

Abstract
Details are presented of the structural analysis of the cytochrome b6f complex from the thermophilic cyanobacterium, Mastigocladus laminosus, in the presence of the electrochemically positive (p)-side quinone analogue inhibitor, 2,5-dibromo-3-methyl-6-isopropylbenzoquinone (DBMIB). One DBMIB binding site was found. This site is peripheral to the quinone binding space defined by the binding sites of other p-side inhibitors previously resolved in cytochrome bc1/b6f complexes. This high-affinity site resides in a p-side interfacial niche bounded by cytochrome f, subunit IV, and cytochrome b6, is close (8 Å) to the p-side heme b, but distant (19 Å) from the [2Fe-2S] cluster. No significant electron density associated with the DBMIB was found elsewhere in the structure. However, the site at which DBMIB can inhibit light-induced redox turnover is within a few Å of the [2Fe-2S] cluster, as shown by the absence of inhibition in mutants of Synechococcus sp. PCC 7002 at iron sulfur protein-Leu-111 near the cluster. The ability of a minimum amount of initially oxidized DBMIB to inhibit turnover of WT complex after a second light flash implies that there is a light-activated movement of DBMIB from the distal peripheral site to an inhibitory site proximal to the [2Fe-2S] cluster. Together with the necessary passage of quinone/quinol through the small Qp portal in the complex, it is seen that transmembrane traffic of quinone-like molecules through the core of cytochrome bc complexes can be labyrinthine.
Keywords: photosynthetic membrane proteins, energy transduction, transmembrane traffic
The cytochrome b6 f complex together with the two reaction center complexes of oxygenic photosynthesis sustain electron transfer and generation of a proton electrochemical potential gradient across thylakoid membranes (1). As in the cytochrome bc1 complex of mitochondria and purple photosynthetic bacteria, the transmembrane proton transfer functions of the b6 f complex are accomplished by oxidization and reduction of lipophilic quinol and quinone, respectively, on the p-(electrochemically positive) and n-(electrochemically negative) sides of the complex (Fig. 1A). X-ray structures of the dimeric b6 f complex have been obtained from the thermophilic cyanobacterium Mastigocladus laminosus (2) in the native state (3.4 Å), in the presence of the p-side quinone analogue inhibitor tridecyl-stigmatellin (TDS) [3.0 Å; Protein Data Bank (PDB) ID code 1VF5] or 2,5-dibromo-3-methyl-6-isopropyl-p-benzoquinone (DBMIB) (3.8 Å; PDB ID code 2D2C), and from the green alga, Chlamydomonas reinhardtii, cocrystallized with TDS (3.1 Å; PDB ID code 1Q90) (3). These structures define binding sites of quinol analogue inhibitors that could be compared with data on the binding sites of p-side inhibitors obtained from x-ray structure analysis of the mitochondrial bc1 complex (4–11).
Fig. 1.

Structure model of the cytochrome b6f complex with bound DBMIB. (A) Model (3.8 Å) of the dimeric b6f complex from M. laminosus with bound DBMIB. To emphasize the prosthetic groups and electron transfer pathways, (Left) all polypeptide components are shown in one monomer with α-helices viewed as cylinders, whereas (Right) only DBMIB and the prosthetic groups are shown in the other monomer. The electron density of DBMIB is shown in yellow with a contour level of 6 σ. The electron transfer pathways (see text for details) in one monomer (Right) are shown schematically. 2Fe2S, iron-sulfur cluster; blue arrows, electron transfer paths; bp, heme bp; bn, heme bn; β-Car, β-carotene; Chl a, chlorophyll a; e–, electron; H+, proton; Q, quinone; QH2, quinol; Qn, n-side quinone binding site (reduction site); Qp, p-side quinone binding site (oxidation site); x, heme x. For clarity, endogenous PQ molecules bound at the Qn site are not shown. (B)(Upper) Chemical structure of DBMIB and (Lower) cross-sectional view of its binding site and position relative to the Qp pocket. To show the position of the Qp pocket, a ring-in TDS molecule (yellow) from the C. reinhardtii structure is superimposed on the structure. The black arrows indicate the putative pathway for the entry of DBMIB to the Qp pocket from the membrane lipid phase. Color code is the same as in A. All protein structural figures were drawn with pymol (http://pymol.sourceforge.net).
The cytochrome b6 f complex is a 220-kDa homodimeric complex composed of eight subunits with 13 transmembrane helices per monomer, two p-side extra-membrane soluble domains, and seven prosthetic groups (Fig. 1 A). The cytochrome b6 subunit contains four transmembrane helices, two b-type hemes (bp and bn), located near the n- and p-sides of the membrane, and a unique covalently bound “heme x” (2), also called ci (3). Subunit IV, with three transmembrane helices, participates in the binding of the single chlorophyll a and β-carotene molecule present in each monomer and in the binding of plastoquinone (PQ). Cytochrome b6 and subunit IV form the core of the dimeric complex. The 3D structure of cytochrome b6 is almost identical to that of the N-terminal half of the cytochrome b subunit in the bc1 complex, and subunit IV is similar to the C-terminal half of the cytochrome b, as predicted by homology of sequences and hydropathy plots (12). Four small hydrophobic subunits (molecular mass = 3.3–4.3 kDa), Pet-G, Pet-L, Pet-M, and Pet-N, each having a single transmembrane helix, form a peripheral “picket fence” around the core. The cytochrome f and iron-sulfur protein (ISP) subunits form a large extra-membrane p-side soluble domain that contains a c-type heme and [2Fe-2S] cluster and are embedded in the membrane by a single C- or N-terminal transmembrane helix, respectively. These two subunits are functionally analogous to cytochrome c1 and the ISP subunit in the cytochrome bc1 complex (13–15).
The proton-motive pathway of the linear electron transfer through the b6 f complex (Fig. 1 A) is believed to be similar in many aspects to that catalyzed by the bc1 complex (14–20). Plastoquinol is oxidized in a bifurcated manner at the Qp site of the b6 f complex. One electron is transferred to the high potential electron transfer chain that consists of the [2Fe-2S] cluster and cytochrome f. Two protons are thereby discharged into the p-side water phase, and the other electron is forced to flow through the low-potential components (hemes bp, bn, and presumably heme x) to reduce PQ on the n-side of the membrane.
Crystallographic analysis of the bc1 and b6 f complexes has defined a large p-side quinone-binding (Qp) space (2–11), in which most of the reactive head groups of these inhibitors reside in a pocket (Qp) and the tails, if present, extend into a narrow portal connecting the pocket to the large intermonomer quinone-exchange cavity. The Qp space is portrayed in Fig. 6, which is published as supporting information on the PNAS web site. The Qp site quinol analogue inhibitors block binding of the physiological quinol at the Qp site and thereby inhibit p-side charge transfer, with the exception that the “ring-out” binding mode of TDS inhibits inefficiently (21). The Qp pocket lies between heme bp and the [2Fe-2S] cluster and is surrounded by the tilted p-side half of the C-transmembrane helix, the N-terminal half of the cd1 helix of cytochrome b6, and the C-terminal end of the ef loop of subunit IV including the Pro-Glu-Trp-Tyr (PEWY) motif and the small ef helix.
DBMIB is an effective p-side inhibitor of the b6 f complex (22). As with other quinone analogue inhibitors, DBMIB is expected to inhibit electron transfer by displacing plastoquinol binding at the Qp site (23–26). However, x-ray crystallographic analysis at a resolution of 3.8 Å of the b6 f complex from M. laminosus, cocrystallized with DBMIB, indicated that this inhibitor is bound in a peripheral site that is outside the Qp pocket and far (19 Å) from the ISP [2Fe-2S] cluster. In the present study, we present structure data for the b6 f–DBMIB complex and investigate whether a DBMIB molecule bound at the peripheral site can function as an effective inhibitor at the [2Fe-2S] cluster.
Materials and Methods
Crystallographic Analysis. Crystallization and structure determination of the M. laminosus b6 f complex was carried out as described (2, 27). For cocrystallization, DBMIB was added at a concentration (0.5 mM) five times that of cytochrome f in the complex, and the insoluble fraction of DBMIB was removed by centrifugation. Two crystal structures at 3.4- and 3.8-Å resolution were originally obtained (2), which were derived from data collected by an image plate and charge-coupled device-based detector, respectively. The bound DBMIB molecule was found in the same position in both crystals, and we considered that the two structures contained similar information. However, it was subsequently found that the 3.8-Å structure suffered less radiation damage, presumably because of a shorter time taken for data collection. Thus, the 3.8-Å data have slightly better crystallographic statistics compared with those obtained at 3.4 Å and are used in the present study. The refinement statistics are summarized in Table 1.
Table 1. Refinement statistics.
| X-ray source | APS-19ID |
|---|---|
| Wavelength, Å | 1.0 |
| Cell constants, P61 | |
| a, b, Å | 156.6 |
| c Å | 361.8 |
| Resolution range, Å | 25-3.8 |
| Completeness, % | 93.1 (79.2) |
| Rcryst | 0.276 |
| Rfree | 0.378 |
| RMSD bonds, Å | 0.012 |
| RMSD angles, ° | 1.9 |
| Mean B values, Å2 | 92.6 |
Values in parentheses apply to the highest-resolution shell. Rcryst and Rfree were calculated from the working and test reflection sets, respectively; the test set comprised 3% of the total reflections not used in refinement. RMSD, rms deviation.
Construction of Mutant Strains and Growth of Cultures. Mutant strains, WTS, ISP-L111A, and ISP-L111Y, of Synechococcus sp. PCC 7002 were constructed and grown in medium A (28) as described (21, 29).
Flash Kinetic Spectroscopy. Flash-induced oxidation and rereduction of cytochrome f/c6 in intact cells of Synechococcus sp. PCC 7002, resuspended at 5 μM chlorophyll in 50 mM Hepes (pH 7.5), 0.1 M NaCl, 10 mM NaHCO3, 10 μM 3-(3′,4′-dichlorophenyl)-1,1-dimethylurea, 10 μM carbonyl cyanide p-trifluoromethoxy-phenylhydrazone, and 1 mM KCN, were monitored in the α-band absorbance region (ΔA556–540nm), using a laboratory-built single-beam spectrometer with an Xe flash actinic source, as described (21, 29, 30). Flash-induced redox changes in cytochrome f in spinach thylakoids were measured under conditions as described in ref. 31 or the figure legends.
EPR Spectroscopy. EPR spectroscopy was performed with a Bruker (Billerica, MA) ESP300 E spectrometer operating at X-band. Instrument parameters were: microwave frequency, 9.4 GHz; temperature, 20 K; microwave power, 5 mW; modulation amplitude, 12.8 G; modulation frequency, 100 kHz; conversion time, 82 ms; time constant, 328 ms; and receiver gain, 1 × 105. Suspensions of the isolated M. laminosus cytochrome b6 f complex (10 μM) were prepared in 70 mM Tris·HCl (pH 7.5), 50 mM NaCl, 50 mM MgCl2, 1 mM EDTA, 0.05% β-d-undecylmaltoside, and 0.1 g/liter dioleolyl-phosphatidylcholine.
Results
Properties of the DBMIB Binding Sites. The overall structure of the M. laminosus b6 f complex cocrystallized with oxidized DBMIB at 3.8-Å resolution is indistinguishable from the structures of the native and TDS–b6 f complex at 3.4 and 3.0 Å. In the difference-Fourier map calculated with the |FDBMIB| – |FNative| coefficient derived from DBMIB–b6 f minus native data and phases from multiple isomorphous replacement, the DBMIB molecule in the M. laminosus b6 f complex (Fig. 1 A) was identified as a circular electron density 5 Å in diameter at contour level of 3 σ and 3.5 Å at 6-σ cut-off. However, this DBMIB binding site is located at an unexpected peripheral site, which is outside the Qp pocket defined by the existing set of Qp-analogue inhibitors (Fig. 6 and Fig. 7, which is published as supporting information on the PNAS web site) and 19 Å from the ISP [2Fe-2S] cluster. In contrast, no such significant electron density (>3 σ) was detected near the [2Fe-2S] cluster where DBMIB is expected to bind and exert its inhibitory effect on photosynthetic electron transport (23–26). This peripheral DBMIB binding site is in a p-side water/membrane interfacial niche, which is accessible from both water and membrane lipid phases (Fig. 1B) and bounded by cytochrome f and the subunit IV ef loop (Fig. 2). This DBMIB binding site is surrounded by residues 61–64 (Met-Val-Gly-Glu) and Leu-76 of the ef loop in subunit IV, Asp-141 and Val-143 at the N-terminal end of the cd1 helix in cytochrome b6, and residues Lys-146, Ala-148, and His-150 of cytochrome f (Fig. 2). The position of the [2Fe-2S] cluster in the M. laminosus structures is shifted outward in the p-side direction by ∼4 Å compared with that in the TDS-C. reinhardtti structure. Because the ISP is not involved in crystal contacts, the lack of interaction between ISP and DBMIB in the crystal structure is unlikely to be a crystallization artifact.
Fig. 2.

Ribbon diagram (stereoview) of the peripheral DBMIB binding environment and its position relative to the [2Fe-2S] cluster and heme bp. As in Fig. 1B, a ring-in TDS molecule (yellow) from C. reinhardtii structure is superimposed on the structure. Residues around the peripheral DBMIB binding site or located between the DBMIB binding niche and the Qp pocket are shown as sticks (see text for details). Color code is the same as in Fig. 1. The black arrow shows the putative pathway for the movement of DBMIB to the Qp pocket.
As inferred from EPR studies of the [2Fe-2S] cluster in spinach b6 f complex, there are two DBMIB binding sites within the complex: one high-affinity site that can be occupied by a stoichiometric amount of DBMIB and exerts a small effect on the EPR signal and one weak site at which excess DBMIB can bind and cause a major shift (from 1.89 to 1.94) of the gy EPR signal (26, 32). A similar result was obtained in the present studies in which addition of one oxidized DBMIB per monomer of M. laminosus b6 f had little effect on the amplitude or position of the gy = 1.90 or shifted 1.94 EPR signals (Fig. 3). However, higher concentrations of DBMIB (>2 molecules per monomer), which allow binding to a lower-affinity site close to the [2Fe-2S] cluster, did perturb the signal. An additional feature of these spectra is the appearance of a g = 1.97 signal that was not seen in spectra of the spinach complex. Therefore, consistent with the EPR results, the observed peripheral binding site in the M. laminosus structure must be a “high-affinity site” for oxidized DBMIB as it is the only site seen in the structure and located distal from the iron-sulfur cluster. Because of the requirement of a high concentration of the b6 f complex during crystallization and the limited solubility of DBMIB in water, a high ratio of DBMIB/b6 f and thereby a detectable DBMIB binding near the iron-sulfur cluster was not found in the crystal structure.
Fig. 3.

Dependence of EPR spectrum from Rieske [2Fe-2S] in M. laminosus b6f complex on DBMIB concentration. Spectra were measured in the absence (black) or the presence of one (red), two (green), or five (blue) molecules of DBMIB per monomer. See Materials and Methods for experimental details.
Effect of DBMIB on Flash-Induced Turnover of the b6f Complex. The inhibitory effect of DBMIB on the electron transfer activity of the b6 f complex was measured in intact cells of the closely related and genetically transformable cyanobacterium, Synechococcus sp. PCC 7002, using the rate of reduction of cytochromes f and c6 after exposure to a light flash (21, 29) (Fig. 4A). A short saturating actinic flash triggered oxidation of photosystem I and subsequent oxidation of cytochromes f and c6 in 100–200 μs. In the absence of inhibitor, oxidation was followed by a reduction in several ms (t1/2 = 3–10 ms) of cytochromes f and c6. In the presence of DBMIB, a “turnover-enhanced inhibition” of the cytochrome f and c6 reduction was observed. Reduction of cytochrome f/c6 after the first flash was relatively insensitive to DBMIB, although with high concentrations of DBMIB (>2 μM), it was almost fully inhibited (Fig. 4A). However, reduction of cytochrome f/c6 after the second flash (dark time between flashes, 100 ms), was >2-fold more sensitive to DBMIB. Further increase in the flash number could only slightly enhance the inhibition (data not shown). This turnover-enhanced inhibition has not been observed with any other inhibitors, e.g., stigmatellin, TDS, dinitrophenylether of iodonitrothymol (DNP-INT), 5-n-heptyl/undecyl-6-hydroxy-4,7-dioxobenzothiazole (UHDBT), 2-nonyl-4-hydroxyquinoline N-oxide (NQNO), and methoxyacrylate (MOA)-stilbene, of the b6 f complex in Synechococcus sp. PCC 7002.
Fig. 4.

Effect of DBMIB on the multiple flash-induced oxidation and reduction of cytochrome f/c6 in Synechococcus sp. PCC 7002 (cells suspended at 5 μM chlorophyll, A and C–E) and cytochrome f in spinach thylakoids (suspended at 40 μM chlorophyll with 50 μM duroquinol, B). (A) WTS of Synechococcus without DBMIB (black) or with 0.1 μM DBMIB (red), 0.3 μM DBMIB (green), 0.5 μM DBMIB (blue), 0.8 μM DBMIB (yellow), or 2 μM DBMIB (magenta). (B) Spinach thylakoids without DBMIB (black) or with 50 nM DBMIB (red) or 100 nM DBMIB (green). (C) WTS of Synechococcus with 0.75 μM DBMIB under oxidizing conditions poised by the addition of 5 μM benzoquinone. (D and E) Mutants of ISP-L111A (D) and ISP-L111Y (E) of Synechococcus, without DBMIB (black) or with 2 μM DBMIB (red) or 50 μM DBMIB (green). The interval between two successive flashes is 100 ms for all measurements in A and C–E and 80 ms for B.
Similar measurements were made with spinach thylakoids. As previously observed in pea thylakoids (33), a small, but progressive, increase in the inhibition of the cytochrome f re-reduction by DBMIB could be observed under multiple flash conditions in the presence of 50 or 100 nM DBMIB (≈0.6 or 1.2 DBMIB per monomer), which gave 73% or 92% inhibition in the rate of cytochrome f reduction after 11 flash-induced turnovers of the complex (Fig. 4B). The requirement of multiple flashes to elicit inhibition by DBMIB implies a relatively inefficient reaction, presumably because DBMIB was reduced to the noninhibitory form, DBMIBH2, by exogenously added duroquinol (≥50 μM) (33). A more efficient light-activated inhibition by DBMIB was seen in intact Synechococcus cells, in the absence of added reductant, at any of the DBMIB concentrations that were used (Fig. 4A). A similar result was obtained under oxidizing conditions in the presence of an excess (7-fold) of benzoquinone (5 μM, Em = +0.28 V), through which the DBMIB (0.75 μM, Em =+0.18 V), added at a low concentration, was kept oxidized (Fig. 4C). Under these oxidizing conditions, the light-activated inhibition by DBMIB cannot result from photo oxidation of reduced DBMIB, as previously proposed to explain the flash-dependent inhibition obtained in the presence of DBMIB and an excess of duroquinol (33).
The DBMIB Binding Site Close to the [2Fe-2S] Cluster Is Responsible for Inhibition. Mutation of the conserved ISP residue Leu-111 (Fig. 2) of the ISP, which is involved in the formation of the Qp site in both b6 f and bc1 complexes (21, 34, 35), to Ala or Tyr led to a complete or an almost complete loss of sensitivity, on both first and second flashes, of the b6 f complex to DBMIB (Fig. 4 D and E). Thus, both the “dark” and “light-activated” DBMIB inhibition must be associated with an inhibitor binding site that is close to the [2Fe-2S] cluster and perturbs its EPR spectrum (Fig. 3 and refs. 25, 26, 32, and 36). This site is a lower-affinity site for binding of oxidized DBMIB, as it was not associated with significant density in the electron density map.
Discussion
Light-Induced Transfer of the Tightly Bound DBMIB Molecule: A Hypothesis. The peripheral high-affinity binding site of the oxidized quinone analogue inhibitor, DBMIB, defined by x-ray structure analysis (Figs. 1 and 2) to be 19 Å from the iron-sulfur cluster, seems anomalous in terms of a possible function as an inhibitor. DBMIB is well outside the domain occupied by the set of distally located quinone analogue inhibitors found in structure analysis of the bc1 complex (Fig. 6). DBMIB bound at this peripheral site does not interact with the [2Fe-2S] cluster (Fig. 3), and the site is not inhibitory (Fig. 4 D and E). The question is whether the peripheral site is a dead end for DBMIB binding or whether DBMIB can be transferred in the light to an inhibitory site close to the ISP [2Fe-2S]. The present data argue for the latter. After the first flash-induced turnover of the complex, the oxidized form of DBMIB initially bound at its peripheral high-affinity site could undergo a guided diffusion to a site near the [2Fe-2S] cluster where it would exert its inhibitory effect for the second flash-induced turnover. The data supporting this inference are: (i) the x-ray structure shows only one DBMIB binding site, the peripheral site; (ii) one molecule/monomer added to the spinach (26) or M. laminosus (Fig. 3) b6 f complex did not perturb the gy EPR signal of the [2Fe-2S] cluster; (iii) one molecule of DBMIB/monomer can inhibit electron transfer, but only after activation by light (Fig. 4B and refs. 24 and 33); and (iv) the light-activated inhibition is more efficient in the absence of exogenous reductant or under oxidizing condition (Fig. 4 A and C vs. B).
The movement of the DBMIB inhibitor through the complex is of interest because the pathway and mechanism by which quinone analogue inhibitors are translocated across b6 f or bc1 complexes is not understood. This problem is made clear by the steric barrier involved in the passage of a long chain (45–50 C) PQ or ubiquinone from the intermonomer lipid-filled exchange cavity through the small 11 × 12-Å portal into the Qp pocket and the guided diffusion of quinone analogue inhibitors into the quinone binding pocket (2, 21).
It was previously proposed that DBMIB may act like a class II inhibitor (e.g., myxothiazol) and bind at a site within the Qp pocket, which is proximal to the heme bp and accessible to DBMIB only after a catalytic cycle of the complex, when PQ is released (37). This model does not apply to the present case for DBMIB, because interaction with the ISP [2Fe-2S] cluster binding region is required for the inhibitory effect of the DBMIB (Fig. 4 D and E). As shown in the bc1 complex, the ISP is not required for the binding of a class II type inhibitor, myxothiazol (38). An alternative interpretation of the light-induced progressive increase in inhibition by DBMIB in chloroplasts of higher plants, when obtained in the presence of an excess of duroquinol, which would reduce DBMIB, is that the effect of light is to oxidize noninhibitory DBMIBH2 to a semiquinone or oxidized form that can bind and inhibit (33). We do not argue against occurrence of the latter mechanism for light-induced inhibition by DBMIB. However, the simplest interpretation of the appearance of inhibition after the second flash obtained with intact cells of Synechococcus (either no reductant added or under oxidizing conditions) (Fig. 4 A and C) is that inhibition results from light-driven movement of DBMIB from its peripheral site presumably after photo reduction to a semiquinone to the [2Fe-2S] cluster.
The peripheral DBMIB binding site seen in the M. laminosus structure is close to heme bp (edge to edge, ≈8 Å) (Figs. 2 and 5). Thus, rapid electron transfer (t1/2 < 1 μs) from heme bp (Em7 =–50 to –150 mV) (39, 40) to the DBMIB 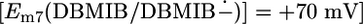 (41) bound at this peripheral site (Fig. 5) is feasible (42, 43). This electron transfer may account for the observation that the amplitude of the flash-induced cytochrome b6 reduction and the slow electrochromic phase were decreased by DBMIB in spinach thylakoids (31) and C. reinhardtii (37). It was suggested from EPR studies that the semiquinone form of DBMIB was stabilized at the Qp site by formation of a complex with the reduced [2Fe-2S] cluster (25, 36), and its binding affinity was significantly higher than that of the fully oxidized form in the spinach b6 f complex (25). It is proposed here that reduction of DBMIB to the semiquinone form by heme bp leads to a switch of the higher-affinity site for DBMIB binding from the peripheral site to the Qp pocket, where it can form a hydrogen bond with the His-129 ligand of the [2Fe-2S] cluster. The dissociation of DBMIB from the peripheral binding site might also be facilitated by electrostatic repulsion between the negatively charged DBMIB semiquinone and the overall negative binding environment that arises from the acidic residues, Asp-141 of cytochrome b6, Glu-64 and Glu-74 of subunit IV, and Asp-243, Asp-244, and Glu-246 of cytochrome f that surround this site.
(41) bound at this peripheral site (Fig. 5) is feasible (42, 43). This electron transfer may account for the observation that the amplitude of the flash-induced cytochrome b6 reduction and the slow electrochromic phase were decreased by DBMIB in spinach thylakoids (31) and C. reinhardtii (37). It was suggested from EPR studies that the semiquinone form of DBMIB was stabilized at the Qp site by formation of a complex with the reduced [2Fe-2S] cluster (25, 36), and its binding affinity was significantly higher than that of the fully oxidized form in the spinach b6 f complex (25). It is proposed here that reduction of DBMIB to the semiquinone form by heme bp leads to a switch of the higher-affinity site for DBMIB binding from the peripheral site to the Qp pocket, where it can form a hydrogen bond with the His-129 ligand of the [2Fe-2S] cluster. The dissociation of DBMIB from the peripheral binding site might also be facilitated by electrostatic repulsion between the negatively charged DBMIB semiquinone and the overall negative binding environment that arises from the acidic residues, Asp-141 of cytochrome b6, Glu-64 and Glu-74 of subunit IV, and Asp-243, Asp-244, and Glu-246 of cytochrome f that surround this site.
Fig. 5.

Schematic diagram of the DBMIB-transfer model. The black arrows show the direction of the electron transfer, and the red dashed arrow shows the direction for the movement of DBMIB (red sticks) to the Qp pocket. Green dotted lines indicate the peripheral DBMIB binding site and the proposed channel to the Qp pocket.
Based on the above considerations, a DBMIB semiquinone transfer model (Fig. 5) is proposed to explain the turnover-enhanced inhibition (Fig. 4 A–C). In dark-adapted cells, DBMIB binds preferentially at the peripheral noninhibitory site. During the first turnover of the complex, electron transfer is only slightly inhibited when the amount of DBMIB is insufficient for significant binding at the [2Fe-2S] site, and the DBMIB semiquinone is generated at the peripheral binding site through reduction by heme bp. The DBMIB semiquinone has a higher binding affinity at the reduced [2Fe-2S] cluster in the Qp pocket, which leads to transfer of the DBMIB semiquinone through a channel to the Qp pocket where it inhibits further turnover of the complex. A possible channel that connects the peripheral DBMIB binding niche to the Qp pocket is identified in the structure (Fig. 1B). This channel is formed by residues 142–144 (QVG) and 146(W) at the N-terminal part of the small cd1 helix in cytochrome b6 and residues 72–76 (PLEIL) of the ef loop in subunit IV (Fig. 2).
Pathway of Transfer Across the Membrane. DBMIB is a smaller and less hydrophobic molecule than PQ and other p-side quinone analogue inhibitors. The peripheral DBMIB site and the channel to the Qp pocket can form an alternative pathway for the transport of DBMIB from membrane lipid to the Qp pocket (Figs. 1B and 5). The observed DBMIB molecule bound at the peripheral binding site then shows the inhibitor trapped during its movement to the Qp pocket. This pathway may not be used by the larger, more hydrophobic PQ molecule. It is, nevertheless, important to understand the passage of DBMIB through the core of the complex, because DBMIB is a frequently used p-side inhibitor, and to understand the movement of nonpolar molecules through the complicated, labyrinthine internal space of an integral membrane protein complex. Together with the binding site of PQ on the n-side of the complex (2), and ring-in, ring-out orientations of TDS seen in the structures from C. reinhardtii (3) and M. laminosus (2, 21), the peripheral high-affinity binding site of DBMIB contributes to a total of four potential sites for quinone/quinol binding that provide markers for their transfer through the b6 f complex.
Supplementary Material
Acknowledgments
We thank H. Zhang for helpful discussions. This study was supported by National Institutes of Health Grant GM-38323 (to W.A.C.) and a Fellowship from the Japanese Ministry of Science and Education (to G.K.). The x-ray structure analysis, on which this study is based, was carried out on beamline SBC-19ID at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL, which is supported by U.S. Department of Energy Grant W31-109-ENG-389, and at Spring-8 BL44XU (Hyogo, Japan).
Author contributions: J.Y., G.K. and W.A.C. designed research, performed research, analyzed data, and wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DBMIB, 2,5-dibromo-3-methyl-6-isopropyl-p-benzoquinone; ISP, iron-sulfur protein; n-, electrochemically negative; p-, electrochemically positive; PQ, plastoquinone; TDS, tridecyl-stigmatellin.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2D2C).
References
- 1.Blankenship, R. E. (2002) Molecular Mechanisms of Photosynthesis (Blackwell, Malden, MA), pp. 124–156.
- 2.Kurisu, G., Zhang, H., Smith, J. L. & Cramer, W. A. (2003) Science 302, 1009–1014. [DOI] [PubMed] [Google Scholar]
- 3.Stroebel, D., Choquet, Y., Popot, J. L. & Picot, D. (2003) Nature 426, 413–418. [DOI] [PubMed] [Google Scholar]
- 4.Xia, D., Yu, C. A., Kim, H., Xia, J. Z., Kachurin, A. M., Zhang, L., Yu, L. & Deisenhofer, J. (1997) Science 277, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang, Z., Huang, L., Shulmeister, V. M., Chi, Y. I., Kim, K. K., Hung, L. W., Crofts, A. R., Berry, E. A. & Kim, S. H. (1998) Nature 392, 677–684. [DOI] [PubMed] [Google Scholar]
- 6.Kim, H., Xia, D., Yu, C. A., Xia, J. Z., Kachurin, A. M., Zhang, L., Yu, L. & Deisenhofer, J. (1998) Proc. Natl. Acad. Sci. USA 95, 8026–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunte, C., Koepke, J., Lange, C., Rossmanith, T. & Michel, H. (2000) Structure (London) 8, 669–684. [DOI] [PubMed] [Google Scholar]
- 8.Gao, X., Wen, X., Yu, C., Esser, L., Tsao, S., Quinn, B., Zhang, L., Yu, L. & Xia, D. (2002) Biochemistry 41, 11692–11702. [DOI] [PubMed] [Google Scholar]
- 9.Gao, X., Wen, X., Esser, L., Quinn, B., Yu, L., Yu, C. A. & Xia, D. (2003) Biochemistry 42, 9067–9080. [DOI] [PubMed] [Google Scholar]
- 10.Palsdottir, H., Lojero, C. G., Trumpower, B. L. & Hunte, C. (2003) J. Biol. Chem. 278, 31303–31311. [DOI] [PubMed] [Google Scholar]
- 11.Esser, L., Quinn, B., Li, Y. F., Zhang, M., Elberry, M., Yu, L., Yu, C. A. & Xia, D. (2004) J. Mol. Biol. 341, 281–302. [DOI] [PubMed] [Google Scholar]
- 12.Widger, W. R., Cramer, W. A., Herrmann, R. G. & Trebst, A. (1984) Proc. Natl. Acad. Sci. USA 81, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith, J. L., Zhang, H., Yan, J., Kurisu, G. & Cramer, W. A. (2004) Curr. Opin. Struct. Biol. 14, 432–439. [DOI] [PubMed] [Google Scholar]
- 14.Berry, E. A., Guergova-Kuras, M., Huang, L. S. & Crofts, A. R. (2000) Annu. Rev. Biochem. 69, 1005–1075. [DOI] [PubMed] [Google Scholar]
- 15.Cramer, W. A., Zhang, H., Yan, J., Kurisu, G. & Smith, J. L. (2004) Biochemistry 43, 5921–5929. [DOI] [PubMed] [Google Scholar]
- 16.Joliot, P. & Joliot, A. (1986) Biochim. Biophys. Acta 849, 211–222. [Google Scholar]
- 17.Joliot, P. & Joliot, A. (1994) Proc. Natl. Acad. Sci. USA 91, 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kallas, T. (1994) in The Molecular Biology of Cyanobacteria, ed. Bryant, D. A. (Kluwer, Dordrecht, The Netherlands), pp. 259–317.
- 19.Crofts, A. R., Shinkarev, V. P., Kolling, D. R. & Hong, S. (2003) J. Biol. Chem. 278, 36191–36201. [DOI] [PubMed] [Google Scholar]
- 20.Crofts, A. R. (2004) Annu. Rev. Physiol. 66, 689–733. [DOI] [PubMed] [Google Scholar]
- 21.Yan, J. & Cramer, W. A. (2004) J. Mol. Biol. 344, 481–493. [DOI] [PubMed] [Google Scholar]
- 22.Trebst, A., Harth, E. & Draber, W. (1970) Z. Naturforsch. B 25, 1157–1159. [DOI] [PubMed] [Google Scholar]
- 23.Malkin, R. (1986) FEBS Lett. 208, 317–320. [DOI] [PubMed] [Google Scholar]
- 24.Graan, T. & Ort, D. R. (1986) Arch. Biochem. Biophys. 248, 445–451. [DOI] [PubMed] [Google Scholar]
- 25.Schoepp, B., Brugna, M., Riedel, A., Nitschke, W. & Kramer, D. M. (1999) FEBS Lett. 450, 245–250. [DOI] [PubMed] [Google Scholar]
- 26.Roberts, A. G. & Kramer, D. M. (2001) Biochemistry 40, 13407–13412. [DOI] [PubMed] [Google Scholar]
- 27.Zhang, H., Kurisu, G., Smith, J. L. & Cramer, W. A. (2003) Proc. Natl. Acad. Sci. USA 100, 5160–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buzby, J. S., Porter, R. D. & Stevens, S. E., Jr. (1985) Science 230, 805–807. [DOI] [PubMed] [Google Scholar]
- 29.Yan, J. & Cramer, W. A. (2003) J. Biol. Chem. 278, 20925–20933. [DOI] [PubMed] [Google Scholar]
- 30.Ponamarev, M. V. & Cramer, W. A. (1998) Biochemistry 37, 17199–17208. [DOI] [PubMed] [Google Scholar]
- 31.Robert, W. J. & Whitmarsh, J. (1988) Biochim. Biophys. Acta 933, 258–268. [Google Scholar]
- 32.Roberts, A. G., Bowman, M. K. & Kramer, D. M. (2004) Biochemistry 43, 7707–7716. [DOI] [PubMed] [Google Scholar]
- 33.Rich, P. R., Madgwick, S. A. & Moss, D. A. (1991) Biochim. Biophys. Acta 1058, 312–328. [Google Scholar]
- 34.Liebl, U., Sled, V., Brasseur, G., Ohnishi, T. & Daldal, F. (1997) Biochemistry 36, 11675–11684. [DOI] [PubMed] [Google Scholar]
- 35.Brasseur, G., Sled, V., Liebl, U., Ohnishi, T. & Daldal, F. (1997) Biochemistry 36, 11685–11696. [DOI] [PubMed] [Google Scholar]
- 36.Malkin, R. (1981) FEBS Lett. 131, 169–172. [Google Scholar]
- 37.Barbagallo, R. P., Finazzi, G. & Forti, G. (1999) Biochemistry 38, 12814–12821. [DOI] [PubMed] [Google Scholar]
- 38.Brandt, U., Haase, U., Schagger, H. & von Jagow, G. (1991) J. Biol. Chem. 266, 19958–19964. [PubMed] [Google Scholar]
- 39.Kramer, D. M. & Crofts, A. R. (1994) Biochim. Biophys. Acta 1184, 193–201. [Google Scholar]
- 40.Girvin, M. E. & Cramer, W. A. (1984) Biochim. Biophys. Acta 767, 29–38. [DOI] [PubMed] [Google Scholar]
- 41.Rich, P. R. & Bendall, D. S. (1980) Biochim. Biophys. Acta 592, 506–518. [DOI] [PubMed] [Google Scholar]
- 42.Page, C. C., Moser, C. C. & Dutton, P. L. (2003) Curr. Opin. Chem. Biol. 7, 551–556. [DOI] [PubMed] [Google Scholar]
- 43.Gray, H. B. & Winkler, J. R. (2005) Proc. Natl. Acad. Sci. USA 102, 3534–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}