

Abstract
Genetic host factors play a substantial role in susceptibility to and severity of malaria, which continues to cause at least one million deaths per year. Recently, members of the toll-like receptor (TLR) family have been shown to be involved in recognition of the etiologic organism Plasmodium falciparum: The glycosylphosphatidylinositol anchor induces signaling in host cells via TLR-2 and -4, whereas hemozoin-induced immune activation involves TLR-9. Binding of microbial ligands to the respective TLRs triggers the release of proinflammatory cytokines via the TLR/IL-1 receptor (TIR) domain and may contribute to the host response in malaria, including cytokine induction and fever. In a case-control study among 870 Ghanaian children, we examined the influence of TLR-2, -4, and -9 polymorphisms in susceptibility to severe malaria. TLR-2 variants common in Caucasians and Asians were completely absent. However, we found a rare previously undescribed mutation (Leu658Pro), which impairs signaling via TLR-2. We failed to detect any polymorphisms within the TLR-9 Toll/IL-1 receptor domain. Two frequent TLR-9 promoter polymorphisms did not show a clear association with malaria severity. In contrast, the TLR-4-Asp299Gly variant occurred at a high rate of 17.6% in healthy controls and was even more frequent in severe malaria patients (24.1%, P < 0.05). Likewise, TLR-4-Thr399Ile was seen in 2.4% of healthy children and in 6.2% of patients (P = 0.02). TLR-4-Asp299Gly and TLR-4-Thr399Ile conferred 1.5- and 2.6-fold increased risks of severe malaria, respectively. These findings suggest TLR4-mediated responses to malaria in vivo and TLR-4 polymorphisms to be associated with disease manifestation.
Keywords: single-nucleotide polymorphisms, innate immunity, Plasmodium falciparum
Plasmodium falciparum malaria affects 300–500 million people annually, but only a minority of patients (1–2%) proceeds to severe and complicated disease (1). Still, one to three million deaths occur per year, most of them among young children in sub-Saharan Africa. Why some children and other nonimmune hosts die while others remain asymptomatic or develop an uncomplicated illness is far from being understood (2). Acquired immunity has been investigated to some extent, but little is known about the role of innate immunity in malaria. In mice, the P. falciparum glycosylphosphatidylinositol (GPI) toxin induces severe malaria symptoms, which can be prevented by a preceding vaccination with GPI (3). In vitro, plasmodial GPI induces the expression of adhesion molecules (intercellular cell adhesion molecule, vascular cell adhesion molecule, E-selectin), proinflammatory cytokines (IL-1, TNF-α), and nitric oxide (4–6). Current evidence shows that innate immune recognition of Plasmodium and subsequent release of cytokines and inflammatory mediators are important for parasite clearance but may also contribute to disease severity (2).
Within the last years, the family of toll-like receptors (TLRs) has been identified as key host molecules in the induction of innate immune responses to microbial ligands (7, 8). TLR-2 (in synergy with TLR-1 and -6) and TLR-4 react to bacterial cell wall compounds (9, 10). TLR-2 is activated by a variety of ligands, such as bacterial lipopeptides, as well as fungal and mycobacterial components (reviewed in ref. 11), and TLR-4 is activated not only by bacterial lipopolysaccharide but apparently also by other ligands, such as viral proteins (12–14). In addition, both TLR-2 and -4 may respond to intrinsic mediators, such as heat-shock proteins, and may be involved in inflammatory or stress hormone reactions (11, 15–17). Regarding protozoa, TLR-2 has first been shown to recognize GPI of Trypanosoma cruzi (18). Very recently, P. falciparum GPI was reported to induce signaling via both TLR-2 and -4 and hemozoin-induced immune activation was reported to involve TLR-9 (19–21).
Frequent single-nucleotide polymorphisms (SNPs) have been described for TLR-2, -4, and -9, altering susceptibility to infectious and inflammatory diseases (reviewed in ref. 22). A TLR-2 SNP Arg753Gln within the intracellular Toll/IL-1 receptor (TIR) domain impairs TLR-2 function (23). This SNP is seen in 9–10% of Caucasians (23, 24) and has been associated with tuberculosis and asthma (25, 26). Another TLR-2 SNP (Arg677Trp) has been described to increase the risk of lepromatous leprosy (27) and tuberculosis (28), respectively. For TLR-4, two frequently cosegregating polymorphisms (Asp299Gly/Thr399Ile) were observed to reduce reactivity to inhaled lipopolysaccharide (29), although findings are partly conflicting (22, 30). Individuals exhibiting Asp299Gly and Thr399Ile SNPs are at increased risk of septic shock (31) and Gram-negative infection (32), respectively. Moreover, other rare TLR-4 mutations have been reported to increase susceptibility to meningococcal meningitis (33, 34). Last, two common TLR9 promoter polymorphisms (T-1237C and T-1486C), assumed to influence transcription regulation, have been described in African Americans, one of them (T-1237C) being potentially associated with asthma (35) and Crohn's disease (36).
Here, we hypothesized that modified recognition or signaling via variants of TLR-2, -4, and -9 could influence susceptibility to and manifestation of malaria. We thus examined known and frequent TLR SNPs leading to a change of function in a case-control study among 290 children with severe malaria and age- and sex-matched control groups of asymptomatic P. falciparum-infected children and healthy children in northern Ghana. These children were screened for known TLR SNPs, and we looked for novel TLR-2 and -9 mutations. We report that TLR-4-Asp299Gly is extraordinarily frequent in this African population and mostly not linked with Thr399Ile. Importantly, children exhibiting these TLR-4 variants have a significantly increased risk of severe malaria.
Materials and Methods
Patients. The present study was conducted between August and November, 2002, i.e., during the rainy season, in Tamale and its vicinity, Northern Region, Ghana. Tamale is the regional capital of ≈350,000 inhabitants but of rural character. Malaria is hyperendemic, and transmission occurs perennially. Tamale Teaching Hospital serves as the region's reference center. For this case-control study, 290 children with severe malaria according to the World Health Organization (2000) criteria, admitted to the hospital, were recruited, as were 290 of each age- and sex-matched asymptomatic P. falciparum-infected and healthy children. Controls were selected from a representative sample of 2,107 children from Tamale and the surrounding districts (37). Clinical characteristics of severe malaria (38) and study design (39) have been described. Venous blood was collected into EDTA, stabilized, and DNA was extracted (AS1 and QIAmp, Qiagen, Hilden, Germany). Malaria parasites were counted on Giemsa-stained thick blood films (38, 39), and P. falciparum was ascertained by specific PCR assays (40). Severe malaria patients received artesunate (Plasmotrim, Mepha, Switzerland) for 5 days, at a dose of 5 mg/kg body weight (double dose on the first day) and supportive care. Parasitemic control children were treated with sulfadoxine–pyrimethamine if parasite density exceeded 5,000/μl. The study was approved by the Ethics Committee, University for Development Studies, Tamale, and participants' parents gave informed written consent.
TLR-2, -4, and -9 Genotyping. The TLR-2 Arg677Trp and Arg753Gln variants were screened for by allele-specific PCR assays (23) and TLR4-Asp299Gly and -Thr399Ile as well as the TLR9 promoter polymorphisms T-1237C and T-1486C by real-time PCR assays (41).
Search for New TLR-2 and -9 Polymorphisms. For assessing TLR-2 polymorphisms, primers used for DNA amplification were sense 5′-CTCGGTGTCGGAATGTCACAG-3′ and antisense 5′-CTAGGACTTTATCGGAGCTCTC-3′ spanning a region of the C-terminal 616 bp including the TLR2/TIR domain (GenBank accession no. U88878). Similarly, the TLR9/TIR domain region was amplified, applying primers 5′-CATGCTGCATCACCTCTGTG-3′ and 5′-GTCAGGGCTCAGGATCACC-3′. Purified DNA was sequenced by applying the CEQ Dye Terminator Cycle Sequencing Quick Start Kit and analyzed on a CEQ 8000 Genetic Analysis System (Beckman Coulter). For investigation of functional relevance, the herein identified Leu658Pro TLR2 mutation was inserted into a plasmid for human TLR2 by using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene). Mutagenesis was performed with the primers sense 5′-TGGGTGGAGAACCCTATGGTCCAGGACC-3′ and antisense 5′-GCTCC TGGACCATAGGGTTCTCCACCCA-3′. Successful insertion was controlled by DNA sequencing. Plasmids were isolated by applying the Invisorb Spin Plasmid Mini Kit (Invitek, Berlin). Human embryonic kidney 293 cells cultured on DMEM-medium (Gibco) supplemented with 10% FCS (Biochrom, Berlin) were transfected by using FuGENE 6 Transfection Reagent (Roche Molecular Biochemicals, Loerrach, Germany). Reporter genes RSV-β-galactosidase and Elam-NF-κB-luciferase were cotransfected. Cells were stimulated with synthetic bacterial lipoprotein analogue (Pam3Cys). As a positive control, wild-type TLR2 was transfected and tested in parallel.
Statistics. Frequencies and proportions were compared by χ2 test, χ2 test for trend, or Fisher exact test. Matched-pair analysis was done by the McNemar test, and odds ratios (ORs) and 95% confidence intervals (CIs) were calculated. A P value <0.05 was considered statistically significant.
Results
The baseline characteristics of the 290 index patients with severe malaria are shown in Table 1. Severe anemia (hemoglobin <5 g/dl), prostration, and respiratory distress were the most common symptoms defining severe malaria. Impaired consciousness was comparatively rare. Of 285 severe malaria patients who could be followed, 11% died. In parasitemic control children, the geometric mean parasite density was 1,732/μl (95% CI, 1,385–2,166/μl). None of these children or of the healthy controls was febrile or exhibited clinical symptoms.
Table 1. Baseline characteristics of 290 children with severe malaria.
| Parameter | Value |
|---|---|
| Age, months (median, range) | 24 (6-102) |
| Female/male | 155:135 |
| Axillary temperature,* C (mean ± SD) | 38.6 ± 1.1 |
| Geometric mean parasite density (/μl; 95% CI) | 29,512 (21,904-39,763) |
| Hemoglobin, g/dl (median, range) | 4.9 (1.5-13.4) |
| Proportion with criterion defining severe | |
| malaria, % (no.) | |
| Severe anemia | 55.2 (160) |
| Prostration | 33.4 (97) |
| Respiratory distress | 22.8 (66) |
| Multiple convulsions | 20.3 (59) |
| Impaired consciousness | 19.3 (56) |
| Jaundice | 11.7 (34) |
| Circulatory collapse | 3.4 (10) |
| Hemoglobinuria | 2.8 (8) |
| Pulmonary edema | 0 (0) |
| Abnormal bleeding | 0 (0) |
| Proportion with other conditions, % (no.) | |
| Hyperparasitaemia (>250,000/μl) | 22.1 (64) |
| Hypoglycemia (glucose < 40 mg/dl) | 16.9 (49) |
| Hyperlactatemia (lactate ≥ 5 mmol/l) | 39.3 (114) |
| Hyperpyrexia* (>40°C) | 8.1 (23) |
| Malnutrition (weight-for-age z score < -2) | 42.4 (123) |
| Case fatality rate, % (no.)† | 11.2 (32) |
, n = 283; †, n = 285.
Because TLR-2 is the TLR most likely responsible for initiating innate immune responses to P. falciparum, we first screened for the common TLR-2 polymorphisms Arg753Gln and Arg677Trp. The former occurs at a frequency of ≈9–10% in Caucasians (23, 24), whereas the latter has been found in Asians (27) and in a North African population (28). To our surprise, both SNPs were completely absent in all cases and controls (data not shown). We next searched for new polymorphisms within the TIR domain of TLR-2 and sequenced this region in 100 randomly selected samples of each, severe malaria patients, and asymptomatically infected and healthy controls. In one child with severe malaria, we found a rare previously undescribed C to A mutation at base pair 2042 exchanging leucine against proline at residue 658. This mutation is located in a highly conserved region of the TLR-2 gene near the known SNP Arg677Trp (Fig. 1). In a functional analysis, we overexpressed TLR-2 containing this Leu658Pro mutation in human embryonic kidney 293 cells and stimulated these with Pam3Cys, a known TLR-2 ligand. Overexpression of the mutated TLR-2 failed to enable the cells for stimulation, supporting the crucial functional role of this region of TLR-2 (Fig. 2).
Fig. 1.

SNPs within the TLR2/TIR domain. (A) Wild-type sequence with locus of the previous described SNPs Arg677Trp. (B) Heterozygous Leu658Pro mutation in one Ghanaian child.
Fig. 2.

Functional analysis of the Leu658Pro mutation within the TLR2/TIR domain. The TLR2 mutation Leu658Pro was inserted into a plasmid for human TLR2. Human embryonic kidney 293 cells were then transfected with mutated as well as wild-type TLR2. Reporter genes RSV-β-galactosidase and Elam-NF-κB-luciferase were cotransfected. Cells were stimulated with synthetic bacterial lipoprotein analogue. As negative control, wild-type TLR2 was tested with medium alone.
Two recent reports suggested that TLR-9 is involved in innate immune responses during malaria (20, 21). We thus also searched for polymorphisms within the TIR domain of TLR-9 and sequenced this region in 100 random samples of severe malaria patients and asymptomatically infected and healthy controls. No TLR-9 mutations were detected (data not shown). The TLR9 promoter variants T-1237C and T-1486C were observed in 68.4% and 53.0%, respectively, of all 870 children examined. Matched-pair analysis revealed no significant differences between cases and controls (Table 2) and no associations with particular symptoms or fatality (data not shown). Yet, the allele frequency of TLR9 T-1486C was lower in parasitemic controls than in patients or healthy children (Table 2).
Table 2. Distribution of TLR9 polymorphisms in severe malaria patients and controls.
| Controls
|
||||
|---|---|---|---|---|
| Severe malaria patients | Asymptomatic, P. falciparum-infected | Healthy | P,* patients vs. healthy controls | |
| Number | 290 | 290 | 290 | |
| Prevalence TLR9 T-1237C, % (no.) | 67.6 (196) | 70.7 (205) | 66.9 (194) | 0.86 |
| Heterozygous, % (no.) | 51.0 (148) | 49.7 (144) | 51.0 (148) | 0.92 |
| Homozygous, % (no.) | 16.6 (48) | 21.0 (61) | 15.9 (46) | 0.81 |
| Allele frequency | 0.421 | 0.459 | 0.414 | 0.81† |
| Prevalence TLR9 T-1486C, % (no.) | 55.5 (161) | 47.6 (138) | 55.9 (162) | 0.94 |
| Heterozygous, % (no.) | 43.4 (126) | 39.3 (114) | 46.2 (134) | 0.67 |
| Homozygous, % (no.) | 12.1 (35) | 8.3 (24) | 9.7 (28) | 0.43 |
| Allele frequency | 0.338 | 0.279‡ | 0.328 | 0.71† |
McNemar test.
χ2 test.
Parasitemic controls vs. severe malaria patients, P = 0.03; parasitemic vs. healthy controls, P = 0.07 (χ2 test).
Next, frequencies of the TLR-4 SNPs Asp299Gly and Thr399Ile were assessed. We found TLR4-Asp299Gly in 21.5% of all individuals studied, which is higher than in all other studies reported before. TLR4-Thr399Ile, commonly cosegregating with the Asp299Gly SNP, was found in 4.3% of the individuals only (Table 3). All children exhibiting the Thr399Ile mutation also possessed the Asp299Gly variant and in 16% (19/117) of children with the Asp299Gly variant, Thr399Ile was detected (P < 0.0001). For both mutations, the allele frequencies were highest in severe malaria patients, lower in parasitemic controls, and lowest in healthy children (Table 3; 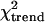 test: Asp299Gly, P = 0.06; Thr399Ile, P = 0.02). Allele and genotype frequencies did not differ significantly between healthy and parasitemic controls or between parasitemic controls and patients. However, comparing healthy children and patients, the risk of severe malaria was increased 1.5-fold (OR, 1.45; 95% CI, 1.01–2.3) in children with TLR4-Asp299Gly and 2.6-fold (OR, 2.57; 95% CI, 1.1–6.0) in children with TLR4-Thr399Ile (Table 3). Adjusting for hemoglobin AS and hemoglobin AC (39) by conditional logistic regression, these risk estimates improved slightly (Asp299Gly, OR, 1.53; 95% CI, 1.0–2.3; P = 0.049; Thr399Ile, OR, 2.78; 95% CI, 1.1–7.4; P = 0.04). In this model, ORs for severe malaria of children with TLR4-Asp299Gly alone and with both TLR4 mutations were 1.33 (95% CI, 0.8–2.1; P = 0.2) and 2.9 (1.1–7.6; P = 0.04), respectively.
test: Asp299Gly, P = 0.06; Thr399Ile, P = 0.02). Allele and genotype frequencies did not differ significantly between healthy and parasitemic controls or between parasitemic controls and patients. However, comparing healthy children and patients, the risk of severe malaria was increased 1.5-fold (OR, 1.45; 95% CI, 1.01–2.3) in children with TLR4-Asp299Gly and 2.6-fold (OR, 2.57; 95% CI, 1.1–6.0) in children with TLR4-Thr399Ile (Table 3). Adjusting for hemoglobin AS and hemoglobin AC (39) by conditional logistic regression, these risk estimates improved slightly (Asp299Gly, OR, 1.53; 95% CI, 1.0–2.3; P = 0.049; Thr399Ile, OR, 2.78; 95% CI, 1.1–7.4; P = 0.04). In this model, ORs for severe malaria of children with TLR4-Asp299Gly alone and with both TLR4 mutations were 1.33 (95% CI, 0.8–2.1; P = 0.2) and 2.9 (1.1–7.6; P = 0.04), respectively.
Table 3. Distribution of TLR4 polymorphisms in severe malaria patients and controls.
| Controls
|
||||
|---|---|---|---|---|
| Severe malaria patients | Asymptomatic, P. falciparum-infected | Healthy | P,* patients vs. healthy controls | |
| Number | 290 | 290 | 290 | |
| Prevalence TLR4 Asp299Gly, % (no.) | 24.1 (70) | 22.8 (66) | 17.6 (51) | 0.046 |
| Heterozygous, % (no.) | 22.4 (65) | 22.1 (64) | 16.2 (47) | 0.04 |
| Homozygous, % (no.) | 1.7 (5) | 0.7 (2) | 1.4 (4) | 1 |
| Allele frequency | 0.129 | 0.117 | 0.095 | 0.06† |
| Prevalence TLR4 Thr399Ile, % (no.) | 6.2 (18) | 4.1 (12) | 2.4 (7) | 0.03 |
| Heterozygous, % (no.) | 5.9 (17) | 3.8 (11) | 2.4 (7) | 0.04 |
| Homozygous, % (no.) | 0.3 (1) | 0.3 (1) | 0 | 1 |
| Allele frequency | 0.033 | 0.022 | 0.012 | 0.02† |
McNemar test.
χ2 test.
In severe malaria patients, the frequency of the TLR-4 Asp299Gly SNP did not differ with the presence of particular symptoms; e.g., in children with severe anemia and in those with impaired consciousness, this polymorphism occurred in 24.4% and 23.2%, respectively. Grouping severe malaria patients into the two predominant syndromes, i.e., severe anemia without cerebral involvement (n = 88) and cerebral involvement (impaired consciousness, prostration, and/or convulsions) without severe anemia (n = 114), these figures were 23.9% and 24.6% (P = 0.9). In these two groups, TLR-4 Thr399Ile was seen in 4.5% and 7.0% (P = 0.5). Correspondingly, in matched-pair analyses, no significant excess of any particular symptom defining severe malaria was discernible in children with TLR4 mutations. Within the group of severe malaria patients, the TLR4 polymorphisms did not correlate with hemoglobin, glucose, lactate, and parasite density (Table 4). Fatality rates were 12.9% (28/217) in TLR4 wild-type children, 8.0% (4/50) in patients with TLR4-Asp299Gly alone, and 0% (0/18) in children with both TLR4 mutations (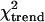 test, P = 0.07).
test, P = 0.07).
Table 4. Laboratory parameters in severe malaria patients separated according to TLR-4 polymorphisms.
|
TLR-4 codon 299
|
TLR-4 codon 399
|
|||
|---|---|---|---|---|
| Wild type (Asp) | Mutation (Gly) | Wild type (Thr) | Mutation (Ile) | |
| Number | 220 | 70 | 272 | 18 |
| Geometric mean parasite density/μl (95% Cl) | 29,242 (20,627-41,454) | 30,269 (17,060-53,706) | 29,923 (22,012-40,676) | 23,714 (6,616-84,991) |
| Hemoglobin, g/dl (median, range) | 4.9 (1.5-13.4) | 4.9 (2.2-13.4) | 4.9 (1.5-13.4) | 4.9 (2.2-10.0) |
| Lactate, mmol/l (median, range) | 4.1 (0.7-21.0) | 4.4 (1.2-16.6) | 4.3 (0.7-21.0) | 3.6 (1.3-15.9) |
| Glucose, mg/dl (median, range) | 76.4 (5-209) | 74.1 (5-168) | 75.3 (5-209) | 64.8 (28-134) |
All comparisons, P > 0.5.
Discussion
The relevance of genetic host factors in malaria has been proposed as early as 50 years ago: The Haldane hypothesis stated that β-thalassemia provided protection against malaria and consequently was selected in endemic areas (42). Strong interindividual variation in susceptibility to and manifestation of malaria is attributable to various host factors. In Sri Lanka, for instance, ≈15% of the variation in malarial infection has been explained by human genetics (43). This portion may be larger in Africa, with its substantially higher burden of potentially fatal falciparum malaria. “Classic” protective factors include the sickle cell trait (HbAS) and other hemoglobin variants (39, 44, 45), as well as glucose-6-phosphate dehydrogenase deficiency (46). Most of these, e.g., the latter, are in a state of balanced polymorphism in malaria-endemic regions (47). In Ghana, we have previously shown that HbAS, HbAC (39), and α+-thalassemia (37) protect from severe malaria. In addition to these red blood cell variants, several polymorphisms of mediators of innate immune response have been shown to protect against, or alternatively predispose to, malaria, including mannose-binding lectin, inducible NO synthase, IFN receptors, TNF-α, and CD36 (48–51).
Several lines of evidence support that the TLRs are involved in recognition of P. falciparum and in malaria pathogenesis. First, in 2001, MyD88, a central mediator of TLR- and IL-1 signaling, was found to be required for IL-12 induction in dendritic cells by Plasmodium berghei parasites (52). Subsequently, TLR-2 has been shown to recognize protozoa (18) and to be a major receptor for P. falciparum GPI (19). Immune responses brought about by GPI from Trypanozoma cruzi, the causative agent of Chagas disease, appear to be mediated by TLR-4 rather than TLR-2 (53). Recent results suggest that both TLR-2 and -4 are involved in the recognition of P. falciparum GPI (19). Moreover, activation of dendritic cells by malaria schizonts was lately demonstrated to involve TLR-9 and to be caused by hemozoin (malaria pigment), a hydrophobic heme polymer formed during the parasite's degradation of red blood cell hemoglobin (20, 21).
So far, few results, if any, have been reported on the frequencies of TLR-gene variants among sub-Saharan Africans or on associations with P. falciparum malaria in humans. Polymorphisms within the TLR-2/TIR domain have previously been associated with various infectious diseases: Although Arg677Trp is not seen in Caucasians, allele frequencies of 47% have been observed in tuberculosis patients in northern Africa (28). Arg753Gln occurs in up to 14% among European populations and inactivates TLR-2 signaling, as does an additional SNP, Pro631His (23, 24, 27). Interestingly, these frequent TLR-2 polymorphisms are completely absent in this population from a malaria-endemic region. A recently described pseudogene (54) associated with Arg677Trp questions previous data and may explain why we could not find this SNP. However, it is surprising that the tuberculosis-related Arg753Gln SNP (25) was also not present, considering the high rates of that disease in the study area (55). Yet, in a recent study in Sudan, the TLR-2 SNPs investigated here were also found to be completely absent (L.H., G. El Ghazali, H. El Turabi, I. El Khidir, and R.R.S., unpublished results). A potential disadvantage in tropical Africa, i.e., inactivated TLR2 signaling in response to P. falciparum, may outweigh a yet-to-be-defined advantage of the TLR-2 polymorphisms, explaining comparatively high frequencies in Europeans, Asians, and Northern Africans. Possibly, these TLR-2 variants are deleterious in malaria and have, therefore, been eliminated from populations in sub-Saharan Africa. The mutation found, Leu659Pro, impairs TLR-2 function in vitro but is too rare to be of epidemiological significance.
TLR-9 appears to be involved in innate immune responses to hemozoin (20, 21). In hemozoin-laden macrophages, increased TNF-α expression has been observed (56), but the pigment may also contribute to immunosuppression (57), potentially depending on the location and degree of accumulation. In our study population, we failed to discover any variants in the TLR-9 TIR domain, pointing to a lack of selection by malaria. This appears to be the case also for the TLR-9 promoter polymorphism T-1237C, considering its equal distribution among severe malaria patients and controls. The functional roles of this promoter SNP and of T-1486C are unclear to date, but modified TRL-9 expression is conceivable. T-1486C was as frequent in patients as in healthy children but less common in P. falciparum-infected asymptomatic controls. The reason for this uneven distribution is unclear; however, it argues against a major role of T-1486C in severe pediatric malaria. Possibly, T-1486C, additional SNPs, and haplotypes (35) show a more clear impact in placental malaria, characterized by excessive localized accumulation of hemozoin. Respective investigations are currently underway in our laboratory.
The findings of the present study on TLRs and malaria in humans indicate that common TLR-4 mutations in African children increase the risk of severe malaria. This supports a recent in vitro study describing that, in addition to TLR-2, TLR-4 mediates innate immune responses to P. falciparum suggesting that TLR-4 is involved in the pathophysiology of malaria (19). In analogy to bacterial infections, TLR-4 polymorphisms may reduce responsiveness to P. falciparum and to GPI in particular. Frequencies of TLR-4 variants were only slightly increased in parasitemic controls but significantly so in severe malaria patients. This argues against a major influence of the TLR-4 variants on susceptibility to P. falciparum infection but points to a critical role in disease progression.
Knowledge of the function of TLRs in malaria is still limited. TLRs are essential for initiating innate immune responses, which are important for early parasite control, but also may aggravate pathophysiology (2, 58). In mice, vaccination with GPI protects against malaria-related acidosis, pulmonary edema, and cerebral symptoms, but not against parasitemic and severe anemia (3). Consequently, deficient GPI recognition and signaling via TLRs could predispose to specific symptoms, although we did not observe this. Reduced GPI responsiveness may cause severe malaria due to both inadequate innate responses at disease onset and insufficient stimulation of specific immunity during preceding infections. In addition, because TLR-4 may also recognize intrinsic mediators such as heat-shock proteins, which are strongly expressed in severe malaria (59), a variation in this reaction pattern is conceivable and may contribute to disease manifestation. Although statistically not significant, patients with TLR-4 variants revealed a rather low fatality, which might result from less excessive proinflammatory mediators contributing to death. One hypothesis is that TLR-4 variants indeed increase the risk of becoming parasitemic and developing malaria but at the same time may prevent progression to fatal disease. In that case, the extraordinary high frequency, particularly of TLR-4 Asp299Gly, could reflect natural selection due to protection not from malaria but from death due to malaria. Larger studies should verify this assumption.
As with other genetic host factors (49), the role of TLR polymorphisms may vary with malaria endemicity, as does disease manifestation. The observed impact of TLR-4 variants on severe malaria among children from a highly endemic area thus needs to be verified in settings of differing malaria transmission and manifestation pattern. Moreover, bacteremic episodes can both complicate and mimic severe malaria (60). In some children with TLR-4 mutations, the observed increased risk of severe malaria could partially reflect an elevated susceptibility to bacterial coinfection (reviewed in ref. 22). We cannot exclude such confounding but thorough clinical examination, and the rather low fatality rate in children with TLR-4 variants argues against a major respective influence.
Our findings may also have other implications. Morbidity and mortality of malaria are increasing in parts of sub-Saharan Africa (61), and efficient control means are urgently needed. Considering signaling via TLR-4, a potential GPI-based vaccine (3) may be impaired in populations with high frequencies of TLR-4 variants. We lack conclusive arguments to explain the extraordinary high frequency of TLR-4 polymorphisms in this African population. In Sudan, a similar prevalence is observed (L.H., G. El Ghazali, H. El Turabi, I. El Khadir, and R.R.S., unpublished work), suggesting that a yet-unknown selective advantage could be involved. As mentioned, one potential benefit of the TLR-4 polymorphisms might consist in a reduced risk of fatality caused by malaria. Nevertheless, regarding vaccine strategies and TLR-2, presumably the major receptor for GPI-related signaling (19), we failed to detect any of the common polymorphisms in several hundred African individuals.
Conclusion
Previously described TLR2/TIR polymorphisms are absent and, thus, have no role in malaria susceptibility in the African population studied here. However, this does not mean that TLR-2 itself does not play an important role in initiating innate immune mechanisms in P. falciparum infection. Irrespective of the pending elucidation of the mechanisms involved, TLR4 polymorphisms, particularly the 399 SNP, predispose to severe malaria. This suggests that TLR-4 contributes to parasite recognition and host responses in vivo.
Acknowledgments
We thank the members of the Northern Region Malaria Project (NORMAP) for patient recruitment; Diana Woellner, Sabine Bobbe, and Susanne Roewer for excellent technical assistance; and Ralf Ignatius for critical comments on the manuscript. Dr. Bruce Beutler (The Scripps Research Institute, La Jolla, CA) is furthermore thanked for critical reading of the manuscript. This work forms part of the doctoral thesis of M.S.S. We acknowledge the Medical Faculty Charité, Berlin (Grant 2003-676) and the German Research Foundation (Grants MO 852/2-1 and SCHR 726/1-1) for financial support.
Author contributions: F.P.M., R.N.O., S.E., U.B., and R.R.S. designed research; F.P.M., J.P.C., L.H., M.S.S., J.E., N.-R.O., R.N.O., S.E., N.W.S., U.B., and R.R.S. performed research; J.P.C., L.H., J.E., N.-R.O., E.D., N.W.S., and R.R.S. contributed new reagents/analytic tools; F.P.M., L.H., M.S.S., R.N.O., E.D., N.W.S., U.B., and R.R.S. analyzed data; and F.P.M., J.P.C., L.H., M.S.S., J.E., N.-R.O., R.N.O., E.D., S.E., N.W.S., U.B., and R.R.S. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GPI, glycosylphosphatidylinositol; TLR, toll-like receptor; SNP, single-nucleotide polymorphism; OR, odds ratio; CI, confidence interval; TIR, Toll/IL-1 receptor.
References
- 1.Greenwood, B. & Mutabingwa, T. (2002) Nature 415, 670–672. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson, M. M. & Riley, E. M. (2004) Nat. Rev. Immunol. 4, 169–180. [DOI] [PubMed] [Google Scholar]
- 3.Schofield, L., Hewitt, M. C., Evans, K., Siomos, M. A. & Seeberger, P. H. (2002) Nature 418, 785–789. [DOI] [PubMed] [Google Scholar]
- 4.Tachado, S. D., Gerold, P., McConville, M. J., Baldwin, T., Quilici, D., Schwarz, R. T. & Schofield, L. (1996) J. Immunol. 156, 1897–1907. [PubMed] [Google Scholar]
- 5.Schofield, L., Novakovic, S., Gerold, P., Schwarz, R. T., McConville, M. J. & Tachado, S. D. (1996) J. Immunol. 156, 1886–1896. [PubMed] [Google Scholar]
- 6.Vijaykumar, M., Naik, R. S. & Gowda, D. C. (2001) J. Biol. Chem. 276, 6909–6912. [DOI] [PubMed] [Google Scholar]
- 7.Janeway, C. A., Jr., & Medzhitov, R. (2002) Annu. Rev. Immunol. 20, 197–216. [DOI] [PubMed] [Google Scholar]
- 8.Beutler, B. (2004) Mol. Immunol. 40, 845–859. [DOI] [PubMed] [Google Scholar]
- 9.Akira, S. & Takeda, K. (2004) Nat. Rev. Immunol. 4, 499–511. [DOI] [PubMed] [Google Scholar]
- 10.Beutler, B. (2004) Nature 430, 257–263. [DOI] [PubMed] [Google Scholar]
- 11.Kirschning, C. J. & Schumann, R. R. (2002) Curr. Top. Microbiol. Immunol. 270, 121–144. [DOI] [PubMed] [Google Scholar]
- 12.Beutler, B. & Rietschel, E. T. (2003) Nat. Rev. Immunol. 3, 169–176. [DOI] [PubMed] [Google Scholar]
- 13.Kurt-Jones, E. A., Popova, L., Kwinn, L., Haynes, L. M., Jones, L. P., Tripp, R. A., Walsh, E. E., Freeman, M. W., Golenbock, D. T., Anderson, L. J., et al.. (2000) Nat. Immunol. 1, 398–401. [DOI] [PubMed] [Google Scholar]
- 14.Tal, G., Mandelberg, A., Dalal, I., Cesar, K., Somekh, E., Tal, A., Oron, A., Itskovich, S., Ballin, A., Houri, S., et al. (2004) J. Infect. Dis. 189, 2057–2063. [DOI] [PubMed] [Google Scholar]
- 15.Tsan, M. F. & Gao, B. (2004) J. Leukocyte Biol. 76, 514–519. [DOI] [PubMed] [Google Scholar]
- 16.Vabulas, R. M., Ahmad-Nejad, P., da Costa, C., Miethke, T., Kirschning, C. J., Hacker, H. & Wagner, H. (2001) J. Biol. Chem. 276, 31332–31339. [DOI] [PubMed] [Google Scholar]
- 17.Bornstein, S. R., Zacharowski, P., Schumann, R. R., Barthel, A., Tran, N., Papewalis, C., Rettori, V., McCann, S. M., Schulze-Osthoff, K., Scherbaum, W. A., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 16695–16700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campos, M. A., Almeida, I. C., Takeuchi, O., Akira, S., Valente, E. P., Procopio, D. O., Travassos, L. R., Smith, J. A., Golenbock, D. T. & Gazzinelli, R. T. (2001) J. Immunol. 167, 416–423. [DOI] [PubMed] [Google Scholar]
- 19.Krishnegowda, G., Hajjar, A. M., Zhu, J., Douglass, E. J., Uematsu, S., Akira, S., Woods, A. S. & Gowda, D. C. (2005) J. Biol. Chem. 280, 8606–8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pichyangkul, S., Yongvanitchit, K., Kum-arb, U., Hemmi, H., Akira, S., Krieg, A. M., Heppner, D. G., Stewart, V. A., Hasegawa, H., Looareesuwan, S., et al. (2004) J. Immunol. 172, 4926–4933. [DOI] [PubMed] [Google Scholar]
- 21.Coban, C., Ishii, K. J., Kawai, T., Hemmi, H., Sato, S., Uematsu, S., Yamamoto, M., Takeuchi, O., Itagaki, S., Kumar, N., et al. (2005) J. Exp. Med. 201, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schröder, N. W. J. & Schumann, R. R. (2005) Lancet Infect. Dis. 5, 156–164. [DOI] [PubMed] [Google Scholar]
- 23.Schröder, N. W. J., Hermann, C., Hamann, L., Göbel, U. B., Hartung, T. & Schumann, R. R. (2003) J. Mol. Med. 81, 368–372. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz, E., Mira, J. P., Cornish, K. L., Arbour, N. C. & Schwartz, D. A. (2000) Infect. Immun. 68, 6398–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogus, A. C., Yoldas, B., Ozdemir, T., Uguz, A., Olcen, S., Keser, I., Coskun, M., Cilli, A. & Yegin, O. (2004) Eur. Respir. J. 23, 219–223. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad-Nejad, P., Mrabet-Dahbi, S., Breuer, K., Klotz, M., Werfel, T., Herz, U., Heeg, K., Neumaier, M. & Renz, H. (2004) J. Allergy. Clin. Immunol. 113, 565–567. [DOI] [PubMed] [Google Scholar]
- 27.Bochud, P. Y., Hawn, T. R. & Aderem, A. (2003) J. Immunol. 170, 3451–3454. [DOI] [PubMed] [Google Scholar]
- 28.Ben-Ali, M., Barbouche, M. R., Bousnina, S., Chabbou, A. & Dellagi, K. (2004) Clin. Diagn. Lab. Immunol. 11, 625–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arbour, N. C., Lorenz, E., Schutte, B. C., Zabner, J., Kline, J. N., Jones, M., Frees, K., Watt, J. L. & Schwartz, D. A. (2000) Nat. Genet. 25, 187–191. [DOI] [PubMed] [Google Scholar]
- 30.Cook, D. N., Pisetsky, D. S. & Schwartz, D. A. (2004) Nat. Immunol. 5, 975–979. [DOI] [PubMed] [Google Scholar]
- 31.Lorenz, E., Mira, J. P., Frees, K. L. & Schwartz, D. A. (2002) Arch. Intern. Med. 162, 1028–1032. [DOI] [PubMed] [Google Scholar]
- 32.Genc, M. R., Vardhana, S., Delaney, M. L., Onderdonk, A., Tuomala, R., Norwitz, E. & Witkin, S. S. (2004) Eur. J. Obstet. Gynecol. Reprod. Biol. 116, 152–156. [DOI] [PubMed] [Google Scholar]
- 33.Read, R. C., Pullin, J., Gregory, S., Borrow, R., Kaczmarski, E. B., di Giovine, F. S., Dower, S. K., Cannings, C. & Wilson, A. G. (2001) J. Infect. Dis. 184, 640–642. [DOI] [PubMed] [Google Scholar]
- 34.Smirnova, I., Mann, N., Dols, A., Derkx, H. H., Hibberd, M. L., Levin, M. & Beutler, B. (2003) Proc. Natl. Acad. Sci. USA 100, 6075–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazarus, R., Klimecki, W. T., Raby, B. A., Vercelli, D., Palmer, L. J., Kwiatkowski, D. J., Silverman, E. K., Martinez, F. & Weiss, S. T. (2003) Genomics 81, 85–91. [DOI] [PubMed] [Google Scholar]
- 36.Torok, H. P., Klimecki W. T., Tonenchi, L., Bruennler, G., Folwaczny, M., Folwaczny C. (2004) Gastroenterology 127, 365–366. [DOI] [PubMed] [Google Scholar]
- 37.Mockenhaupt, F. P., Ehrhardt, S., Gellert, S., Otchwemah, R. N., Dietz, E., Anemana, S. D. & Bienzle, U. (2004) Blood 104, 2003–2006. [DOI] [PubMed] [Google Scholar]
- 38.Mockenhaupt, F. P., Ehrhardt, S., Burkhardt, J., Bosomtwe, S. Y., Laryea, S., Anemana, S. D., Otchwemah, R. N., Cramer, J. P., Dietz, E., Gellert, S., et al. (2004) Am. J. Trop. Med. Hyg. 71, 167–172. [PubMed] [Google Scholar]
- 39.Mockenhaupt, F. P., Ehrhardt, S., Cramer, J. P., Otchwemah, R. N., Anemana, S. D., Goltz, K., Mylius, F., Dietz, E., Eggelte, T. A. & Bienzle, U. (2004) J. Infect. Dis. 190, 1006–1009. [DOI] [PubMed] [Google Scholar]
- 40.Djimde, A., Doumbo, O. K., Cortese, J. F., Kayentao, K., Doumbo, S., Diourte, Y., Dicko, A., Su, X. Z., Nomura, T., Fidock, D. A., et al. (2001) N. Engl. J. Med. 344, 257–263. [DOI] [PubMed] [Google Scholar]
- 41.Hamann, L., Hamprecht, A., Gomma, A. & Schumann, R. R. (2004) J. Immunol. Methods 285, 281–291. [DOI] [PubMed] [Google Scholar]
- 42.Haldane, J. B. S. (1949) Hereditas 35, 267–272. [Google Scholar]
- 43.Mackinnon, M. J., Gunawardena, D. M., Rajakaruna, J., Weerasingha, S., Mendis, K. N. & Carter, R. (2000) Proc. Natl. Acad. Sci. USA 97, 12661–12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aidoo, M., Terlouw, D. J., Kolczak, M. S., McElroy, P. D., ter Kuile, F. O., Kariuki, S., Nahlen, B. L., Lal, A. A. & Udhayakumar, V. (2002) Lancet 359, 1311–1312. [DOI] [PubMed] [Google Scholar]
- 45.Chotivanich, K., Udomsangpetch, R., Pattanapanyasat, K., Chierakul, W., Simpson, J., Looareesuwan, S. & White, N. (2002) Blood 100, 1172–1176. [PubMed] [Google Scholar]
- 46.Bienzle, U., Ayeni, O., Lucas, A. O. & Luzzatto, L. (1972) Lancet 1, 107–110. [DOI] [PubMed] [Google Scholar]
- 47.Beutler, E. (1994) Blood 84, 3613–3636. [PubMed] [Google Scholar]
- 48.Luty, A. J., Kun, J. F. & Kremsner, P. G. (1998) J. Infect. Dis. 178, 1221–1224. [DOI] [PubMed] [Google Scholar]
- 49.Cramer, J. P., Mockenhaupt, F. P., Ehrhardt, S., Burkhardt, J., Otchwemah, R. N., Dietz, E., Gellert, S. & Bienzle, U. (2004) Trop. Med. Int. Health 9, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 50.Koch, O., Awomoyi, A., Usen, S., Jallow, M., Richardson, A., Hull, J., Pinder, M., Newport, M. & Kwiatkowski, D. (2002) J. Infect. Dis. 185, 1684–1687. [DOI] [PubMed] [Google Scholar]
- 51.Aitman, T. J., Cooper, L. D., Norsworthy, P. J., Wahid, F. N., Gray, J. K., Curtis, B. R., McKeigue, P. M., Kwiatkowski, D., Greenwood, B. M., Snow, R. W., et al. (2000) Nature 405, 1015–1016. [DOI] [PubMed] [Google Scholar]
- 52.Adachi, K., Tsutsui, H., Kashiwamura, S., Seki, E., Nakano, H., Takeuchi, O., Takeda, K., Okumura, K., Van Kaer, L., Okamura, H., et al. (2001) J. Immunol. 167, 5928–5934. [DOI] [PubMed] [Google Scholar]
- 53.Oliveira, A. C., Peixoto, J. R., de Arruda, L. B., Campos, M. A., Gazzinelli, R. T., Golenbock, D. T., Akira, S., Previato, J. O., Mendonca-Previato, L., Nobrega, A., et al. (2004) J. Immunol. 173, 5688–5696. [DOI] [PubMed] [Google Scholar]
- 54.Malhotra, D., Relhan, V., Reddy, B. S. & Bamezai, R. (2005) Hum. Genet. 116, 413–415. [DOI] [PubMed] [Google Scholar]
- 55.Lawn, S. D. (2000) Int. J. Tuberc. Lung Dis. 4, 1190–1191. [PubMed] [Google Scholar]
- 56.Moormann, A. M., Sullivan, A. D., Rochford, R. A., Chensue, S. W., Bock, P. J., Nyirenda, T. & Meshnick, S. R. (1999) J. Infect. Dis. 180, 1987–1993. [DOI] [PubMed] [Google Scholar]
- 57.Deshpande, P. & Shastry, P. (2004) Cytokine 28, 205–213. [DOI] [PubMed] [Google Scholar]
- 58.Gazzinelli, R. T., Ropert, C. & Campos, M. A. (2004) Immunol. Rev. 201, 9–25. [DOI] [PubMed] [Google Scholar]
- 59.Medana, I. M., Mai, N. T., Day, N. P., Hien, T. T., Bethell, D., Phu, N. H., Farrar, J., White, N. J. & Turner, G. D. (2001) Neuropathol Appl. Neurobiol. 27, 421–433. [DOI] [PubMed] [Google Scholar]
- 60.Evans, J. A., Adusei, A., Timann, C., May, J., Mack, D., Agbenyega, T. Horstmann, R. D. & Frimpong, E. (2004) QJM 97, 591–597. [DOI] [PubMed] [Google Scholar]
- 61.Trape, J. F. (2001) Am. J. Trop. Med. Hyg. 64, 12–17. [DOI] [PubMed] [Google Scholar]