

Abstract
Reconciliation of apparently contradictory experimental results obtained on the quinol:fumarate reductase, a diheme-containing respiratory membrane protein complex from Wolinella succinogenes, was previously obtained by the proposal of the so-called “E pathway hypothesis.” According to this hypothesis, transmembrane electron transfer via the heme groups is strictly coupled to cotransfer of protons via a transiently established pathway thought to contain the side chain of residue Glu-C180 as the most prominent component. Here we demonstrate that, after replacement of Glu-C180 with Gln or Ile by site-directed mutagenesis, the resulting mutants are unable to grow on fumarate, and the membrane-bound variant enzymes lack quinol oxidation activity. Upon solubilization, however, the purified enzymes display ≈1/10 of the specific quinol oxidation activity of the wild-type enzyme and unchanged quinol Michaelis constants, Km. The refined x-ray crystal structures at 2.19 Å and 2.76 Å resolution, respectively, rule out major structural changes to account for these experimental observations. Changes in the oxidation–reduction heme midpoint potential allow the conclusion that deprotonation of Glu-C180 in the wild-type enzyme facilitates the reoxidation of the reduced high-potential heme. Comparison of solvent isotope effects indicates that a rate-limiting proton transfer step in the wild-type enzyme is lost in the Glu-C180 → Gln variant. The results provide experimental evidence for the validity of the E pathway hypothesis and for a crucial functional role of Glu-C180.
Keywords: anaerobic respiration, atomic model, bioenergetics, membrane protein, succinate:quinone oxidoreductase
According to Peter Mitchell's chemiosmotic theory (1), the energy released upon the oxidation of electron donor substrates in both aerobic and anaerobic respiration is transiently stored in the form of an electrochemical proton potential (Δp) across the energy-transducing membranes, which can then be used by the ATP synthase for ADP phosphorylation with inorganic phosphate. Fundamentally, there are two mechanisms by which integral membrane proteins can act as catalysts in this coupling of electron transfer reactions to the generation of a transmembrane Δp: the redox loop mechanism and the proton pump mechanism (2). The redox loop mechanism essentially involves transmembrane electron transfer. Reduction reactions on one side of the energy-transducing membrane are associated with proton binding, whereas oxidation reactions on the opposite side of the membrane are associated with proton release. In a simple form, this mechanism is represented by the formate dehydrogenase (3) and membrane-bound nitrate reductase (4) enzymes of anaerobic respiration and, in a more complicated form, by the Q-cycle of the cytochrome bc1 complex, complex III of the aerobic respiratory chain (5). The proton pump mechanism involves the actual translocation of protons across the membrane. Transfer of electrons between sites of catalysis and prosthetic groups of the membrane protein complex results in conformational changes, which in turn lead to changes in the pKa values of amino acid side chains and other protonatable groups. Additionally or alternatively, the degree of exposure of these groups to one or the other side of the membrane may also change. Such a mechanism is thought to be operative, e.g., in cytochrome c oxidase, complex IV of the respiratory chain (6). Here we present experimental evidence of both of these mechanisms being harnessed together in a single respiratory membrane protein complex to ensure the counterbalancing of the effects of both processes for energetic reasons. The membrane protein in question is the diheme-containing quinol:fumarate reductase (QFR) from the anaerobic ε-proteobacterium Wolinella succinogenes. QFR is the terminal enzyme of fumarate respiration (7, 8), a form of anaerobic respiration that allows anaerobic bacteria to use fumarate instead of dioxygen as the terminal electron acceptor. QFR couples the two-electron reduction of fumarate to succinate (reaction 1) to the two-electron oxidation of hydroquinone (quinol) to quinone (reaction 2).
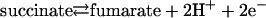 |
[1] |
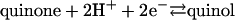 |
[2] |
This reaction is part of an electron transfer chain that enables the bacterium to grow with various electron donor substrates such as formate or hydrogen (Fig. 1a). QFR from W. succinogenes consists of two hydrophilic subunits, A and B, and one hydrophobic, membrane-embedded subunit, C. The larger hydrophilic subunit A is associated with a covalently bound flavin adenine dinucleotide (FAD), the smaller hydrophilic subunit B contains three iron–sulfur clusters ([2Fe–2S], [4Fe–4S], and [3Fe–4S]), and the hydrophobic subunit C harbors two heme b groups (Fig. 1b). Based on their relative distance to the hydrophilic subunits, these heme groups are referred to as the proximal heme (bP) and the distal heme (bD), respectively. Although it has long been known (9) that the two heme groups have different oxidation-reduction midpoint potentials, it has only recently been possible to assign the “high-potential” heme to bP and the “low-potential” heme to bD (10).
Fig. 1.

Electron and proton transfer in fumarate respiration (a) and W. succinogenes QFR (b and c). Positive and negative sides of the membrane are the periplasm and the cytoplasm, respectively. Figs. 1, 2, and 3 were prepared with a version of molscript (39) modified for color ramping (40) and map drawing (41) capabilities. (a) The key enzymes involved in fumarate respiration are indicated. (b) Hypothetical transmembrane electrochemical potential generation as suggested by the essential role of Glu-C66 for menaquinol oxidation by W. succinogenes QFR (16). The prosthetic groups of the W. succinogenes QFR dimer are displayed (coordinate set 1QLA, ref. 14). Distances between prosthetic groups are edge-to-edge distances in Å as defined by Page et al. (42). Also indicated are the side chain of Glu-C66 and a model of menaquinol (MKH2) binding. The position of bound fumarate (Fum) is taken from PDB ID code 1QLB (14). (c) Hypothetical cotransfer of one H+ per electron across the membrane (E pathway hypothesis). The two protons that are liberated upon oxidation of menaquinol (MKH2) are released to the periplasm (bottom) via the residue Glu-C66. In compensation, coupled to electron transfer via the two heme groups, protons are transferred from the periplasm via the ring C propionate of the distal heme bD and the residue Glu-C180 to the cytoplasm (top), where they replace those protons that are bound during fumarate reduction. In the oxidized state of the enzyme, the E pathway is blocked.
Although electrophysiological experiments performed with inverted vesicles and proteoliposomes containing QFR demonstrated that the reaction catalyzed by the diheme-containing QFR from W. succinogenes is not directly associated with the generation of a transmembrane electrochemical proton potential (11–13), the three-dimensional structure of this membrane protein complex, initially solved at 2.2-Å resolution (14), revealed locations of the active sites of fumarate reduction (15) and of menaquinol oxidation (16) that are oriented toward opposite sides of the membrane (Fig. 1b). Because the binding of two protons upon fumarate reduction invariably occurs from the cytoplasm, and the release of two protons associated with menaquinol oxidation invariably occurs to the periplasm, this arrangement of catalytic sites indicated that menaquinol oxidation by fumarate, as catalyzed by W. succinogenes QFR, should be associated directly with the establishment of an electrochemical proton potential across the membrane, in contrast with the results of the functional measurements.
To reconcile these apparently conflicting experimental observations, the so-called “E pathway hypothesis” (17) has been proposed (Fig. 1c). According to this hypothesis, the transfer of two electrons via the two QFR heme groups is strictly coupled to a compensatory, parallel transfer of two protons across the membrane via a proton transfer pathway that is transiently open during the reduction of the two hemes and closed in the oxidized state of the enzyme. The two most prominent constituents of the proposed pathway were suggested to be the ring C propionate of the distal heme bD and, in particular, the amino acid residue Glu-C180, after which the E pathway was named. This residue is conserved in all ε-proteobacterial diheme-containing QFR enzymes of known sequence (17). Removal of 2H+ from the periplasm balances those released upon menaquinol oxidation, whereas supply of 2H+ to the cytoplasm compensates for those bound upon fumarate reduction. This unprecedented transmembrane proton transfer, as predicted by the E pathway hypothesis, therefore can explain why W. succinogenes QFR operates electroneutrally despite the orientation of the catalytic sites to opposite sides of the membrane.
The role of Glu-C180 proposed within the context of this hypothesis has recently received support from the results of multiconformer continuum electrostatic calculations (10), which indicate that the side chain of this residue undergoes a combination of a conformational change and protonation upon heme reduction. This finding prompted us to investigate the possible role of Glu-C180 for W. succinogenes QFR. Here we describe the effects of replacing Glu-C180 with Gln or Ile by site-directed mutagenesis. The consequences of these mutations for the viability of the resulting mutants and the structural and functional characteristics of the corresponding variant enzymes are presented and discussed. We conclude that Glu-C180 is an essential constituent of the membrane-bound enzyme in a way that is fully compatible with its proposed role in the context of the E pathway hypothesis.
Materials and Methods
Mutagenesis. The mutants W. succinogenes FrdC-E180Q and FrdC-E180I were constructed by transforming W. succinogenes ΔfrdCAB with derivatives of pFrdcat2 (18). Plasmid pFrdcat2 contains the entire frdCAB operon and integrates into the genome of W. succinogenes ΔfrdCAB by homologous recombination (18). Plasmids pFrdcat2-E180Q and pFrdcat2-E180I were generated by using the QuikChange site-directed mutagenesis kit (Stratagene) with pFrdcat2 as template and specifically synthesized complementary primer pairs [forward primers: 5′-3338CTTCTTTTTGCCGTTCAGCTTCATGGCTCTGTAGG-3′ (E180Q), 5′-3338CTTCTTTTTGCCGTTATCCTTCATGGCTCTGTAGG-3′ (E180I); numbered according to the sequence deposited in the DNA Data Base in Japan/European Molecular Biology Laboratory/GenBank databases under accession no. AJ000662]. The altered nucleotides are printed in bold, and the resulting glutamine and isoleucine codons are underlined. Cells of W. succinogenes ΔfrdCAB grown with formate and nitrate were used for transformation as previously described in ref. 18. Transformants were selected on agar plates with a medium containing kanamycin (25 mg/liter) and chloramphenicol (12.5 mg/liter). The respective integration of plasmids pFrdcat2-E180Q and pFrdcat2-E180I into the genome of W. succinogenes ΔfrdCAB was confirmed by Southern blot analysis as previously described in ref. 18. The mutations were confirmed, and unwanted mutations were ruled out by sequencing a PCR product containing the frdC gene that was obtained by using genomic DNA of the mutant strains as template and specifically synthesized primer pairs.
Cell Growth and Purification of QFR. W. succinogenes was grown with formate as electron donor and either fumarate or nitrate as electron acceptor as described in refs. 19 and 20. The latter medium was supplemented with brain–heart infusion broth (0.5% mass/vol, GIBCO/BRL). Fumarate reductase activities were measured with cell homogenates from bacteria grown with formate and nitrate (18). Protein was determined by using the biuret method with KCN (21). QFR was isolated as described in ref. 22 with modifications previously reported in refs. 14 and 23. The enzymatic activities listed in Table 1 were measured at 37°C according to Unden and Kröger (24). Solvent isotope effects were measured with the following modifications: The buffer stock solutions were diluted in either 2H2O or 1H2O to respective concentrations of 100% or 0% 2H, 20 mM Mops, pH 7.0, T = 310 K. The amount of heme b reduced by 2,3-dimethyl-1,4-naphthoquinol (DMNH2) or by dithionite was calculated from the absorbance difference between the reduced and the oxidized sample at 565 nm minus that at 575 nm by using the molar extinction coefficient of 23.4 mM–1·cm–1 (9).
Table 1. Growth and enzymatic activities of W. succinogenes FrdC-E180Q and FrdC-E180I cells and properties of the isolated QFR enzymes.
| Property | Wild type | FrdC-E66Q | FrdC-E1801 | FrdC-E180Q |
|---|---|---|---|---|
| Growth with | ||||
| Formate + fumarate | + | - | - | - |
| Formate + nitrate | + | + | + | + |
| Specific activity of membrane-bound enzyme in cell homogenate, U per mg of cell protein | ||||
| Succinate → methylene blue | 0.31 | 0.17 | 0.31 | 0.35 |
| DMNH2 → fumarate | 0.24 | ≤0.01 | ≤0.01 | ≤0.01 |
| Specific activity of isolated enzyme, U per mg of protein | ||||
| Succinate → methylene blue | 28.8 | 16.9 | 26.0 | 24.9 |
| DMNH2 → fumarate | 7.4 | ≤0.01 | 0.96 | 0.80 |
| Km (DMNH2), mM | 0.1 | 0.1 | 0.1 | |
| Heme b of isolated enzyme reduced by, μmol per g of protein | ||||
| 200 μM DMNH2 | 3.8 | 0.5 | 6.5 | 7.4 |
| Dithionite | 7.2 | 7.0 | 11.8 | 14.2 |
| Heme oxidation—reduction midpoint potentials EM7, mV | ||||
| Heme bP | -10 | -16 | +38 | +39 |
| Heme bD | -149 | -145 | -151 | -144 |
| Solvent isotope effect, k(1H2O)obs/k(2H2O)obs | ||||
| On DMNH2 → fumarate | 3.3 | 1.1 |
Values for wild type and FrdC-E66Q (16) are shown for comparison (in italics). One unit (U) corresponds to 1 μmol of substrate turned over per min.
Synthesis of 2,3-Dimethyl-1,4-naphthoquinone (DMN). For the synthesis of DMN, we followed the procedure of Wurm and Geres (25). The crude product was purified by HPLC on a Nucleoprep 20-μm, 700-mm × 50-mm column with a flow rate of 0.1 liter/min and hexane/ethyl acetate (10 + 1) as solvent.
Crystallization of QFR. Before crystallization, Glu-C180 → Gln QFR and Glu-C180 → Ile QFR were further purified by preparative flat-bed isoelectric focusing as previously described in refs. 14 and 23. Monoclinic crystals, space group P21, were grown by sitting-drop vapor diffusion as described earlier (14, 23).
Data Collection and Analysis. X-ray data collection was carried out at the European Synchrotron Radiation Facility beamline ID14 EH1 (λ = 0.934 Å, T = 2–4°C), Grenoble. Intensity data were obtained by using a charge-coupled device detector (Area Detector Systems Corp., Poway, CA). Only one crystal each was required for the datasets listed in Table 2. Data were processed with the hkl programs denzo and scalepack (26). After the removal of all solvent atoms from the coordinate set, a new, high-resolution (1.78 Å) structural model for wild-type QFR was used [C.R.D.L., unpublished data; PDB ID code 2BS2; derived from the original coordinates 1QLA (14)], for phasing the variant enzyme data by using the difference Fourier technique with the program cns (27). Simulated annealing, followed by conventional positional refinement and restrained individual B-factor refinement, were performed by using the program cns. The atomic model of QFR was rebuilt by using o (28). Oxygen atoms of water molecules were placed according to criteria described earlier (29). Strict noncrystallographic symmetry constraints were applied between the two QFR monomers in the asymmetric unit for the refinement of the lower resolution Glu-C180 → Ile QFR structure, and in the case of the 2.2-Å resolution Glu-C180 → Gln QFR structure, the same noncrystallographic symmetry restraints as previously described for the wild-type enzyme (14) were applied.
Table 2. Data collection and refinement statistics of the Glu-C180 → Gln and Glu-C180 → Ile QFR crystals.
| Characteristic | Glu-C180 → Gln QFR | Glu-C180 → Ile QFR | ||
|---|---|---|---|---|
| Resolution range, Å | 38.6-2.19 | 2.24-2.19 | 29.7-2.76 | 2.93-2.76 |
| Rsym,* % | 9.2 | 35.6 | 9.6 | 34.5 |
| I/σ(I) | 9.5 | 7.7 | 6.0 | 1.5 |
| No. of reflections used | 181,351 | 10,430 | 79,362 | 11,606 |
| In working set | 180,351 | 10,372 | 78,362 | 11,458 |
| In test set | 1,000 | 58 | 1,000 | 148 |
| Completeness, % | ||||
| Reflections used | 99.0 | 85.4 | 86.0 | 76.7 |
| In working set | 98.5 | 84.8 | 84.7 | 75.4 |
| In test set | 0.5 | 0.6 | 1.3 | 1.3 |
| Rfree,† % | 19.8 | 25.7 | 21.6 | 29.1 |
| Rcryst,‡ % | 18.3 | 22.0 | 20.0 | 28.0 |
| Cross-validated | ||||
| Luzzati coor. error,§ Å | 0.26 | 0.33 | ||
| No. of nonhydrogen atoms in the model | ||||
| Protein atoms | 18,306 | 9,068 | ||
| Heterogen atoms | 440 | 221 | ||
| Solvent atoms | 992 | 236 | ||
| B factor from Wilson plot, Å2 | 28.6 | 51.6 | ||
| Average B factor, Å2 | 34.0 | 38.3 | ||
| nobs/npar¶ | 2.3 | 2.1 | ||
| rms deviations from ideal values | ||||
| Bonds, Å | 0.007 | 0.008 | ||
| Bond angles, ° | 1.3 | 1.4 | ||
| Torsional angles, ° | 21.3 | 21.4 | ||
| Improper torsional angles, ° | 1.63 | 1.66 | ||
∥Based on protein parameter files (46), heme cofactor parameter files (29), and parameter files generated for the other prosthetic groups (14).
Rsym = Σi,hkl| 〈I(hkl) 〉 — Ii(hkl)| /Σi,hklIi(hkl).
Rfree = Σ(hkl)εT| |Fo| — |Fc| |/Σ(hkl)εT|Fo|, where T is the test set (44).
Rcryst = Σ(hkl)| |Fo| — |Fc| |/Σ(hkl)| |Fo|.
Estimate of the mean coordinate error from a Luzzati plot (45) using Rfree.
nobs = number of observed unique reflections used in the working set; npar = number of parameters necessary to define the model. This includes four parameters (x, y, and z coordinates plus isotropic atomic B factor) per atom.
Spectroelectrochemical Redox Titration. The QFR samples were concentrated to ≈0.8 mM, and the redox titrations were performed as previously described in refs. 16, 30, and 31. The applied potentials were measured with an Ag/AgCl/3 M KCl reference electrode, but all quoted values are potentials versus standard hydrogen electrode at pH 7. Absorbance difference spectra in the range of 400 nm to 700 nm were recorded at 5°C as previously described in refs. 16, 31, and 32. The titration curves were generated on the basis of the redox dependence of the amplitudes of the Soret and α band, respectively. To obtain values for the midpoint potentials of the high-potential heme b and the low-potential heme b, iterative fitting of a calculated Nernst function (33) was performed.
Results and Discussion
Construction and Properties of the Strains W. succinogenes FrdC-E180Q and FrdC-E180I and the Isolated Glu-C180 → Gln and Glu-C180 → Ile QFR Variant Enzymes. The mutant strains FrdC-E180Q and FrdC-E180I are compared in Table 1 to the wild type and the previously characterized strain FrdC-E66Q, where the essential residue Glu-C66 (Fig. 1b) at the menaquinol oxidation site was replaced with a Gln (16). The mutants did not grow with fumarate as the terminal electron acceptor; however, they did grow when nitrate (and brain–heart infusion broth) replaced fumarate. As assayed with succinate oxidation by methylene blue, activities of the mutant cell homogenate and the isolated enzyme were comparable to those of the wild type. The activity monitored by this assay is independent of the diheme subunit C (22). However, when fumarate reductase activities were assayed by monitoring fumarate reduction by DMNH2, which is diheme-subunit C-dependent (22), it was not detectable in the case of the membrane-bound variant enzymes. Upon solubilization of the isolated membranes with Triton X-100 (14, 22, 23), however, the activities of fumarate reduction by DMNH2 for the Glu-C180 variants were detectable and corresponded to approximately 1/10 of that of the wild-type enzyme. In this respect, the Glu-C180 QFR variants differ significantly from the Glu-C66 → Gln variant (16). The detergent-solubilized Glu-C180 variants are similar to the wild-type enzyme considering the fraction (1/2) of the dithionite-reducible heme b that could be reduced by DMNH2 (Table 1). The higher heme b content determined for the Glu-C180 variants is explained by the fact that the heme b reduction assay for the Glu-C180 variants had to be performed at a later stage of the purification procedure, i.e., after isoelectric focusing (14, 23), whereas it had previously been performed before this step for the wild-type enzyme and the Glu-C66 → Gln variant. The Glu-C180 variants are also similar to the wild-type enzyme with respect to the unchanged Michaelis constant Km for DMNH2 of 0.1 mM (Table 1).
Structure Determination. To investigate the nature of these intriguing effects, the Glu-C180 → Ile QFR and Glu-C180 → Gln QFR variants were crystallized, and x-ray diffraction data were collected as described in Materials and Methods. Under the reported crystallization conditions, three different crystal forms have been observed for the wild-type enzyme in its ferricyanide-oxidized state, all of them monoclinic, space group P21 (14–16, and reviewed in ref. 34). Both of the Glu-C180 variants crystallized in crystal form “A” (a = 85.2 Å, b = 189.0 Å, c = 117.9 Å, and β = 104.5°).
In the case of the Glu-C180 → Ile variant QFR, refinement of a model containing one heterotrimer of subunits A, B, and C (9,068 protein atoms with nonzero occupancy, 221 heterogen atoms, and 236 solvent atoms) was performed using the working data set between 29.71- and 2.76-Å resolution (84.7% complete, compare Table 2). Strict non-noncrystallographic symmetry constraints were imposed, relating the two heterotrimers in the asymmetric unit, which resulted in a ratio of the number of independent observations, nobs, to the number of parameters required for the model, npar, of 2.1. The crystallographic R factor (Rcryst) and the free R factor (Rfree) of the refined model were 20.0% and 21.6%, respectively.
In the case of the Glu-C180 → Gln variant QFR, the availability of a larger number of independent and higher resolution data allowed the refinement of a model containing both heterotrimers of subunits A, B, and C (18,306 protein atoms with nonzero occupancy, 440 heterogen atoms for the prosthetic groups, and 992 solvent atoms), by using the working data set between 38.6- and 2.19-Å resolution (84.8% complete, compare Table 2). As described earlier for the refinement at 2.2-Å resolution of the structure of wild-type QFR (14), noncrystallographic symmetry restraints were applied to the two heterotrimers in the asymmetric unit. The resulting nobs/npar ratio was 2.3, and Rcryst and Rfree of the refined model were 18.3% and 19.8%, respectively.
In the |Fvar| – |Fwt| difference electron density maps of both variants, there is only one peak with an amplitude larger than 6 standard deviations (σ) above the mean density of the map. This signal is a negative peak at –6.8σ in the case of the Glu-C180 → Ile variant QFR at the position occupied by the alternate conformers of the Glu-C180 side chain and an adjacent water molecule in the wild-type QFR structure 2BS2. In all of these new structures of crystal form “A,” extended electron density at the dicarboxylate-binding site was observed and was interpreted to indicate the binding of the tricarboxylate citrate, a component of the crystallization buffer (to be discussed elsewhere). With the coordinate error estimated to be 0.26 Å (Glu-C180 → Gln QFR) and 0.33 Å (Glu-C180 → Ile QFR), respectively, no significant differences compared with the wild-type enzyme are observed (Figs. 2c and 3), as is reflected by rms deviations from the respective Cα positions of the wild-type enzyme of 0.20 Å for the 2,296 Cα atoms of Glu-C180 → Gln QFR and 0.19 Å for the 1,148 Cα atoms of Glu-C180 → Ile QFR. In summary, the crystal structure determinations of the oxidized forms of the enzymes rule out that any major structural changes relative to the wild-type QFR could explain the observed functional effects in Glu-C180 → Glu and Glu-C180 → Ile variant enzymes.
Fig. 2.

Stereoviews of the crystal structures of Glu-C180 → Ile and Glu-C180→Gln QFR. (a and b) Representative sections of the 2|Fo| – |Fc| composite-omit (43) electron density maps, contoured at 1.0 standard deviations (σ) above the mean density of the map for the Glu-C180 → Ile (a) and Glu-C180 → Gln (b) variant enzymes. Shown are the proximal heme group, bP, in subunit C, as well as the residues between Val-C179 and Gly-C183. Also displayed are the |Fvar| – |Fwt| difference electron density maps contoured at 3σ (in green) and –3σ (in red). (a) The structure shown is that of Glu-C180 → Ile QFR with carbon atoms in yellow, nitrogen atoms in blue, oxygen atoms in red, and the heme iron in orange. Superimposed from the structure of wild-type QFR (PDB ID code 2BS2) are the two Glu-C180 side chain conformers (with carbon atoms in black and gray, respectively) and the oxygen atom of an adjacent water molecule (in light blue). (b) The structure shown is that of Glu-C180 → Gln QFR with atomic color coding for a. The second Gln-C180 side-chain conformer is shown with carbon atoms colored in orange. (c) Superposition for the region displayed in a and b of the structures of wild-type QFR (black, PDB ID code 2BS2, with the second Glu-C180 displayed in gray), Glu-C180 → Gln QFR (orange, with the second Gln-C180 conformer displayed in red) and Glu-C180 → Ile (green).
Fig. 3.

Stereoview comparison of the heme positions and of the Cα traces for the C subunits of wild-type QFR (black, PDB ID code 2BS2), Glu-C180 → Gln QFR (orange), and Glu-C180 → Ile (green). The position of the respective residue C180 is also indicated.
Redox Titration. A role of Glu-C180 in the proposed coupling of transmembrane proton transfer to transmembrane electron transfer via the heme groups should be reflected in a change of the oxidation–reduction midpoint potential of at least one of the two heme groups upon replacement of Glu-C180 with either Gln or Ile. Analysis of electrochemically induced absorbance difference spectra at the wavelengths of 428 nm (Soret band) and 561 nm (α band) for both variants and the comparison to the previously determined data for the wild-type enzyme (Table 1) indicated that this expectation was indeed the case, because the midpoint potential for the high-potential, proximal heme was increased by almost 50 mV in both variants. Considering that the (3Fe–4S) cluster (Fig. 1b) has been reported to have an oxidation–reduction midpoint potential of –24 mV (35), and assuming that this property is unchanged in the Glu-C180 variants, such an increase in the midpoint potential of bP makes electron transfer from bP to the (3Fe–4S) cluster at least 4 times more energetically unfavorable than the corresponding process in the wild-type enzyme. If both Gln and Ile are considered to mimic the protonated, neutral glutamic acid side chain rather than the deprotonated, anionic glutamate side chain, then these results indicate that a Glu-C180 side chain in the wild-type enzyme that was fully protonated for all combinations of heme redox states would make it harder to reoxidize a reduced heme bP by 50 mV. Conversely, we conclude that, in the wild-type enzyme, the reoxidation of the reduced proximal heme is facilitated by proton release from the (protonated) Glu-C180 side chain. This feature is fully consistent with the E pathway hypothesis.
Solvent Isotope Effects. A key role of residue Glu-C180 is also indicated by the comparison of the solvent isotope effects on the specific activities of DMNH2 oxidation by fumarate as catalyzed by the wild-type enzyme and the E180Q variant (Table 1). Although a significant solvent isotope effect can be measured for the wild-type enzyme, such an effect is negligible for the E180Q variant, which is a strong indication for Glu-C180 in the wild-type enzyme participating in a rate-limiting proton-transfer step.
Conclusions
Taken together, the results indicate an important, but indirect, role of Glu-C180 for events linked to the quinol oxidation process. This role becomes essential when the enzyme is membrane-bound. Together with the previously published results from electrostatic calculations (10), these experimental results provide strong support for the E pathway hypothesis and for an essential role of Glu-C180 within the context of this hypothesis. Although the E pathway hypothesis is currently the only working model that adequately explains all experimental and theoretical data obtained so far, its ultimate proof will require the unequivocal identification of further constituents of this pathway. The ring C propionate of heme bD has already been suggested to be such a participant because of its unusual orientation, and the structure provides suggestions of other polar and titratable amino acid side chains and water molecules as candidate constituents of such a proton transfer pathway. However, based on the theoretical calculations (10) and its position in the structure, it is very likely that Glu-C180 will turn out to be the central “switch” in the coupling of transmembrane electron and proton transfer.
One may ask why W. succinogenes possesses such an apparently complicated enzyme to perform a task electroneutrally (Fig. 4a) that, e.g., Escherichia coli performs with a much simpler enzyme. In E. coli QFR, the membrane anchor contains no heme b groups and the menaquinol oxidation is oriented toward the cytoplasmic, proximal side of the membrane (36), allowing the protons released by the menaquinol oxidation reaction to directly balance those consumed upon fumarate reduction (Fig. 4b). It appears reasonable to assume that the predecessor of present-day QFR originally lacked the E pathway and was used as a diheme-containing succinate:menaquinone reductase (SQR), catalyzing the reaction in the opposite direction in an aerobically living predecessor of W. succinogenes. As illustrated in Fig. 4c, this enzyme used the transmembrane Δp to drive the succinate oxidation by menaquinone, an endergonic reaction under standard conditions, in a manner similar to that proposed for the SQR of Gram-positive positive bacteria, e.g., Bacillus subtilis (37, 38), and very different from that of mitochondrial complex II, which reduces ubiquinone (Fig. 4d). Upon W. succinogenes adopting an anaerobic metabolism in the bovine rumen, the diheme-containing SQR could only operate as a QFR if, for energetic reasons, the overall electroneutrality of the catalyzed reaction is preserved. Although the oxidation of menaquinol by fumarate is, under standard conditions, an exergonic reaction, it is apparently not exergonic enough to support the generation of a transmembrane Δp (17). This case is elegantly demonstrated by the inability of the FrdC-E180Q mutant to grow with fumarate as the terminal electron acceptor, a fact that also illustrates how vital the inferred pathway is for the viability of the microorganism. We conclude that the establishment of an E pathway as a transmembrane proton transfer pathway has been evolutionarily preferred over a relocation of the menaquinol oxidation site from its distal position in W. succinogenes QFR to a proximal position similar to that found in E. coli QFR.
Fig. 4.

The coupling of electron and proton flow in anaerobic (a and b) and aerobic SQRs respiration (c and d), respectively. Positive and negative sides of the membrane are described for Fig. 1. (a and b) Electroneutral reactions as catalyzed by diheme-containing QFR from W. succinogenes (a) and the hemeless QFR from E. coli. (b) MK and MKH2, menaquinone and menaquinol, respectively. (c) Utilization of a transmembrane electrochemical potential as possibly catalyzed by diheme-containing SQR enzymes. (d) Electroneutral reaction as catalyzed by monoheme-containing SQR enzymes (complex II). Q and QH2, ubiquinone and ubiquinol, respectively.
Acknowledgments
We thank Annette Roth, Nicole Hilgendorff (both of the Max Planck Institute of Biophysics), Oliver Schürmann, and Monica Sänger (both of the Institut für Mikrobiologie) for technical assistance, and Drs. Germaine Sainz and Stéphanie Monaco (both of the European Synchrotron Radiation Facility, Grenoble) for serving as local contacts during data acquisition at European Synchrotron Radiation Facility beamline ID14-EH1. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 472, “Molecular Bioenergetics”) and the Max-Planck-Gesellschaft.
Author contributions: C.R.D.L., U.S.S., R.G., A.H.H., and M.G.M. performed research; J.G., H.S., W.M., and J.S. contributed new reagents/analytic tools; and C.R.D.L. designed research, analyzed data, and wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: bD, distal heme; bP, proximal heme; DMNH2, 2,3-dimethyl-1,4-naphthoquinol; QFR, quinol:fumarate reductase; SQR, succinate:quinone reductase.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2BS3 and 2BS4).
References
- 1.Mitchell, P. (1979) Science 206, 1148–1159. [DOI] [PubMed] [Google Scholar]
- 2.Richardson, D. J. & Sawers, G. (2002) Science 295, 1842–1843. [DOI] [PubMed] [Google Scholar]
- 3.Jormakka, M., Törnroth, S., Byrne, B. & Iwata, S. (2002) Science 295, 1863–1868. [DOI] [PubMed] [Google Scholar]
- 4.Bertero, M. G., Rothery, R. A., Palak, M., Hou, C., Lim, D., Blasco, F., Weiner, J. H. & Strynadka, N. C. J. (2003) Nat. Struct. Biol. 10, 681–687. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell, P. (1976) J. Theor. Biol. 62, 327–367. [DOI] [PubMed] [Google Scholar]
- 6.Michel, H. (1999) Nature 402, 602–603. [DOI] [PubMed] [Google Scholar]
- 7.Kröger, A. (1978) Biochim. Biophys. Acta 505, 129–145. [DOI] [PubMed] [Google Scholar]
- 8.Lancaster, C. R. D. (2004) in Diversity of Prokaryotic Electron Transport Carriers, Respiration in Archaea and Bacteria, ed. Zannoni, D. (Kluwer, Dordrecht, The Netherlands), Vol. 1, pp. 57–85. [Google Scholar]
- 9.Kröger, A. & Innerhofer, A. (1976) Eur. J. Biochem. 69, 497–506. [Google Scholar]
- 10.Haas, A. H. & Lancaster, C. R. D. (2004) Biophys. J. 87, 4298–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geisler, V., Ullmann, R. & Kröger, A. (1994) Biochim. Biophys. Acta 1184, 219–226. [Google Scholar]
- 12.Kröger, A., Biel, S., Simon, J., Groß, R., Unden, G. & Lancaster, C. R. D. (2002) Biochim. Biophys. Acta 1553, 23–38. [DOI] [PubMed] [Google Scholar]
- 13.Biel, S., Simon, J., Groß, R., Ruiz, T., Ruitenberg, M. & Kröger, A. (2002) Eur. J. Biochem. 269, 1974–1983. [DOI] [PubMed] [Google Scholar]
- 14.Lancaster, C. R. D., Kröger, A., Auer, M. & Michel, H. (1999) Nature 402, 377–385. [DOI] [PubMed] [Google Scholar]
- 15.Lancaster, C. R. D., Groß, R. & Simon, J. (2001) Eur. J. Biochem. 268, 1820–1827. [PubMed] [Google Scholar]
- 16.Lancaster, C. R. D., Groß, R., Haas, A., Ritter, M., Mäntele, W., Simon, J. & Kröger, A. (2000) Proc. Natl. Acad. Sci. USA 97, 13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancaster, C. R. D. (2002) Biochim. Biophys. Acta 1565, 215–231. [DOI] [PubMed] [Google Scholar]
- 18.Simon, J., Groß, R., Ringel, M., Schmidt, E. & Kröger, A. (1998) Eur. J. Biochem. 251, 418–426. [DOI] [PubMed] [Google Scholar]
- 19.Bronder, M., Mell, H., Stupperich, E. & Kröger, A. (1982) Arch. Microbiol. 131, 213–223. [DOI] [PubMed] [Google Scholar]
- 20.Lorenzen, J. P., Kröger, A. & Unden, G. (1993) Arch. Microbiol. 159, 477–483. [Google Scholar]
- 21.Bode, C., Goebell, H. & Stähler, E. (1968) Z. Klin. Chem. Klin. Biochem. 6, 419–422. [PubMed] [Google Scholar]
- 22.Unden, G., Hackenberg, H. & Kröger, A. (1980) Biochim. Biophys. Acta 591, 275–288. [DOI] [PubMed] [Google Scholar]
- 23.Lancaster, C. R. D. (2003) in Membrane Protein Purification and Crystallization: A Practical Guide, eds. Hunte, C., Schägger, H. & von Jagow, G. (Academic, San Diego), 2nd Ed., pp. 219–228.
- 24.Unden, G. & Kröger, A. (1981) Eur. J. Biochem. 120, 577–584. [DOI] [PubMed] [Google Scholar]
- 25.Wurm, G. & Geres, U. (1984) Arch. Pharm. 317, 606–609. [Google Scholar]
- 26.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 27.Brünger, A. T., Adams, P. T., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 28.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 29.Lancaster, C. R. D. & Michel, H. (1997) Structure 5, 1339–1359. [DOI] [PubMed] [Google Scholar]
- 30.Moss, D. A., Nabedryk, E., Breton, J. & Mäntele, W. (1990) Eur. J. Biochem. 187, 565–572. [DOI] [PubMed] [Google Scholar]
- 31.Mäntele, W. (1996) in Biophysical Techniques in Photosynthesis, eds. Hoff, A. J. & Amesz, J. (Kluwer, Dordrecht, The Netherlands), pp. 137–160.
- 32.Mäntele, W. (1993) Trends Biochem. Sci. 18, 197–202. [DOI] [PubMed] [Google Scholar]
- 33.Baymann, F., Moss, D. A. & Mäntele, W. (1991) Anal. Biochem. 199, 269–274. [DOI] [PubMed] [Google Scholar]
- 34.Lancaster, C. R. D. (2003) in Methods and Results in Membrane Protein Crystallization, ed. Iwata, S. (International University Line, La Jolla, CA), pp. 177–192.
- 35.Unden, G., Albracht, S. P. J. & Kröger, A. (1984) Biochim. Biophys. Acta 767, 460–469. [DOI] [PubMed] [Google Scholar]
- 36.Rothery, R. A., Seime, A. M., Spiers, A.-M. C., Maklashina, E., Schröder, I., Gunsalus, R. P., Cecchini, G. & Weiner, J. H. (2005) FEBS J. 272, 313–326. [DOI] [PubMed] [Google Scholar]
- 37.Schirawski, J. & Unden, G. (1998) Eur. J. Biochem. 257, 210–215. [DOI] [PubMed] [Google Scholar]
- 38.Matsson, M., Tolstoy, D., Aasa, R. & Hederstedt, L. (2000) Biochemistry 39, 8617–8624. [DOI] [PubMed] [Google Scholar]
- 39.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946–950. [Google Scholar]
- 40.Esnouf, R. M. (1997) J. Mol. Graphics Model. 15, 132–134. [DOI] [PubMed] [Google Scholar]
- 41.Esnouf, R. M. (1999) Acta Crystallogr. D 55, 938–940. [DOI] [PubMed] [Google Scholar]
- 42.Page, C. C., Moser, C. C., Chen, X. & Dutton, P. L. (1999) Nature 402, 47–52. [DOI] [PubMed] [Google Scholar]
- 43.Hodel, A., Kim, S.-H. & Brünger, A. T. (1992) Acta Crystallogr. A 48, 851–859. [Google Scholar]
- 44.Brünger, A. T. (1992) Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 45.Luzzati, P. V. (1952) Acta Crystallogr. 5, 802–810. [Google Scholar]
- 46.Engh, R. A. & Huber, R. (1991) Acta Crystallogr. A 47, 392–400. [Google Scholar]