

Abstract
We present a detailed structural characterization of the intermediate state populated during the folding and unfolding of the bacterial immunity protein Im7. We achieve this result by incorporating a variety of experimental data available for this species in molecular dynamics simulations. First, we define the structure of the exchange-competent intermediate state of Im7 by using equilibrium hydrogen-exchange protection factors. Second, we use this ensemble to predict Φ-values and compare the results with the experimentally determined Φ-values of the kinetic refolding intermediate. Third, we predict chemical-shift measurements and compare them with the measured chemical shifts of a mutational variant of Im7 for which the kinetic folding intermediate is the most stable state populated at equilibrium. Remarkably, we found that the properties of the latter two species are predicted with high accuracy from the exchange-competent intermediate that we determined, suggesting that these three states are characterized by a similar architecture in which helices I, II, and IV are aligned in a native-like, but reorganized, manner. Furthermore, the structural ensemble that we obtained enabled us to rationalize the results of tryptophan fluorescence experiments in the WT protein and a series of mutational variants. The results show that the integration of diverse sets of experimental data at relatively low structural resolution is a powerful approach that can provide insights into the structural organization of this conformationally heterogeneous three-helix intermediate with unprecedented detail and highlight the importance of both native and non-native interactions in stabilizing its structure.
Keywords: hydrogen exchange, protein folding, protein intermediates
Intermediate states have been identified in the folding and unfolding processes of many proteins (1–11). Owing to their transient nature and conformational heterogeneity, however, the experimental determination of their structure is an enormously challenging task (6, 7, 9–11).
The bacterial immunity protein Im7 is a four-helical protein that has been shown to fold through an on-pathway kinetic intermediate (12). The structural properties of this kinetic intermediate state (KIS) have been characterized experimentally by Φ-value analysis (4) and shown to comprise three of the four native helices (I, II, and IV), stabilized by both native and non-native interactions. In addition, equilibrium hydrogen-exchange experiments (7) on the WT protein and the variant I72V showed that amides in these three helices, but not in helix III, exchange slowly with a free energy similar to that associated with global unfolding, suggesting that they are substantially formed in the exchange-competent intermediate state (EIS), whereas helix III is not significantly protected from hydrogen exchange in this state. Furthermore, the double-mutant L53AI54A (13) has been designed to trap the kinetic intermediate as a stable intermediate state (SIS) at equilibrium. Various biophysical methods suggested that the SIS closely resembles the KIS and EIS (7, 13). However, the extent to which these three intermediate states are structurally similar is still unresolved, and, importantly, the orientation of the three helices and the nature and extent of the non-native interactions that stabilize this state could not be determined with mutational analysis, as it is too unstable for double-mutant cycles (14), and conformational dynamics precludes a detailed structural analysis by using standard NMR methods.
As intermediate states are more heterogeneous than their native counterparts, their characterization involves the accurate determination of the relatively broad ensembles of structures that represent the conformational fluctuations of the protein in these states. Thus, by using equilibrium amide hydrogen-exchange protection factors (7) as restraints in molecular dynamics simulations, we determined an ensemble of structures representing the EIS. We then verified whether the structures determined for the EIS are consistent with the experimental knowledge of the SIS, by measuring the deviations of the chemical shifts predicted for the EIS structures from the experimentally derived chemical shifts of the double-mutant L53AI54A. In addition, we used the structures of the EIS to make predictions about the Φ-value measurements of the KIS. The remarkable accuracy (Δδtot = 0.40 ± 0.01 ppm for the chemical shifts and correlation coefficient 0.74 for the Φ-values) of these predictions suggests that the intermediates detected by each experimental method have a similar overall architecture, characterized by the docking of helices I, II, and IV in a non-native manner, but also show that they differ locally. The considerable rearrangement of the structure of the intermediate of Im7 relative to the native structure contrasts with a mutational variant of cytochrome b562 recently characterized (9), whose backbone was found to be much more native-like, and for which the detection of nuclear Overhauser effects was possible.
Our results highlight the importance of both native and non-native interactions in stabilizing intermediates of Im7 and demonstrate the power of combining computational and experimental approaches for the elucidation of the structural propensities of non-native states of proteins that display substantial conformational heterogeneity.
Methods
Determination of the EIS. Twenty six protection factors determined from the amide exchange rates of Im7*, a hexa-histidine-tagged variant of Im7 (7), were used as restraints in molecular dynamics simulations (15). In these simulations the EIS of Im7 was sampled by adding a pseudoenergy term (16) to the charmm force field (17). In this study, the reaction coordinate ρ used in the pseudoenergy term is defined as (18)
 |
[1] |
where we defined the average over M replicas (M = 10) as
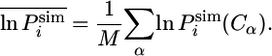 |
[2] |
In this expression, we defined the natural logarithm of the simulated protection factor 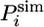 of the amide hydrogen of residue i in conformation C as
of the amide hydrogen of residue i in conformation C as
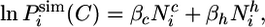 |
[3] |
where 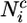 is the number of native contacts of residue i in conformation C and
is the number of native contacts of residue i in conformation C and 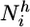 is the number of hydrogen bonds of the amide hydrogen of residue i. The two parameters βc and βh in Eq. 3 were determined as 0.3 and 0.1, respectively, in a best-fit procedure using a set of seven proteins, which did not include Im7, for which the experimental structure and equilibrium hydrogen-exchange data were available (15). Closely related interpretations of protection factors in terms of structural propensities have been proposed by several other authors (19–25). Their relevance for the proposed method and a more detailed discussion about the structural interpretation of the protection factors are provided in Supporting Text, which is published as supporting information on the PNAS web site.
is the number of hydrogen bonds of the amide hydrogen of residue i. The two parameters βc and βh in Eq. 3 were determined as 0.3 and 0.1, respectively, in a best-fit procedure using a set of seven proteins, which did not include Im7, for which the experimental structure and equilibrium hydrogen-exchange data were available (15). Closely related interpretations of protection factors in terms of structural propensities have been proposed by several other authors (19–25). Their relevance for the proposed method and a more detailed discussion about the structural interpretation of the protection factors are provided in Supporting Text, which is published as supporting information on the PNAS web site.
In the simulations the velocities were randomly assigned from a Maxwell–Boltzmann distribution at 300 K with a different random seed for each of the 10 replicas. The force constant that controls the weight of the restraint term ρ with respect to the force field was increased during the first 1.2 ns at 300 K to improve progressively the agreement between experimental and calculated protection factors. Subsequently, a series of 220 1-ns cycles of simulated annealing between 300 and 500 K were carried out to sample effectively the conformational space. The structural evolution of the ensemble was monitored by clustering the conformations according to their Cα-rms deviation (26) (Fig. 6A, which is published as supporting information on the PNAS web site). To minimize the correlation with the initial structure, the first 20 annealing cycles were not used for the analysis (Fig. 6B).
As controls, three other structural ensembles were determined by following the same protocol but using as restraints different subsets (leaving out 3, 7, and 13 protection factors, respectively) extracted from the list of 26 protection factors. Using the resulting ensembles, the subset of protection factors left out in the calculations were predicted with good accuracy, thus validating the definition used for 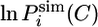 (Fig. 7, which is published as supporting information on the PNAS web site). To further test the robustness of the methods used, we also carried out additional controls with two different types of artificial protection factors: reshuffled protection factors (Fig. 8, which is published as supporting information on the PNAS web site) and very large artificial protection factors (Fig. 9, which is published as supporting information on the PNAS web site). The resulting ensembles were then tested for their ability to predict the properties observed experimentally for the KIS and SIS. In all cases, the predictions were very inaccurate (see Supporting Text for a more quantitative comparison), indicating that the control ensembles represented very poor models for the KIS and SIS, and thereby validating the use of experimentally derived protection factors for defining with good accuracy the EIS.
(Fig. 7, which is published as supporting information on the PNAS web site). To further test the robustness of the methods used, we also carried out additional controls with two different types of artificial protection factors: reshuffled protection factors (Fig. 8, which is published as supporting information on the PNAS web site) and very large artificial protection factors (Fig. 9, which is published as supporting information on the PNAS web site). The resulting ensembles were then tested for their ability to predict the properties observed experimentally for the KIS and SIS. In all cases, the predictions were very inaccurate (see Supporting Text for a more quantitative comparison), indicating that the control ensembles represented very poor models for the KIS and SIS, and thereby validating the use of experimentally derived protection factors for defining with good accuracy the EIS.
Chemical-Shift Measurements. Chemical shifts of the double-mutant L53AI54A were measured at 25°C, pH 7.0 (50 mM phosphate and 0.4 M sodium sulfate) in 10% D2O/90% H2O. Resonance assignments were obtained from standard 3D triple-resonance 1H/13C/15N spectra acquired on a Varian Inova 500 with pulse sequences from the Proteinpack library of experiments (data not shown). 1H chemical shifts were referenced directly to external 2,2-(dimethylsilyl)propanesulfonic acid (DSS) and the 13C and 15N chemical shifts indirectly to DSS (27).
Prediction of Chemical Shifts. Chemical shifts for HN, N, CO, Cα and Cβ atoms were calculated by using the program shiftx (28). The total deviation, Δδtot, of the HN, N, CO, Cα, and Cβ chemical shifts (29) of the EIS structures from the experimental chemical shifts of the double-mutant L53AI54A were determined by using the weighted sum
 |
[4] |
where WI denotes the weight factor for each type of nucleus relative to HN.
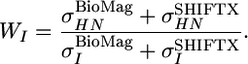 |
[5] |
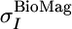 is the average variance of the chemical shifts of nuclei HN, N,CO, Cα, and Cβ as reported in the BioMagResBank (www.bmrb.wisc.edu) and
is the average variance of the chemical shifts of nuclei HN, N,CO, Cα, and Cβ as reported in the BioMagResBank (www.bmrb.wisc.edu) and 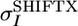 is the average rms deviation of the chemical shifts of nuclei HN, N, CO, Cα, and Cβ calculated by shiftx from the experimental chemical shifts. The values used here are: WHN = 1, WN = 0.16, WCO = 0.35, WCα = 0.33, and WCβ = 0.34.
is the average rms deviation of the chemical shifts of nuclei HN, N, CO, Cα, and Cβ calculated by shiftx from the experimental chemical shifts. The values used here are: WHN = 1, WN = 0.16, WCO = 0.35, WCα = 0.33, and WCβ = 0.34.
A weighted-shift difference Δδtot = 0.40 ± 0.01 ppm (ranging from 0.37 to 0.42) was found for the EIS. This value should be compared with Δδtot = 0.47 ± 0.02 ppm in the native-state ensemble. These results indicate that structures consistent with equilibrium hydrogen-exchange data are also characterized by chemical shifts close to those of the L53AI54A mutant. For further analysis, the subset of the EIS ensemble with Δδtot < 0.39 ppm (30 structures) was selected and refined in explicit water in the charmm22 force field (30) for 2 ns (see Supporting Text for further details on the refinement procedure). The analysis reported in the following is based on this refined subset of structures.
Prediction of Φ-Values. The native and EIS structures were used as inputs to the program fold-x. By averaging over ensembles of native and intermediate structures, prediction of 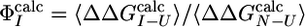 can be obtained (31). Only mutations for which the relative error in
can be obtained (31). Only mutations for which the relative error in 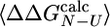 is < 0.3 (i.e.,
is < 0.3 (i.e., 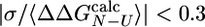 , where σ is the standard deviation of
, where σ is the standard deviation of 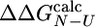 ) are appropriate for this type of analysis (31). A similar criterion has been discussed in the selection of experimental Φ-values (5, 32). In the ensemble of structures that we determined, 12 of the 24 mutations investigated by Capaldi et al. (4) satisfy the former criterion and have been used in the analysis.
) are appropriate for this type of analysis (31). A similar criterion has been discussed in the selection of experimental Φ-values (5, 32). In the ensemble of structures that we determined, 12 of the 24 mutations investigated by Capaldi et al. (4) satisfy the former criterion and have been used in the analysis.
Results
Analysis of the EIS. To determine the conformational properties of the EIS of Im7, hydrogen-exchange protection factors were used as restraints in molecular dynamics simulations (see Methods). The resulting ensemble of structures (Fig. 1) demonstrates that this species is structurally heterogeneous. We identified the most relevant conformational substates by performing a cluster analysis (see Methods). The structures of the two most populated clusters, comprising 38% and 20% of the conformations in the ensemble, respectively, are shown in Fig. 1. The distance between helices II and IV is significantly different in the two substates shown, whereas their overall architecture is very similar and involves a reorganization of the helices, whereby helices I and II move away from each other with respect to the native state, thus allowing the docking of helix IV onto them (see below). To characterize further the heterogeneity of the EIS, pairwise rms deviations were calculated for each α-helix, after a least-squares superposition of all backbone atoms of the protein. Helices II and III show a much broader distribution of pairwise rms deviations than helices I and IV (Fig. 10, which is published as supporting information on the PNAS web site). The increased heterogeneity of helices II and III appears not to be an artifact of the low number of protection factors measured for these secondary structural elements, as helix I has only one more measured protection factor than helix II (five and four, respectively) and yet is structurally much more homogeneous. Moreover, for residues 39–49 that form part of helix II and its connecting loop with helix III, no resonances could be assigned for the L53AI54A variant. These results support the view that the poor spectral quality of regions representing side chains of L53AI54A, which effectively rules out a direct and detailed characterization of the SIS by NMR spectroscopy, is a direct result of its structural heterogeneity (see below).
Fig. 1.

Intermediate state ensemble of Im7. (A) X-ray structure of native Im7 (Protein Data Bank ID code 1AYI). The native helices I (red), II (green), III (magenta), and IV (yellow) include residues 12–24, 32–45, 51–56, and 66–79, respectively. (B and C) Representative structures of the two most populated clusters in the EIS ensemble. Side view (Upper) and top view (90° rotation) (Lower) of the structures.
In Fig. 2A we compare the calculated protection factors of the EIS ensemble with the experimental values. For some residues, no protection factor was determined experimentally because resonances were either unassigned, not well resolved, or exchanged too rapidly. For the majority of these we calculate 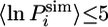 . The exceptions are residues 19, 20, and 22 in helix I, residues 39, 40, and 43–45 in helix II, and residues 68, 70, 71 and 79 in helix IV. High protection factors for these residues can be rationalized by the almost complete formation of helices I, II, and IV in the EIS ensemble despite the lack of experimental restraints for these residues (Fig. 2B). Helix III, by contrast, is not protected in almost all members of the EIS ensemble, and all residues in this helix, apart from residue 56, have a lnP < 5. Helix III is of particular interest because equilibrium hydrogen-exchange experiments on WT Im7* and the variant I72V indicate that hydrogen exchange from this α-helix may occur by a subglobal transition involving a state in which helix III is unfolded. Residue 54, although restrained by a single
. The exceptions are residues 19, 20, and 22 in helix I, residues 39, 40, and 43–45 in helix II, and residues 68, 70, 71 and 79 in helix IV. High protection factors for these residues can be rationalized by the almost complete formation of helices I, II, and IV in the EIS ensemble despite the lack of experimental restraints for these residues (Fig. 2B). Helix III, by contrast, is not protected in almost all members of the EIS ensemble, and all residues in this helix, apart from residue 56, have a lnP < 5. Helix III is of particular interest because equilibrium hydrogen-exchange experiments on WT Im7* and the variant I72V indicate that hydrogen exchange from this α-helix may occur by a subglobal transition involving a state in which helix III is unfolded. Residue 54, although restrained by a single 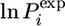 value, shows a wide distribution of
value, shows a wide distribution of 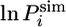 in the EIS (Fig. 3A). Similar distributions are observed for the other residues of helix III (data not shown). Importantly, the protection factors of residues corresponding to the native helix III are highly correlated in the different members of the EIS (Fig. 3B), and individual residues throughout this region have similar
in the EIS (Fig. 3A). Similar distributions are observed for the other residues of helix III (data not shown). Importantly, the protection factors of residues corresponding to the native helix III are highly correlated in the different members of the EIS (Fig. 3B), and individual residues throughout this region have similar  values in the same conformation of the EIS. Moreover, in conformations with low
values in the same conformation of the EIS. Moreover, in conformations with low  values, a significant distance is observed between the hydrogen and oxygen atoms that form a hydrogen bond in the native state (Fig. 3C). By contrast, the hydrogen bond is still present in structures with a high
values, a significant distance is observed between the hydrogen and oxygen atoms that form a hydrogen bond in the native state (Fig. 3C). By contrast, the hydrogen bond is still present in structures with a high 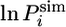 . Overall, these analyses suggest that helix III is acting as a single locally cooperative structural entity, either mostly folded or mostly unfolded in the EIS (Fig. 3D), in which all residues behave in a similar manner.
. Overall, these analyses suggest that helix III is acting as a single locally cooperative structural entity, either mostly folded or mostly unfolded in the EIS (Fig. 3D), in which all residues behave in a similar manner.
Fig. 2.

Prediction of protection factors and secondary structure content. (A) Comparison of ln(Pexp) (blue dots) with ln(Psim) (red line) calculated in the EIS ensemble (see also Fig. 13, which is published as supporting information on the PNAS web site). Protection factors for which ln(Pexp) < 5 are usually not measurable experimentally, and they are shown as zeros. (B) α-Helical propensities in EIS ensemble.
Fig. 3.

Structural analysis of the helical regions. (A) Probability distribution of the ln(Psim) values of residue 54 in the EIS ensemble. (B) Correlation between ln(Psim) of residues 53 and 54 (black), 54 and 55 (red), and 55 and 56 (green). (C) Anticorrelation between the ln(Psim) values and the distance between the hydrogen and the oxygen atoms that form a hydrogen bond in the native structure for residues 54 (black), 55 (red), and 56 (blue). (D) Probability of formation of helix III as a function of the ln(Psim) value of residue 54. (E) Probability distribution of the distance between different pairs of helices in the EIS ensemble. For comparison, the distance between each helix pair in the native state is indicated by a vertical dashed line.
Although the degree of formation of helices I, II, and IV in the EIS and native states is very similar, their packing is different. We calculated the distances between the centers of mass of each of the helices present in the EIS ensemble. Helix IV and the region corresponding to helix III in the native state are closer to helix II in >50% of the EIS structures, whereas the remaining structures maintain their native orientation (Fig. 3E). Helices I and II have moved apart from each other in virtually all structures to allow the docking of helix IV onto them. This type of analysis indicates clearly that helix IV, in most of the EIS structures, has a different position than in the native state. While staying in close contact with helix I, which enables numerous stabilizing interactions between them, helix IV moves closer toward helix II. This change in the disposition of the helices is realized through the formation of many non-native side-chain interactions (Fig. 11 and Table 1, which are published as supporting information on the PNAS web site). The presence of non-native interactions, however, does not imply that the intermediate state is characterized by a non-native topology. As illustrated in Fig. 1, the non-native interactions in the intermediate are topologically conserving and the overall native architecture, although distorted, is still present. Significant contributions to the stability of the EIS result from non-native interactions between residues in helix II and helix IV, as well as between the unfolded helix III and helix IV. Overall, the analysis of the secondary and tertiary structural features indicates that the EIS not only contains stable hydrogen-bonded structure in helices I, II, and IV, but also that its structure is stabilized by both native and non-native specific tertiary interactions (Fig. 11).
The formation of non-native contacts between side chains in the EIS of Im7 could not be anticipated from the direct analysis of the protection factors (7), which only probe the main chain, but is provided by the present analysis that uses simultaneously experimental information about individual residues within a detailed structural model of the protein. This approach, therefore, enables us to define the structure of the species responsible for the native-state exchange, including the description of the interactions involving both secondary and tertiary structure that define the structural properties of this state, as well as the probability of different locally and subglobally unfolded species.
Prediction of the Properties of the Kinetic Refolding Intermediate. Having obtained a description of the structural properties of the EIS, we next investigated whether the properties of these species are related to those of the KIS populated transiently after dilution of the denatured state from urea (4). Two properties are considered:
β-Tanford. The Tanford β-value, βT, in the EIS was predicted from the relative solvent-accessible surface area of the native state, EIS, and denatured states (see Supporting Text) to be in the range between 0.71 and 0.84. These values indicate that the EIS structures have a compactness very similar to the experimentally determined βT value of 0.77 for the KIS (4).
Φ-values. Protein engineering techniques have provided information about the structural properties of the KIS transiently populated during the folding of Im7* (4). We used the EIS structures to predict Φ-values for this ensemble (31) and compared them with the experimentally available measurements of the Φ-values for the KIS (4) (see Methods). The calculated values are in good agreement with the experimental Φ-values (correlation coefficient 0.74, Fig. 4A), despite the sizable statistical error on the predicted values, whereas the errors on the Φexp values of these residues are ≈0.1 (4). We excluded from the correlation the I72V variant for which the change in
 is small and thus the estimated error for the
is small and thus the estimated error for the  value is large (4). We also excluded the V69A variant, which has a
value is large (4). We also excluded the V69A variant, which has a 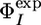 value of 0.12, whereas the predicted
value of 0.12, whereas the predicted 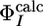 value is 1.0. This difference may be caused by the large protection factor of V69 (lnP = 8.8), which results in highly conserved interactions in the EIS ensemble for this residue.
value is 1.0. This difference may be caused by the large protection factor of V69 (lnP = 8.8), which results in highly conserved interactions in the EIS ensemble for this residue.
Fig. 4.

Prediction of chemical shifts and Φ values. (A) Comparison of the Φ-values predicted from the EIS structures (Φcalc) with the experimental values (Φexp) measured for the kinetic folding intermediate (4). (B) Total number of native (black) and non-native (red) atomic contacts per residue present in >25% of the EIS structures. (C) Differences between the experimentally determined chemical shifts in the native state of WT Im7* and the L53AI54A double mutant (black bars). For residues 39–49 no resonances could be assigned. Differences between the predicted chemical shifts of the native state of Im7 and the EIS structures are shown (red bars).
Kinetic folding experiments showed that the destabilizing mutations F15A, L18A, L19A, L34A, L37A, L38A, F41L, and V42A cause an increase in the rate constant kIN (4). Moreover, all of these residues, as well as I68, have higher Φ-values for the KIS than the rate-limiting transition state for folding. In particular, Φ-values for residues 34, 38, and 41 in the KIS exceed the ones of the rate-limiting transition state by 0.6, 0.4, and 0.9, respectively. These experimental data suggested that substantial non-native interactions are present in the KIS, although their identity remained unresolved because the KIS is too unstable to permit double mutant analysis (4). We analyze here the presence of non-native contacts in the EIS ensemble (Fig. 11 and Table 1), by calculating the number of specific long-range non-native contacts, i.e., atom pairs separated by >10 Å in the native state that become as close as 6 Å in at least 25% of the EIS structures (Fig. 4B). Consistent with the experimental data, segments 10–27, 34–44, and 66–75, which constitute major parts of helices I, II, and IV in the native state, form a significant number of specific non-native contacts with each other (see also Fig. 11). In particular, residues in the segment 34–44 form nearly as many non-native as native contacts in the EIS ensemble. In the region of the unfolded helix III, residues Y55 and Y56 form many non-native contacts, especially with W75 (see below).
In summary, therefore, the consistency between the structural features derived from the EIS probed by native-state hydrogen exchange and the kinetic intermediate defined by the Φ-value analysis suggest that these two species are highly similar.
Prediction of the Conformational Properties of a Stable Intermediate Populated at Equilibrium
Prediction of Chemical-Shift Measurements. In a recent study (13) the kinetic refolding intermediate of Im7 was trapped as a stable state (SIS) by creating the double-mutant L53AI54A. To determine whether the conformational properties of the EIS are also consistent with the SIS, we compared the experimentally determined chemical shift differences between WT Im7* and the double-mutant L53AI54A with the corresponding differences calculated between the native state of Im7 and the EIS structures that we determined (Fig. 4C). The small magnitude of chemical-shift changes measured experimentally for WT Im7* and L53AI54A in segments 12–24 (helix I), 32–38 (N-terminal part of helix II), and 66–79 (helix IV) indicate the presence of major parts of these secondary structural elements in the equilibrium intermediate of Im7. Segment 51–56 (helix III), by contrast, shows significantly different chemical shifts in L53AI54A, which indicates substantial structural alterations after accounting for the difference in primary chemical shifts resulting from the mutations. For most regions of the sequence, the changes calculated between the native state of Im7 and the EIS are compatible with those observed experimentally, especially in the region of helix III where the changes in chemical shifts are also particularly significant. The only region for which experimental and predicted chemical shift differences deviate considerably is the C-terminal part of helix I (residues 23–25). The similarity in the Cα chemical shifts measured experimentally for WT Im7* and the L53AI54A variant suggests that helix I is entirely formed in the equilibrium intermediate of Im7. The very low protection factors for residues in the C terminus of helix I are responsible for the loss of the last turn of this helix in the EIS structures that we determined and hence in the differences observed for the chemical shifts. However, as the time scale for the hydrogen-exchange process is much longer than that probed by chemical shifts, it is possible that rapid hydrogen exchange could occur without significant perturbation of the chemical shifts.
CD and β-Tanford Measurements. In addition to the double-mutant L53AI54A, three other variants have been designed recently to trap the KIS at equilibrium by disrupting the interactions between helix III and the rest of the protein (13). By comparing the mean residue ellipticity at 222 nm of the WT protein and the variants, a α-helical content between 40% and 45% was estimated for the equilibrium intermediate of the WT protein compared with 50% for the native state. Consistent with these data, the average α-helical content in the EIS ensemble that we determined is 40%.
Diffusion experiments (13) on these variants also indicated that these species are ≈20–30% expanded compared with the native state of WT Im7* (13). Consistent with these data, the estimated βT value of the EIS ensemble ranges between 0.71 and 0.84 (see above).
Fluorescence Measurements. Recent UV resonance Raman and time-resolved fluorescence measurements suggested that the structural changes in the L53AI54A variant compared with WT Im7* result in an increase in hydrophobicity of the local environment and a decrease in solvent-accessible surface area of W75 in the SIS (33). Consistent with these observations, we found that W75 is more buried in the EIS structures than in the native state of WT Im7 (Fig. 12, which is published as supporting information on the PNAS web site). In addition, the hydrophilic residue H47, which quenches the fluorescence emission of W75 in native Im7 (13, 34), moves away from W75, whereas the aromatic residues Y55, Y56, and F41 are significantly closer to W75 in the EIS compared with the native state (Fig. 5). The non-native interactions between W75 and specific hydrophobic residues and its burial from the solvent may explain the increased fluorescence emission observed experimentally for the KIS and SIS (4, 12, 33). It is important to note that variants in which the six residues comprising the native helix III are substituted simultaneously with three or six glycine residues are less fluorescent than their L53AI54A counterpart (13). Furthermore, the Y55A and Y56A variants show a reduction of the fluorescence emission intensity of the KIS (S.E.R. and S. Knowling, unpublished work). The spatial proximity of W75 and the two tyrosine residues Y55 and Y56 in the EIS ensemble, therefore, may suggest that a tyrosine–tryptophan energy transfer contributes to the increased tryptophan emission of the intermediate, although indirect effects could also explain the difference in fluorescent quantum yield observed (33).
Fig. 5.

Comparison with fluorescence measurements. (A) Distribution of distances between W75 and other selected residues in the native state (blue) and EIS (green) of Im7. (B) Structures illustrating the distance of residues W75 (dark blue), Y55 (cyan), and Y56 (green) in native Im7 (Left) and EIS (Right). Helix IV is shown in yellow, and residues in the region corresponding to the native helix III are in pink.
Taken together, chemical shifts, α-helical content, β-Tanford values, and fluorescence measurements indicate that the structural properties of the L53AI54A variant resemble closely those of both the exchange-competent state of Im7 and its kinetic refolding intermediate.
Conclusions
We have determined an ensemble of conformations representing the EIS of the immunity protein Im7 by using equilibrium hydrogen-exchange protection factors as restraints in molecular dynamics simulations and compared the resulting structures with the intermediate states studied experimentally by trapping this species at equilibrium by mutagenesis, as well as the kinetic intermediate transiently populated during refolding, characterized by Φ-value analysis.
A crucial aspect of the comparison of the various intermediate states of Im7 is that different experimental measurements were carried out under identical conditions (pH 7, 10°C, 0.4 M Na2SO4) and for different variants. The protection factors were measured under native conditions and report the backbone structural properties of the EIS of Im7. The Φ-values were measured from kinetic denaturant dilution experiments and probe the importance of different side chains in stabilizing the transient on-pathway kinetic folding intermediate of Im7. Chemical shifts were measured under equilibrium conditions for the L53AI54A variant, for which the native state is only 1% populated. Predicting with good accuracy the results of these measurements from the structures of the EIS of Im7 indicates that the equilibrium and kinetic intermediates have a similar overall architecture and share several structural features.
A detailed analysis of the structural features of the EIS of Im7 has revealed that it is a highly structured species, containing three of the four native helices (I, II, and IV). The structural ensembles that we present here provide detailed information about the interactions between the helices that could not be obtained directly from the experimental data. We have found that helices I and II are on average >3 Å further apart than in the native state, thus allowing the docking of helix IV onto them in the majority of the EIS structures. While having interactions with helix I that are as close as those in the native state, helix IV forms numerous non-native interactions with helix II and the region that corresponds to helix III in the native state. In particular, W75 is more buried in the EIS than in native Im7 through the formation of specific non-native interactions with the aromatic residues Y55 and Y56, a result that suggests a rational basis for the hyperfluorescence of the different intermediates found experimentally.
In conclusion, the high level of compatibility of the structures determined from hydrogen exchange with the measurements of chemical shifts, Φ-values, and fluorescence indicate that the intermediate states of Im7 populated under both kinetic and equilibrium conditions are structurally closely related species. The combination of experimental and computational techniques that we present enabled us to obtain detailed structural information about the intermediate state of Im7 that was not possible to access experimentally, as the conformational heterogeneity of this species rendered a detailed structural analysis by standard NMR or x-ray methods impossible. Most importantly, the remarkable agreement between the structural ensembles characterized by using very different approaches enabled us to provide a uniquely detailed description of the transient conformations that this protein populates during folding, revealing the structural reorganization that separates this species from the native state and providing information about the role of individual side chains, involving both native-like and non-native contacts, in stabilizing this state.
Supplementary Material
Acknowledgments
We thank Stuart Knowling, Claire Friel, Victoria Morton, and Eva Sanchez-Cobas for critical reading of the manuscript and many helpful discussions. J.G. is supported by the Swiss National Science Foundation. H.H. is supported by the Finnish Cultural Foundation, the Helsingin Sanomat Centennial Foundation, and the Instrumentarium Science Foundation. G.R.S. and S.B.-M.W. are supported by the Biotechnology and Biological Sciences Research Council. S.E.R. is a Biotechnology and Biological Sciences Research Council Professorial Fellow. M.V. is supported by the Royal Society and the Leverhulme Trust.
Author contributions: J.G. and M.V. designed research; J.G., H.H., and S.B.-M.W. performed research; J.G., H.H., S.B.-M.W., G.R.S., G.R.M., E.P., S.E.R., and M.V. analyzed data; and J.G., S.E.R., and M.V. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: KIS, kinetic intermediate state; EIS, exchange-competent intermediate state; SIS, stable intermediate state.
References
- 1.Rumbley, J., Hoang, L., Mayne, L. & Englander, S. W. (2001) Proc. Natl. Acad. Sci. USA 98, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker, M. J. & Marqusee, S. (2001) J. Mol. Biol. 305, 593–602. [DOI] [PubMed] [Google Scholar]
- 3.Teilum, K., Maki, K., Kragelund, B. B., Poulsen, F. M. & Roder, H. (2002) Proc. Natl. Acad. Sci. USA 99, 9807–9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capaldi, A. P., Kleanthous, C. & Radford, S. E. (2002) Nat. Struct. Biol. 9, 209–216. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez, I. E. & Kiefhaber, T. (2003) J. Mol. Biol. 325, 367–376. [DOI] [PubMed] [Google Scholar]
- 6.Jemth, P., Gianni, S., Day, R., Li, B., Johnson, C. M., Daggett, V. & Fersht, A. R. (2004) Proc. Natl. Acad. Sci. USA 101, 6450–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorski, S. A., Le Duff, C. S., Capaldi, A. P., Kalverda, A. P., Beddard, G. S., Moore, G. R. & Radford, S. E. (2004) J. Mol. Biol. 337, 183–193. [DOI] [PubMed] [Google Scholar]
- 8.Cliff, M. J., Higgins, L. D., Sessions, R. B., Waltho, J. P. & Clarke, A. R. (2004) J. Mol. Biol. 336, 497–508. [DOI] [PubMed] [Google Scholar]
- 9.Feng, F. H. Q., Zhou, Z. & Bai, Y. W. (2005) Proc. Natl. Acad. Sci. USA 102, 5026–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maity, H., Maity, M., Krishna, M. M. G., Mayne, L. & Englander, S. W. (2005) Proc. Natl. Acad. Sci. USA 102, 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimura, C., Dyson, J. H. & Wright, P. E. (2005) Proc. Natl. Acad. Sci. USA 102, 4765–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capaldi, A. P., Shastry, M. C., Kleanthous, C., Roder, H. & Radford, S. E. (2001) Nat. Struct. Biol. 8, 68–72. [DOI] [PubMed] [Google Scholar]
- 13.Spence, G. R., Capaldi, A. P. & Radford, S. E. (2004) J. Mol. Biol. 341, 215–226. [DOI] [PubMed] [Google Scholar]
- 14.Horovitz, A. & Fersht, A. R. (1992) J. Mol. Biol. 214, 613–617. [DOI] [PubMed] [Google Scholar]
- 15.Best, R. B. & Vendruscolo, M. (2005) Structure (London), in press.
- 16.Paci, E. & Karplus, M. (1999) J. Mol. Biol. 288, 441–459. [DOI] [PubMed] [Google Scholar]
- 17.Lazaridis, T. & Karplus, M. (1999) Proteins 35, 133–152. [DOI] [PubMed] [Google Scholar]
- 18.Vendruscolo, M., Paci, E., Dobson, C. M. & Karplus, M. (2003) J. Am. Chem. Soc. 125, 15686–15687. [DOI] [PubMed] [Google Scholar]
- 19.Miller, D. W. & Dill, K. A. (1987) Protein Sci. 196, 641–656. [Google Scholar]
- 20.Hilser, V. J. & Freire, E. (1996) J. Mol. Biol. 262, 765–772. [DOI] [PubMed] [Google Scholar]
- 21.Sheinerman, F. B. & Brooks, C. L. (1998) Proc. Natl. Acad. Sci. USA 95, 1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bahar, I., Wallqvist, A., Covell, D. G. & Jernigan, R. L. (1998) Biochemistry 37, 1067–1075. [DOI] [PubMed] [Google Scholar]
- 23.Garcia, A. E. & Hummer, G. (1999) Proteins 36, 175–191. [PubMed] [Google Scholar]
- 24.Viguera, A. R. & Serrano, L. (2003) Proc. Natl. Acad. Sci. USA 100, 5730–5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinkam, P., Zong, C. & Wolynes, P. G. (2005) Proc. Natl. Acad. Sci. USA 102, 12401–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paci, E., Vendruscolo, M., Dobson, C. M. & Karplus, M. (2002) J. Mol. Biol. 324, 151–163. [DOI] [PubMed] [Google Scholar]
- 27.Wishart, D. S., Bigam, C. G., Yao, J., Abildgaard, F., Dyson, H. J., Oldfield, E., Markley, J. L. & Sykes, B. D. (1995) J. Biomol. NMR 6, 135–140. [DOI] [PubMed] [Google Scholar]
- 28.Neal, S., Nip, A. M., Zhang, H. & Wishart, D. S. (2003) J. Biomol. NMR 26, 215–240. [DOI] [PubMed] [Google Scholar]
- 29.Ayed, A., Mulder, F. A. A., Yi, G. S., Lu, Y., Kay, L. E. & Arrowsmith, C. H. (2001) Nat. Struct. Biol. 9, 756–760. [DOI] [PubMed] [Google Scholar]
- 30.MacKerell, A. D., Jr., Bashford, D., Bellot, M., Dunbrack, R. L. J., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 31.Lindorff-Larsen, K., Paci, E., Serrano, L., Dobson, C. M. & Vendruscolo, M. (2003) Biophys. J. 85, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fersht, A. R. (2004) Proc. Natl. Acad. Sci. USA 101, 14338–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Mendieta, I. R., Spence, G. R., Gell, C., Radford, S. E. & Smith, D. A. (2005) Biochemistry 44, 3306–3315. [DOI] [PubMed] [Google Scholar]
- 34.Dennis, C. A., Videler, H., Pauptit, R. A., Wallis, R., James, R., Kleanthous, C. & Moore, G. R. (1998) Biochem. J. 333, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.