foci were identified on “sandwich” slides by dotting the tumor periphery and then extrapolated to the frozen slice so that tumor tissue of ≈90% purity could be dissected. The histologically normal tissues surrounding the tumors showed no indication of contamination from tumor or other abnormal cells [e.g., prostatic intraepithelial neoplasia (10)] and were thus essentially pure.
DNA Extraction. DNA (≈50 μg) was obtained from each frozen tissue (70-100 mg) with Qiagen (Chatsworth, CA) 100/G Genomic-tips by using a modification of the Qiagen extraction procedure. The DNA was passed through a 5.0-μm Cameo 30N filter (Osmonics, Minnetonka, MN) before precipitation. In preparation for FT-IR spectroscopy, the DNA was dissolved in 10-40 μl (depending on the size of the pellet) of optima grade distilled water (Fisher Scientific). All samples were selected randomly for analysis to avoid batch effects.
FT-IR Spectroscopy. FT-IR spectral analysis was undertaken with a microscope spectrometer (System 2000, PerkinElmer), as reported previously (1, 2, 4). A 0.2-μl aliquot of the DNA solution was spotted directly on a BaF2 plate, forming an outer ring containing the DNA. Two separate rings were created for each DNA sample. To completely dry the DNA, the plate was placed in a lyophilizer for 1 h. By using the microscope spectrometer, 10 spectral determinations were made around each of the two rings per sample, and the percentage transmittance was converted (Fourier-transformed) to absorbance. Each spectrum was baselined and normalized to adjust for the optical characteristics of the sample before obtaining the mean spectrum. Structural assignments for spectral differences were based on previous reports (11-13). The FT-IR statistical models have a high sensitivity for identifying differences between groups of DNA (1, 3), as demonstrated by the ability to readily distinguish a 25-base oligonucleotide with a single 8-oxo substituent (6).
Statistical Analyses. A t test was performed at each wavenumber to determine the statistical differences (P values) between DNA groups (1, 2, 14). Principal components analysis (6, 15), which involves ≈106 correlations between spectral absorbances, integrates different properties of the spectra, such as changes in peak heights and peak locations. This analysis resulted in 10 principal component (PC) scores per sample (1, 2, 14). A t test established statistical differences between groups for each PC score. PCs with significant differences (P ≤ 0.05) were used to construct two-dimensional scatter plots. Structural differences between the groups were indicated by the separation of sample clusters in the plots (1, 2, 14), whereas the tightness of the points within the clusters was evidence for structural similarity.
Results and Discussion
The cancer DNA phenotype found in some older men is strikingly different from the DNA structure of the younger men (Fig. 1). In this comparison, ≈42% of the spectral range was significantly different between the groups. These differences were evident in various aspects of the base and backbone structures, suggesting that complex age-related changes potentially threatening the integrity of transcription and replication (6) had occurred in the evolution of the DNA structure from that of younger men to the cancer DNA phenotype in the older men.
Fig. 1.

Significant differences in the base and backbone structures exist between the DNA of the younger men and that of the older men with the cancer DNA phenotype. (A) Comparison of FT-IR spectra of the DNA of younger men (black line; n = 9) and the cancer DNA phenotype in the older men (magenta line; n = 5). (B) Corresponding P values.
The many structural differences between the DNA of younger men and the cancer DNA phenotype in the older men are presented in Table 1. These differences include structural alterations in functional groups (e.g., C=O and NH2 of guanine and C=N of adenine) and in the ring-stretching vibration of thymine. Differences between the phosphodiester-deoxyribose structures of the DNA of younger men and the cancer DNA phenotype in the older men are also evident. Some of these differences (e.g., those involving the 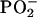 group) result from alterations in vertical base stacking, which, in turn, induce changes in torsion angles associated with the conformation of the backbone (6). Additional structural changes in the DNA backbone may arise from reactions of hydroxyl radicals with the sugar moiety (16). In all likelihood, the age-related DNA differences found in this study would also be reflected in genetic alterations, such as those recently identified in prostate tissues by using gene expression analysis (9).
group) result from alterations in vertical base stacking, which, in turn, induce changes in torsion angles associated with the conformation of the backbone (6). Additional structural changes in the DNA backbone may arise from reactions of hydroxyl radicals with the sugar moiety (16). In all likelihood, the age-related DNA differences found in this study would also be reflected in genetic alterations, such as those recently identified in prostate tissues by using gene expression analysis (9).
Table 1. Spectral peaks identifying structural differences (P ≤ 0.05) between the DNA of the younger normal men (ages 16–36 years; n = 9) and the cancer DNA phenotype of the older healthy men (ages 55–80 years; n = 5).
| Wavenumber, cm–1 | Peak strength | Significant differences in peak amplitudes, P values | Structural assignments (vibrations) |
|---|---|---|---|
| 1689 | Strong | 0.01 | Guanine residue: C=O stretching and NH2 scissoring |
| 1647 | Strong | 0.05 | Cytosine residue: in-plane ring |
| 1605 | Strong | <0.01 | Adenine residue: NH2 bending and C=N stretching |
| 1577 | Weak | <0.01 | Thymine residue: ring stretching |
| 1531 | Weak | 0.03 | Base residues: in-plane ring vibrations of NH and CH deformation modes |
| 1482 | Weak | <0.01 | Base residues: in-plane ring vibrations of NH and CH deformation modes |
| 1415 | Weak | 0.04 | Base residues: in-plane ring vibrations of NH and CH deformation modes |
| 1369 | Weak | 0.04 | Base residues: in-plane ring vibrations of NH and CH deformation modes |
| 1231 | Strong | 0.04 | PO2– antisymmetric stretching |
| 1088 | Strong | <0.01 | PO2– symmetric stretching |
| 1021 | Medium and broad | <0.01 | Deoxyribose residue: in-plane |
| 1020 | Medium and broad | <0.01 | Deoxyribose residue: in-plane |
| 964 | Strong | <0.01 | C–C stretching |
| 886 | Weak | 0.01 | Base residues: out-of-plane |
| 884 | Weak | 0.02 | Base residues: out-of-plane |
The PC plot in Fig. 2A shows a complete separation between the cancer DNA phenotype in the older men and the DNA of the older men without the phenotype, demonstrating that the structures are distinctly different. The cluster of points representing the phenotype is tightly packed, suggesting a close similarity between the component DNA structures. However, the points for the DNA of the older men without the phenotype are highly diverse. These findings imply that the evolution of the phenotype in normal cells involves a substantial focusing of DNA to form stable and ordered structures that potentially constitute a basis for altered biological function, such as the initiation of carcinogenesis (17).
Fig. 2.

PC plots of FT-IR spectra of DNA show complete discrimination between prostate groups. (A) PC4 (P < 0.01) vs. PC10 (P < 0.01) for the cancer DNA phenotype in the older men (▴; n = 5) and the older men without the phenotype (▵; n = 7). (B) PC7 (P = 0.03) vs. PC9 (P = 0.02) for the cancer DNA phenotype in the older men (▴; n = 5), the older men without the phenotype (▵; n = 7), and the younger men (•; n = 9).
Fig. 2B compares the DNA of the following groups (PCs different from those in Fig. 2 A were used): the younger men, the older men without the phenotype, and the older men with the phenotype. Each of these three groups of DNA is completely distinct. The seemingly linear pattern of separated clusters suggests that the DNA of some individuals in the diverse group of younger men will be damaged with age. As these men grow older, this damage leads to the formation of a group of DNA structures constituting the cancer DNA phenotype that exhibits little structural diversity.
Fig. 3A shows the P values from a comparison of the spectra representing the cancer DNA phenotype in the older men and the spectra for the prostate tumors over the entire spectral range. The total absence of significant differences between these spectra means that the structures are virtually identical. Fig. 3B shows a similar comparison between the DNA from normal tissues adjacent to the tumor and the DNA phenotype in certain older men, demonstrating that these structures are also indistinguishable. Significant P values (≤ 0.05) are confined to 0.9% of the total spectral range; <5% is considered statistically insignificant (6). The results from each of these spectral comparisons imply that the phenotype is biologically conserved in that it apparently survives from the point of formation in normal cells of healthy men through tumor development. In each of these comparisons, PC analysis, involving ≈106 correlations between numerous spectral properties, yielded no significant PC scores. This finding further supports the conclusion that the DNA structures in normal tissues of some older men and the normal tissues surrounding tumors are essentially identical and thus represent the same phenotype.
Fig. 3.

P values resulting from comparisons of FT-IR spectra of DNA from the following prostate tissues show no significant differences (P < 0.05), thus establishing the structural similarity between the groups: (A) tumor (n = 9) vs. the cancer DNA phenotype in the older men (n = 5); and (B) histologically normal tissues surrounding tumors (n = 9) vs. the cancer DNA phenotype in the older men.
The evidence suggests that the phenotype arises from disordered molecules derived from reactions of various factors in the cellular environment, such as oxidation (18-20), methylation (21, 22), and depurination (23). The structural stability of the phenotype may well be the key to its survival in normal prostate cells through tumor formation, as well as to its proposed relationship to carcinogenesis.
It is now possible to identify a cancer DNA phenotype, identical to the DNA structure of tumors, in normal tissues in some older men, as well as in histologically normal tissues adjacent to tumors, thus suggesting that the presence of this phenotype in apparently normal prostates is a promising early indicator of cancer risk. If additional evidence confirms that this phenotype is mechanistically linked to tumor development, it may be a convenient target for early intervention with therapeutic agents that inhibit its development or even eliminate it once formed.
No clinical evidence was found in the present study to indicate that the prostate tumors had metastasized. However, we have reported previously (2, 5, 24) the ability to differentiate the DNA of prostate tumors with evidence of metastasis from those without such evidence. In this regard, a metastatic cancer DNA phenotype (structurally indistinguishable from the tumor DNA) was identified in histologically normal tissues surrounding prostate tumors having confirmed distant metastases (5). This metastatic phenotype was structurally distinguishable from the presently described cancer DNA phenotype. For the future, it may be possible to use these two distinct phenotypes, identified via the FT-IR statistical models of DNA from biopsy tissues, for predicting the likelihood of developing a benign tumor as opposed to a tumor with metastatic potential. In addition, the possibility exists for distinguishing between the risk for benign tumors vs. tumors having varying degrees of aggressiveness, such as revealed in the Gleason grading system (25).
Acknowledgments
We thank Drs. Miral Dizdaroglu, Ramesh C. Gupta, Karl Erik Hellström, and Edward L. Weber for helpful comments and Callie Ramsey and Mark Vinson for technical assistance. We also thank Washington Pathology Consultants and the Specialized Program of Research Excellence at Baylor College of Medicine (funded by National Cancer Institute Grant CA58204) for providing tissues and pathology data. This study was supported by National Cancer Institute Grant CA79690 and the Pacific Northwest Research Institute.
Author contributions: D.C.M., E.A.B., and K.M.A. designed research; N.K.G. and K.M.A. performed research; E.A.B. and T.M.W. contributed new reagents/analytic tools; D.C.M., N.K.G., V.M.G., E.A.B., and K.M.A. analyzed data; and D.C.M., V.M.G., and K.M.A. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: FT-IR, Fourier transform-infrared; PC, principal component.
References
- 1.Garcia-Closas, M., Hankinson, S. E., Ho, S., Malins, D. C., Polissar, N. L., Schaefer, S. N., Su, Y. & Vinson, M. A. (2000) J. Natl. Cancer Inst. Monogr. 27, 147-156. [DOI] [PubMed] [Google Scholar]
- 2.Malins, D. C., Johnson, P. M., Barker, E. A., Polissar, N. L., Wheeler, T. M. & Anderson, K. M. (2003) Proc. Natl. Acad. Sci. USA 100, 5401-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malins, D. C., Anderson, K. M., Polissar, N. L., Ostrander, G. K., Knobbe, E. T., Green, V. M., Gilman, N. K. & Spivak, J. L. (2004) Proc. Natl. Acad. Sci. USA 101, 5008-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malins, D. C., Anderson, K. M., Gilman, N. K., Green, V. M., Barker, E. A. & Hellström, K. E. (2004) Proc. Natl. Acad. Sci. USA 101, 10721-10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malins, D. C., Gilman, N. K., Green, V. M., Wheeler, T. M., Barker, E. A., Vinson, M. A., Sayeeduddin, M., Hellström, K. E. & Anderson, K. M. (2004) Proc. Natl. Acad. Sci. USA 101, 11428-11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malins, D. C., Polissar, N. L., Ostrander, G. K. & Vinson, M. A. (2000) Proc. Natl. Acad. Sci. USA 97, 12442-12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigelt, B. & van't Veer, L. J. (2004) Cell Cycle 3, 756-757. [DOI] [PubMed] [Google Scholar]
- 8.Bernards, R. & Weinberg, R. A. (2002) Nature 418, 823. [DOI] [PubMed] [Google Scholar]
- 9.Yu, Y. P., Landsittel, D., Jing, L., Nelson, J., Ren, B., Liu, L., McDonald, C., Thomas, R., Dhir, R., Finkelstein, S., Michalopoulos, G., Becich, M. & Luo, J. H. (2004) J. Clin. Oncol. 22, 2790-2799. [DOI] [PubMed] [Google Scholar]
- 10.Bostwick, D. G. & Qian, J. (2004) Mod. Pathol. 17, 360-379. [DOI] [PubMed] [Google Scholar]
- 11.Tsuboi, M. (1969) Appl. Spectrosc. Rev. 3, 45-90. [Google Scholar]
- 12.Tsuboi, M. (1974) in Basic Principles in Nucleic Acid Chemistry, ed. Ts'o, P. O. P. (Academic, New York), pp. 399-452.
- 13.Shimanouchi, T., Tsuboi, M. & Kyogoku, Y. (1964) in Advances in Chemical Physics (Wiley Interscience, New York), Vol. VII, pp. 436-498. [Google Scholar]
- 14.Malins, D. C., Polissar, N. L., Su, Y., Gardner, H. S. & Gunselman, S. J. (1997) Nat. Med. 3, 927-930. [DOI] [PubMed] [Google Scholar]
- 15.Fisher, L. D. & van Belle, G. (1993) Biostatistics: A Methodology for the Health Sciences (Wiley, New York).
- 16.von Sonntag, C. (1987) The Chemical Basis of Radiation Biology (Taylor and Francis, New York).
- 17.Prigogine, I. & Stengers, I. (1984) Order Out of Chaos (New Science Library, Boulder, CO).
- 18.Cooke, M. S., Evans, M. D., Dizdaroglu, M. & Lunec, J. (2003) FASEB J. 17, 1195-1214. [DOI] [PubMed] [Google Scholar]
- 19.Dizdaroglu, M. (1992) Mutat. Res. 275, 331-342. [DOI] [PubMed] [Google Scholar]
- 20.Dizdaroglu, M., Jaruga, P., Birincioglu, M. & Rodriguez, H. (2002) Free Radical Biol. Med. 32, 1102-1115. [DOI] [PubMed] [Google Scholar]
- 21.Wachsman, J. T. (1997) Mutat. Res. 375, 1-8. [DOI] [PubMed] [Google Scholar]
- 22.Szyf, M. (2003) Drug Resist. Updat. 6, 341-353. [DOI] [PubMed] [Google Scholar]
- 23.Cavalieri, E. L., Li, K. M., Balu, N., Saeed, M., Devanesan, P., Higginbotham, S., Zhao, J., Gross, M. L. & Rogan, E. G. (2002) Carcinogenesis 23, 1071-1077. [DOI] [PubMed] [Google Scholar]
- 24.Malins, D. C. (2004) Cell Cycle 3, e45-e46. [DOI] [PubMed] [Google Scholar]
- 25.Kirby, R. S., Christmas, T. J. & Brawer, M. K. (1996) Prostate Cancer (Mosby, London).