

Abstract
Endothelial dysfunction (ED) is an early feature of cardiovascular risk and diabetes. Hyperglycemia and hyperlipidemia are causative factors. Excessive endothelial mitochondrial superoxide (ROS) production with hyperglycemia and hyperlipidemia is a key mechanism. Inositol components of an insulin inositol glycan mediator, d-chiro-inositol (DCI) and 3-O-methyl DCI (pinitol), decrease hyperglycemia and hyperlipidemia. We tested whether these, myoinositol and dibutyryl DCI (db-DCI), would prevent or reverse ED in diabetic rats and rabbits. Oral inositols reduced hyperglycemia and hypertriglyceridemia with different potencies and prevented ED in rat aortic rings and mesenteric beds. Inositols added in vitro to five diabetic tissues reversed ED. Relaxation by Ach, NO, and electrical field stimulation was potentiated by inositols in vitro in rabbit penile corpus cavernosa. Inositols in vitro restored impaired contraction by the eNOS inhibitor l-NAME and increased NO effectiveness. DCI and db-DCI decreased elevated ROS in endothelial cells in high glucose and db-DCI reduced PKC activation, hexosamine pathway activity, and advanced glycation end products to basal levels. Xanthine/xanthine oxidase generated superoxide was reduced by superoxide dismutase or inositols, with db-DCI efficacious in a mechanism requiring chelated Fe3+. Histochemical examination of rat aortic rings for protein SNO demonstrated a decrease in diabetic rings with restoration by inositols. In summary, inositols prevented and reversed ED in rat and rabbit vessels, reduced elevated ROS in endothelial cells, potentiated nitrergic or vasculo-myogenic relaxations, and preserved NO signaling. These effects are related to their metabolic actions, direct superoxide scavenging, and enhancing and protecting NO signaling. Of the inositols tested, db-DCI was most effective.
Keywords: d-chiro-inositol, diabetes, NO, insulin, mimetic
Inositol phosphoglycans (IPGs) are potentially important putative intracellular mediators of insulin action (1, 2). Separate classes have been identified (3), one containing d-chiro-inositol (DCI) and galactosamine (4) and a second containing myo-inositol (myo-INS) and glucosamine (1–5). Generated rapidly from lipid and/or protein precursors in response to insulin, they have insulin-like effects in vitro and in vivo (1, 2, 4, 6, 7). Myo-INS phospho-glycans have been prepared by enzymatic cleavage of protein precursors and chemically synthesized (7–9). They have dose-dependent insulin-like effects on isolated adipocytes, cardiomyocytes, and diaphragms including increased glucose transport (7–9). One DCI containing IPG has been isolated from liver, and its structure has been determined and chemically synthesized (10). It is active in vivo; lowering elevated blood glucose when administered intravenously to diabetic rats (10–12). In vitro, it enhances insulin to stimulate [14C]glucose incorporation into glycogen in H4IIE hepatoma cells (10). It activates pyruvate dehydrogenase (PDH) phosphatase (10), phosphoprotein phosphatase PP2C directly (13), and PP1 indirectly (4, 14). Both PDH and glycogen synthase are activated in vivo by insulin via dephosphorylation (15, 16).
To assess the physiological significance of chiro-inositol glycans, the contents of the rarer chiro-inositol and the more common myo-INS were measured in urine and tissues of diabetic subjects and compared with controls. In human urine (17), monkey urine (18), genetic G/K type 2 diabetic rat urine (19), G/K rat muscle, liver, and kidney (20), and human type 2 diabetic muscle (21), the level of chiro-inositol is decreased and myo-INS content increased compared to controls. The decrease in urinary chiro-inositol in humans (22) and monkeys (18) was inversely correlated to the degree of insulin resistance. Administration of DCI to streptozotocin type 2 diabetic rats (23), monkeys (24), and humans (25, 26) effectively decreased hyperglycemia and hypertriglyceridemia (HTG). It was also effective in human polycystic ovarian syndrome, where insulin resistance is an underlying factor, in restoring ovulation and metabolic balance (27).
Diabetes is associated with vascular disease. A much higher risk of myocardial infarction, stroke, and limb amputation (28) is related to endothelial dysfunction (ED) (29, 30). Hyperglycemia and hyperlipidemia are risk factors for ED and can induce ED in vitro and in vivo (29, 31). An underlying mechanism is the enhanced generation of endothelial mitochondrial superoxide (ROS) (29, 32–35). Pathophysiological mechanisms for hyperglycemia-induced ED via generation of ROS involve: (i) protein kinase C activation, (ii) increased generation of advanced glycation end products, (iii) increased glucose flux through the aldose reductase pathway, (iv) increased activity of glutamine:fructose-6-phosphate amidotransferase leading to insulin resistance, and (v) activation of NFκB leading to nuclear transcription of cytokines and inflammatory mediators. Additional factors include decreased NO production (35) and effectiveness (36).
We tested the hypothesis that inositol components of inositol glycan putative insulin mediators and one synthetic derivative, dibutyryl DCI (db-DCI), could prevent or reverse ED. Inositols were metabolically effective chronically in vivo reducing hyperglycemia and HTG and preventing ED. Furthermore, inositols were also effective acutely when administered in vitro to diabetic tissues to reverse ED. Inositols reduced ROS dose dependently in endothelial cells incubated in high glucose and scavenged superoxide in an in vitro xanthine/xanthine oxidase system. Furthermore, they enhanced NO action and protected NO signaling. Separate mechanisms underlying the beneficial chronic and acute effects are proposed.
Results
Orally administered inositols significantly decreased plasma glucose in alloxan-diabetic rats after 4 weeks. Percentage decreases were 8.8 ± 1.4% (Myo-INS NS vs. vehicle); 28.7 ± 3.1% DCI; (P < 0.05 vs. diabetes plus vehicle); 26.7 ± 2.5% 3-O-methyl DCI (me-DCI) (P < 0.05 vs. diabetes plus vehicle) and 31.1 ± 3.7% db-DCI; (P < 0.05 vs. diabetes plus vehicle). A slight increase of 11 ± 2.6% was observed in the diabetic group treated with saline. (Table 1, which is published as supporting information on the PNAS web site). All four inositols effected more pronounced decreases in plasma triglycerides compared to the diabetic saline-treated rats. HTG values decreased by 60.9 ± 5.8%, 51.7 ± 4.7%, 49.7 ± 4.3%, and 48.1 ± 5.1% (P < 0.01 vs. internal control; with me-DCI alone, the decrease did not achieve statistical significance compared to diabetic controls; Table 1) in diabetic animals gavaged with Myo-INS, DCI, me-DCI, db-DCI, respectively. In controls, a moderate decrease of 19 ± 4.7% was observed. Thus, inositols acted chronically to restore metabolic balance.
Endothelial-dependent relaxation induced by acetylcholine was strongly impaired in aortic rings and vascular mesenteric beds of 4-week alloxan-diabetic rats. Maximal relaxant response to acetylcholine in aortic rings from saline gavaged diabetic rats was 43.7 ± 7.6% with a PD2 of 7.2 (range, 6.8–7.6) compared to 88.3 ± 3.5% relaxation in rings from normoglycemic rats with a PD2 value of 7.5 (range, 7.4–7.6) (Fig. 1A). Chronic oral administration of inositols (20 mg/kg per 12 h; orally) for 4 weeks prevented ED observed in the untreated diabetic rats. The relaxant response induced by acetylcholine in aortas of inositol-treated diabetic rats was 38.7 ± 4.5% (Myo-INS), 54.7 ± 3.6% (DCI), 60.6 ± 3.6% (me-DCI), and 64.3 ± 4.9% (db-DCI) (P < 0.05, compared to saline-treated controls).
Fig. 1.

Effects of 1-month oral treatment with 20 mg/kg per 12 h myo-INS, DCI, 3-O-methyl DCI (me-DCI), or db-DCI in endothelium-dependent responses with acetylcholine (Ach) in aortic rings (A) or arteriolar mesenteric bed (B) of alloxan-diabetic rats (Diab). Responses were compared with either euglycemic (Eugly) or diabetic rats treated with saline (vehicle). (C and D) The effect of 1 h in vitro incubation with 1 μM inositols in 4-week saline treated diabetic rats. (C) Aortic rings. (D) Mesenteric arteriolar beds. Note the improvement in ED with both chronic (A and B) and acute (C and D) inositol treatment. Data are expressed as mean ± SEM of seven animals. *, P < 0.05 inositol treated vs. Diab plus vehicle; #, P < 0.05 inositol treated vs. Eugly plus vehicle; ANOVA followed by Tukey–Kramer.
Similarly, endothelium-dependent vasorelaxant response with acetylcholine in the vascular mesenteric bed was impaired in tissues from diabetic rats. Maximal decrease in perfusion pressure of the arteriolar mesenteric bed of normoglycemic rats was 96.9 ± 2.7% (PD2 = 5.4; range, 5.2–5.8) vs. 32.1 ± 4.3% (PD2 = 6.7; range, 6.3–7.1) in beds from 4-week alloxan-diabetic rats (Fig. 1B). Beds from alloxan-diabetic rats treated with inositols had higher maximal decreases in perfusion pressure to acetylcholine than control hyperglycemic rats treated with saline. Maximal relaxant response to ACh in mesenteric beds from inositol-treated diabetic rats was 54.4 ± 3.2% (Myo-INS), 58.7 ± 6.0% (DCI), 55.4 ± 3.2 (me-DCI), and 68.8 ± 3.3% (db-DCI) (P < 0.05 vs. diabetes plus vehicle) (Fig. 1B). Interestingly, maximal relaxant response and sensitivity of aortic rings to the endothelium-independent agonist sodium nitroprusside was not affected in any of the groups (Table 2, which is published as supporting information on the PNAS web site), indicating that the endothelium, not the smooth muscle, was defective.
To determine whether inositols could have a direct action on ED, we tested their acute “curative” effect on diabetic tissues from diabetic rats administered saline. The maximal relaxant response to Ach attained in aortic rings from euglycemic rats was 94.4 ± 4.6% compared to 43.6 ± 2.5% in aortic rings from diabetic rats treated with and incubated with saline (Fig. 1C). After 1-h incubation with 1 μM inositols, the maximal response was increased to 50.4 ± 3.6% (Myo-INS), 52.7 ± 3.6% (DCI), 58.8 ± 3.1% (me-DCI), and 67.4 ± 5.4% (db-DCI) (P < 0.05 vs. vehicle) (Fig. 1C). The same response pattern was observed in the arteriolar mesenteric bed acute incubations (Fig. 1D). Maximal decrease in perfusion pressure induced by Ach in tissues obtained from euglycemic rats was 95.1 ± 2.9% compared to 48.3 ± 5.7% in diabetic beds (Fig. 1D). When beds were perfused with inositols, the maximal response was increased to 50.3 ± 5.6% (Myo-INS), 60.2 ± 4.9% (DCI), 68.8 ± 4.7% (me-DCI), and 70.7 ± 4.6% (db-DCI) (Fig. 1D). The decrease in perfusion pressure induced by endothelial-independent vasodilator sodium nitroprusside added to aortic rings and mesenteric beds was no different between treated and untreated groups (Table 3, which is published as supporting information on the PNAS web site).
Acute experiments were performed in tissues from diabetic rabbits to test whether inositols would restore ED in another species. Endothelial relaxation in aortic rings from diabetic rabbits was also blunted as measured by the maximal response to Ach. In diabetic rings it was 41.3 ± 2.5% compared to 97.9 ± 1.3% (P < 0.005) in rings from euglycemic nondiabetics (Fig. 6A, which is published as supporting information on the PNAS web site). Incubation of aortic rings for 1 h with 1 μM inositols increased the endothelial response to 53.3 ± 5.6% (Myo-INS), 56.8 ± 3.4% (DCI), 63.5 ± 5.4% (me-DCI), and 85.4 ± 4.3% (db-DCI) (Fig. 6A). Similar acute “curative” results were demonstrated in the kidney vascular bed where the maximal endothelium-dependent response was restored from 22.9 ± 3.6% in diabetic kidneys to 36.7 ± 3.1% (Myo-INS), 42.7 ± 4.9% (DCI), 51.3 ± 4.6% (me-DCI), and 58.3 ± 5.9% (db-DCI) (Fig. 6B). The maximal response induced by Ach in kidneys isolated from euglycemic rabbits was 63.6 ± 4.9%. In contrast, the pattern of response to sodium nitroprusside was unchanged in the tissues of any group (Fig. 7 A and B, which is published as supporting information on the PNAS web site).
Relaxation of rabbit penile corpus cavernosa (RbCC) to 1 μM Ach was blunted in alloxan-diabetic rabbits (27.8 ± 3.8%) compared to the response from euglycemic rabbits (87 ± 8.0%) (P < 0.05). Incubation of RbCC from diabetic rabbits with inositols (1 μM for 1 h) restored the relaxant response to Ach to 33.3 ± 3.4% (Myo-INS), 37.6 ± 3.2% (DCI), 41.3 ± 5.4% (me-DCI), and 57.1 ± 4.4% (db-DCI) (Fig. 2A). The relaxant response to exogenous NO was also impaired in diabetic RbCC. RbCC from diabetic animals had a maximal relaxant response to 30 μM NO of 46.5 ± 3.4% compared to 84.6 ± 5.7% from euglycemic RbCC. Prior incubation of tissues with inositols restored this relaxation to 49.8 ± 6.3% (Myo-INS), 59.6 ± 3.4 (DCI), 61.3 ± 6.4% (me-DCI), and 68.5 ± 4.2% (db-DCI) (Fig. 2A). Nitrergic relaxation with transmural electrical field stimulation (EFS) was also impaired in RbCC from diabetic animals. Maximal relaxation to EFS was 89.6 ± 6.8% in euglycemic tissues compared to 37 ± 4.0% (P < 0.05; n = 7) in diabetic RbCC. Incubation with Myo-INS, DCI, me-DCI and db-DCI reversed the nitrergic response to 38.7 ± 4.3%, 42.5 ± 3.6%, 51.7 ± 4.4%, and 65.3 ± 3.7%, respectively (Fig. 2B).
Fig. 2.

Effects of in vitro incubation for 1 h in 1 μM myo-INS, DCI, 3-O-methyl DCI (me-DCI), or db-DCI in relaxation induced by 1 μM acetylcholine (ACh), 1–30 μMNO(A) or nitrergic relaxation induced by transmural electrical field stimulation (B) in strips of RbCC. Responses were compared with responses attained in tissues from euglycemic (Eugly) or diabetic rabbits (Diab) treated isovolumetrically with vehicle. Note the improvement in ED with acute inositol treatment enhancing NO action (A) and electrical field stimulation action (B). Data are expressed as mean ± SEM of seven animals. *, P < 0.05 inositol treated vs. Diab plus vehicle; #, P < 0.05 inositol treated vs. Eugly plus vehicle; ANOVA followed by Tukey–Kramer.
These studies (Figs. 1, 2, and 6) strongly indicate that the inositols act on the endothelial cells to reverse defective endothelial function in diabetes. Diabetic smooth muscle responds normally to chemically generated NO (Fig. 7 A and B and Tables 2 and 3, which are published as supporting information on the PNAS web site). Because endothelial ROS is a key underlying mechanism in diabetic complications including ED, we next examined the direct action of the inositols to reduce elevated ROS generated in the presence of high glucose in endothelial cells, an established protocol.
DCI and db-DCI (Fig. 3), added in the concentration range of 1–10 μM, reduced elevated ROS generated in the presence of high glucose (30 mM; Fig. 3) with db-DCI more effective than DCI. Baseline ROS production at 5 mM glucose concentration was 52.4 ± 1.8 nmol/ml and, in a high glucose medium (30 mM), increased to 121.5 ± 3.4 nmol/ml. DCI (10 μM) reduced ROS production to 58.2 ± 1.3 nmol/ml (P < 0.001 vs. 30 mM glucose alone), and db-DCI at the same concentration reduced ROS production to 54.5 ± 1.8 nmol/ml (P < 0.001 vs. 30 mM glucose alone). These data clearly demonstrate that inositols reduced the concentration of ROS within endothelial cells either through reduced generation via glycolytic metabolism or by increased scavenging.
Fig. 3.

Suppression by DCI and db-DCI of ROS in bovine aortic endothelial cells incubated in the presence of 30 mM glucose. Note the increased effect of 3,4-dibutyryl DCI (db-DCI) over DCI (DCI). Data are expressed as mean ± SEM of six experiments. *, P < 0.001 vs. 30 mM glucose (hg) alone; **, P < 0.01 vs. 30 mM glucose alone; #, P < 0.05 vs. 5 μM DCI plus 30 mM glucose; ANOVA followed by Tukey–Kramer. lg, 5 mM glucose.
We examined three sequelae of increased ROS in endothelial cells incubated in high glucose in the absence and presence of 20 μM db-DCI. Addition of db-DCI completely blocked the increased PKC activity seen in the presence of high glucose. Similarly, db-DCI addition completely blocked increased hexosamine accumulation and db-DCI completely blocked the generation of advanced glycation products (see Fig. 8, which is published as supporting information on the PNAS web site).
To investigate whether inositols might act directly as antioxidants, we tested their action on a ROS, superoxide, generated in vitro. As shown in Fig. 4, all four inositols showed dose-dependent decreases in the rate of reduction of nitroblue tetrazolium (NBT) induced by superoxide, generated by oxidizing xanthine with xanthine oxidase. The rate of reduction was unchanged in saline controls (not shown), whereas superoxide dismutase (SOD) (1 unit/ml) also effectively reduced the rate (not shown). Therefore, inositols themselves are effective scavengers of superoxide generated in vitro.
Fig. 4.

Effect of inositols on the NBT reduction rate induced by superoxide generated in a xanthine/xanthine oxidase (XA/XO) system compared with isovolumetric addition of vehicle. Note the increased effect of dibutyryl DCI (db-DCI). Data are expressed as mean ± SEM (n = 12). *, P < 0.05 inositols vs. vehicle; #, P < 0.05 db-DCI vs. other inositols; ANOVA followed by Tukey–Kramer.
To investigate the mechanism of the inositol's superoxide scavenging action, we tested the possible role of Fe3+, based on the known antioxidant action of phytate, myo-INS hexaphosphate (Fe3+)4 chelate. Addition of the iron chelator desferrioxamine completely reversed the effect of db-DCI (see Fig. 9, which is published as supporting information on the PNAS web site). Furthermore, the addition of excess Fe3+ partially restored the effect of db-DCI. This finding clearly demonstrates that db-DCI acts as a superoxide scavenger by a mechanism similar to phytate requiring the presence of Fe3+.
Superoxide reacts rapidly with NO and thus by scavenging superoxide inositols may increase the bioavailability of NO. To assess the effects of inositols on in vivo generated NO by eNOS, the increase in tone was measured after the addition of 100 μM l-NAME to phenylephrine precontracted aortic rings from diabetic rats. Maximal increase in tonus induced by l-NAME was 67.3 ± 4.9% in euglycemic rings compared to 8.6 ± 3.8% (P < 0.05) from diabetic rings (Fig. 5). Acute incubation with inositols (1 μM for 1 h) restored this l-NAME-induced increase in tonus to 14 ± 2.7% (Myo-INS), 23.5 ± 6.4% (DCI), 26.3 ± 5.6% (me-DCI), and 43.5 ± 6.4% (db-DCI) (Fig. 5) demonstrating enhanced eNOS activity in response to inositols.
Fig. 5.

Effects of incubation of 1 μM inositols for 1 h to increase tonus induced by 100 μM l-NAME in diabetic rat aortic rings. Note the acute effect of inositols to enhance eNOS activity. #, P < 0.05 vs. euglycemic (Eugly); *#, P < 0.05 vs. diabetic control (C) and euglycemic (Eugly); ANOVA followed by Bonferroni.
Similarly, vasodilatation induced by exogenous NO was blunted in diabetic rat aortic rings, but restored in inositol and SOD acutely treated tissues (Fig. 10, which is published as supporting information on the PNAS web site). All inositols increased the bioavailability of NO dose dependently to enhance NO induced relaxation, with db-DCI and me-DCI being most effective.
The formation of protein S-nitrosothiol (SNO) within both the endothelial surface and the smooth muscle has been shown to be associated with NO-mediated vasorelaxation. We assessed protein SNO formation in aortic rings from diabetic hyperglycemic, euglycemic, and diabetic rats gavaged with myo-INS and db-DCI for 4 weeks by immunohistochemistry (Fig. 11, which is published as supporting information on the PNAS web site). Protein SNO staining was reduced in untreated diabetic rings compared to euglycemic controls. Chronic treatment with Myo-INS and db-DCI clearly enhanced protein SNO staining over that seen in diabetic animals. Nonparametric scoring of the endothelial protein SNO revealed a significant increase in staining within the inositol-treated animals over the hyperglycemic diabetics (hyperglycemic 1, n = 4; euglycemic 2.5, n = 6; db-DCI 3, n = 5, Myo-INS 4, n = 5). Interestingly, staining in normal and treated tissues extended into subendothelial sites as well as endothelial as has been previously seen in mouse vessel rings. In summary, inositols chronically metabolically prevented ED, acutely reversed ED in diabetic tissues, acutely reduced ROS and sequelae in endothelial cells, acutely scavenged superoxide in vitro, acutely enhanced eNOS and NO action in diabetic tissues, and chronically maintained NO signaling.
Discussion
Decreased endothelial-dependent relaxation, a common assay for ED in aortic rings (37–39) and mesenteric microvessels (40, 41), indicates that both conductance and resistance vessels are affected. In the diabetic mesentery the vascular tree undergoes hypertrophy preceded by attenuated endothelial dependent relaxation (42, 43). Oxygen radicals contribute to enhanced basal vascular tone, tubuloglomerular feedback, monocyte/macrophage infiltration and impaired endothelium-dependent relaxation in diabetic kidney (38, 39). SOD reverses the impaired activity of nitric oxide in renal arterioles (44). Clearly, superoxide is responsible, at least in part, for endothelial dysfunction in the renal vasculature (45, 46). Also related to ED are blunted neurogenic (nitrergic) and endothelium-dependent relaxant responses in diabetic men with erectile dysfunction (47), also observed in alloxan-diabetic rabbits (48).
Aortic rings, arteriolar mesenteric beds, vascular kidney beds, and penile corpora cavernosa from both rats and rabbits demonstrated impaired relaxations to acetylcholine in a 4-week alloxan induced diabetes model. There was significant improvement in relaxation with chronic and/or acute inositol administration.
Separate mechanisms are proposed to explain the chronic metabolic and the acute ROS scavenging effects. DCI and me-DCI have already been shown to have insulin-like metabolic effects in diabetic animals, primates, and humans, decreasing hyperglycemia and hyperlipidemia (1, 11, 12, 23, 24, 27). Clearly, the inositols were active in correcting hyperglycemia and HTG, key components known to induce ED in normal and diabetic tissues. Our hypothesis is that they act metabolically by first being incorporated into precursor IPG phospholipids and/or proteins, then subsequently cleaved into biologically active IPGs. Insulin-like actions of both biologically derived (1–3, 5–7) and chemically synthesized Myo-INS and me-DCI containing IPGs have been clearly documented (9, 10). Most recently, we have demonstrated the mechanism of action of a novel galactosamine pinitol pseudodisaccharide to activate PP2C (49).
Beneficial effects in response to low concentrations of Ach suggests that the inositols enhanced tissue sensitivity to Ach. Relaxation by sodium nitroprusside, an endothelium-independent agonist, was not affected by diabetes or by inositol treatment. Thus, (i) the cGMP pathway itself is not impaired by diabetes, but the endothelium is possibly compromised as a source of NO, (ii) the effect of the inositols is likely not myogenic, and (iii) improvement of endothelial function is probably due to an effect of the inositols (direct or indirect) on the defective endothelial cells.
Potential mechanisms by which the inositols might act acutely to reverse endothelial based ED include decreasing ROS and enhancing endothelial NOS and NO bioactivity. Oxidative damage has been demonstrated to play a pivotal role in the development of diabetic complications (50–52). A causal relationship between oxidative stress and endothelial dysfunction in diabetes is supported by the decreased expression of SOD in aortas of diabetic animals (53) and by the demonstration that increased expression of SOD restores ED in diabetic vessels (54). Tempol, an SOD mimetic, ameliorates the endothelium-dependent response to ACh in renal afferent arterioles of diabetic rabbits (55, 56).
We next demonstrated that DCI and db-DCI dose-dependently reduced ROS to basal, with db-DCI more effective than DCI in endothelial cells incubated in high glucose.
Furthermore, Db-DCI restored to normal three sequelae of increased ROS, namely increased PKC, increased hexosamine pathway activity, and increased advanced glycation end products.
Inositols added to superoxide generated in an in vitro xanthine/xanthine oxidase system eliminated dose-dependently ROS. Again, db-DCI was more effective than the other three. Clearly inositols act here as well as on endothelial cells as superoxide scavengers.
It is well recognized that phytate, as an Fe3+ chelate, is a powerful antioxidant. Studies have shown that phytate (Fe3+)4 blocks ·OH formation by virtue of its lack of an available water coordination site and by its ability to accelerate 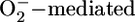 Fe2+ depletion while inhibiting the Fenton reaction (57, 58).
Fe2+ depletion while inhibiting the Fenton reaction (57, 58).
Myo-INS was recently suggested as a free radical scavenger (59). Although no effect to reduce ·OH was observed in a xanthine/xanthine oxidase system where phytate clearly was effective, myo-INS did protect intact closed circular DNA from strand breaks suggesting an antioxidant effect.
Studies with added desferrioxamine, an Fe chelator, demonstrate that it effectively blocked the action of db-DCI and that added Fe3+ in turn reversed the effect of desferrioxamine. The mechanism of action of the free inositol db-DCI is thus similar to action of phytate because both require the presence of Fe3+ for their antioxidant action. The detailed mechanism will require further investigation.
Superoxide reacts chemically with NO to produce peroxynitrite, a powerful oxidant (60). NO and eNOS are known to be impaired in ED and in diabetes (35, 36, 44). The eNOS inhibitor l-NAME acts to induce contraction. The improvement in the impaired eNOS and NO system was shown by the inositol addition enhanced contraction in the presence of l-NAME and the enhanced relaxation in the presence of NO.
To investigate inositols protecting NO signaling systems, we examined protein-SNO-stained rat aortic rings from controls, diabetics administered saline, and diabetics administered inositols. The abundant staining seen in the endothelium of control rings was markedly reduced in the untreated diabetic rings. Gavaged inositols resulted in enhanced staining seen in the rings from the treated animals with staining essentially restored to normal. Of interest, subendothelial staining seen in the control was also seen in the treated with subendothelial staining as previously noted (61). Thus, inositols act chronically to preserve NO signaling by maintaining protein SNO.
Recently, three cellular transporters for myo-INS have been identified, two Na+ coupled, SMIT1 and SMIT2, and one proton-coupled, HMIT (62). Furthermore, SMIT2 has been shown to transport DCI (63).
The present studies demonstrate that inositols, particularly DCI derivatives such as db-DCI, deserve consideration as therapeutic agents for preventing and treating metabolic syndrome, ED, and erectile dysfunction by virtue of both their chronic metabolic, acute ROS scavenging, and NO protective beneficial effects.
Furthermore, in connection with a possible therapeutic application, we have demonstrated a decrease in chiro-inositol in muscle, hemodialysate, and urine of type 2 diabetics (17, 21).
Materials and Methods
Animals. Diabetic rats and rabbits were prepared as detailed in Supporting Text, which is published as supporting information on the PNAS web site (64). Diabetic rats were gavaged with saline or inositols for 4 weeks, and tissues were removed and tested for ED. Diabetic tissues from the saline-gavaged animals were used for the “acute” treatment protocols.
Drugs and Chemicals. Acetylcholine and sodium nitroprusside were from Merck; sodium nitrate and indomethacin were from Sigma. Inositols were from Allomed Pharmaceuticals and Sigma.
Blood Sampling and Biochemical Assays. Venous blood was obtained by infraorbital plexus puncture. Plasma triglycerides and total cholesterol were measured by standard enzymatic methods with commercial kits.
Blood Pressure Measurements. The right carotid artery was cannulated, and systemic blood pressure was recorded via a pressure transducer and polygraph.
Tissue Preparation. Diabetic rat and rabbit aortic thoracic rings, rat arteriolar mesenteric beds, rabbit renal vascular beds, and rabbit penile corpus cavernosum were prepared surgically under anesthesia and perfused by standard procedures.
Concentration dose–response curves were performed to vasodilators, i.e., acetylcholine or sodium nitroprusside. Experiments were performed in the presence of indomethacin (10–5 M) to avoid interference of cyclooxygenase products.
To test the acute effect of inositol administration, concentration dose–response curves to both endothelium-dependent and -independent agonists were performed in the absence and presence of inositols (1 μM for 1 h).
For rabbit RbCC, transmural EFS was performed. Stimuli were studied in the absence and presence of 1 μM inositols.
Endothelial Cell ROS Production. Bovine aortic endothelial cells were cultured in normal glucose and high glucose for 4–6 h. ROS was determined fluorometrically by using the probe CM-H2DCFDDA (34, 35).
PKC Activity. Assay was performed by using the Protein Kinase C Assay System (Invitrogen).
Hexosamine Pathway Activity. Hexosamine pathway activity was determined by measuring UDP-GlcNAc concentration (65).
Advanced Glycation End Products. Methylglyoxal-derived imidizole advanced glycation end products were detected with monoclonal Ab 1H7G5 at 1:10,000 dilution (66, 67).
Protein SNO. Aortic rings were fixed, embedded in paraffin, sectioned, and stained for SNO (61, 68).
NO-Potentiation and NBT Reduction Test. NO-induced (1 μM) relaxation was evaluated in diabetic rat aortic rings. Superoxide anions (O–2) were generated by xanthine (1 mM) oxidized by xanthine oxidase (10 milliunits/ml). Rate of NBT reduction by superoxide anion was determined spectrophotometrically.
Statistical Analysis. Biochemical parameters were analyzed by two-tailed paired Student's t test and considered significantly different if the probability of the occurrence of the null hypothesis was <5%.
Supplementary Material
Acknowledgments
We thank Dr. David L. Brautigan for review of and comments on the manuscript.
Author contributions: J.L. and M.C.F. designed research; N.R.F.N., L.M.A.L., M.R.K., C.M.S., R.S.A., M.G.R.Q., J.P., D.B.H., X.D., M.B., A.G., C.D., and M.C.F. performed research; J.L. and M.C.F. contributed new reagents/analytic tools; N.R.F.N., L.M.A.L., M.R.K., C.M.S., R.S.A., M.G.R.Q., J.P., D.B.H., J.L., X.D., M.B., A.G., C.D., and M.C.F. analyzed data; and N.R.F.N., L.M.A.L., M.R.K., C.M.S., R.S.A., M.G.R.Q., J.P., D.B.H., J.L., X.D., M.B., A.G., C.D., and M.C.F. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: IPG, inositol phosphoglycan; DCI, d-chiro-inositol; HTG, hypertriglyceridemia; ED, endothelial dysfunction; me-DCI, 3-O-methyl DCI; db-DCI, dibutyryl DCI; RbCC, rabbit penile corpus cavernosa; EFS, electrical field stimulation; NBT, nitroblue tetrazolium; SOD, superoxide dismutase; SNO, S-nitrosothiol.
References
- 1.Larner, J. & Huang, L. C. (1999) Diabetes Rev. 7, 217–231. [Google Scholar]
- 2.Caro, H. N., Kunjara, S., Rademacher, T. W., León, Y., Jones, D. R., Avila, M. A. & Varela-Nieto, I. (1997) Biochem. Mol. Med. 61, 214–228. [DOI] [PubMed] [Google Scholar]
- 3.Larner, J., Huang, L. C., Tang, G., Susuki, S., Schwartz, C. F. W., Romero, G., Roulidis, Z., Zeller, K, Shen, T. Y., Oswald, A. S., et al. (1988) Cold Spring Harbor Symp. Quant. Biol. 53, 965–971. [DOI] [PubMed] [Google Scholar]
- 4.Huang, L. C., Heimark, D., Linko, J., Nolan, R. & Larner, J. (1999) Biochem. Biophys. Res. Commun. 255, 150–156. [DOI] [PubMed] [Google Scholar]
- 5.Malchoff, C. D., Huang, L., Gillespie, N., Villar-Palasi, C., Schwartz, C. F. W., Cheng, K., Hewlett, E. L. & Larner, J. (1987) Endocrinology 120, 1327–1337. [DOI] [PubMed] [Google Scholar]
- 6.Romero, G., Luttrell, L., Rogol, A., Zeller, K., Hewlett, E. & Larner, J. (1988) Science 240, 509–511. [DOI] [PubMed] [Google Scholar]
- 7.Müller, G., Wied, S., Crecelius, A., Kessler, A. & Eckel, J. (1997) Endocrinology 138, 3459–3475. [DOI] [PubMed] [Google Scholar]
- 8.Müller, G., Wied, S., Piossek, C., Bauer, J. & Frick, W. (1998) Mol. Med. 4, 299–323. [PMC free article] [PubMed] [Google Scholar]
- 9.Frick, W., Bauer, A., Bauer, J., Wied, S. & Müller, G. (1998) Biochem. J. 336, 163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larner, J., Price, J. D., Heimark, D., Smith, L., Rule, G., Piccariello, T., Fonteles, M. C., Pontes, C., Vale, D. & Huang, L. (2003) J. Med. Chem. 46, 3283–3291. [DOI] [PubMed] [Google Scholar]
- 11.Huang, L. C., Fonteles, M. C., Houston, D. B., Zhang, C. & Larner, J. (1993) Endocrinology 132, 652–657. [DOI] [PubMed] [Google Scholar]
- 12.Fonteles, M. C., Huang, L. C. & Larner, J. (1996) Diabetologia 39, 731–734. [DOI] [PubMed] [Google Scholar]
- 13.Abe, S., Huang, L. C. & Larner, J. (1996) in Alpha-keto Acid Dehydrogenase Complex, eds. Patel, M. S., Roche, T. E. & Harris, R. A. (Birkhauser, Basel), pp. 187–195.
- 14.Larner, J. (2001) Int. Union Biochem. Mol. Biol. Life 51, 139–148. [Google Scholar]
- 15.Villar-Palasi, C. & Larner, J. (1960) Biochem. Biophys. Acta 39, 171–173. [DOI] [PubMed] [Google Scholar]
- 16.Moule, S. K. & Denton, R. M. (1977) Am. J. Cardiol. 80, 41A–49A. [DOI] [PubMed] [Google Scholar]
- 17.Kennington, A. S., Hill, C. R., Craig, J., Bogardus, C., Raz, I., Ortmeyer, H. K., Hansen, B. C., Romero, G. & Larner, J. (1990) N. Eng. J. Med. 323, 373–378. [DOI] [PubMed] [Google Scholar]
- 18.Ortmeyer, H. K., Bodkin, N. L., Lilley, K., Larner, J. & Hansen, B. C. (1993) Endocrinology 132, 640–645. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki, S., Taneda, Y., Hirai, S., Abe, S., Sasaki, A., Auzuki, K. & Toyota, T. (1991) in New Directions in Research and Clinical Works for Obesity and Diabetes Mellitus, eds. Angel, A. & Hotta, N. (Elsevier, Amsterdam), pp. 197–203.
- 20.Sun, T., Heimark, D. B., Nguygen, T., Nadler, J. L. & Larner, J. (2002) Biochem. Biophys. Res. Commun. 293, 1092–1098. [DOI] [PubMed] [Google Scholar]
- 21.Asplin, I., Galasko, G. & Larner, J. (1993) Proc. Natl. Acad. Sci. USA 90, 5924–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki, S., Kawasaki, H., Satoh, Y., Ohtomo, M., Hirai, M., Hirai, A., Hirai, S., Oneda, M., Matsumoto, M., Hinokio, Y., et al. (1994) Diabetes Care 17, 1465–1468. [DOI] [PubMed] [Google Scholar]
- 23.Ortmeyer, H. K., Huang, L. C., Zhang, L., Hansen, B. C. & Larner, J. (1993) Endocrinology 132, 646–651. [DOI] [PubMed] [Google Scholar]
- 24.Ortmeyer, H. K., Bodkin, N. L., Hansen, B. C. & Larner, J. (1995) J. Nutr. Biochem. 6, 499–503. [Google Scholar]
- 25.Larner, J., Allan, G., Kessler, C., Reamer, P., Gunn, R. & Huang, L. C. (1998) J. Basic Clin. Physiol. Pharmacol. 9, 127–137. [DOI] [PubMed] [Google Scholar]
- 26.Larner, J. (2002) Int. J. Exp. Diabetes Res. 3, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nestler, J. E., Jakubowicz, D. J., Reamer, P., Gunn, R. D. & Allan, G. (1999) N. Engl. J. Med. 340, 1314–1320. [DOI] [PubMed] [Google Scholar]
- 28.Ostlund, R. E., Jr., McGill, J. B., Hershkowitz, I., Kipnis, D. M., Santiago, J. V. & Sherman, W. R. (1993) Proc. Natl. Acad. Sci. USA 90, 9988–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brownlee, M. (2001) Nature 414, 813–820. [DOI] [PubMed] [Google Scholar]
- 30.Tesfamariam, B., Brown, M. L. & Cohen, R. A. (1991) J. Clin. Invest. 87, 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinberg, H. O., Tarshoby, M., Monestel, R., Hook, G., Cronin, J., Johnson, A., Bayazeed, B. & Baron, A. D. (1997) J. Clin. Invest. 100, 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundman, P., Eriksson, M. J., Schenk-Gustafsson, K., Karpe, F. & Tornvall, P. (1997) Circulation 96, 3266–3268. [DOI] [PubMed] [Google Scholar]
- 33.Tesfamariam, B. (1994) Free Radical Biol. Med. 16, 383–391. [DOI] [PubMed] [Google Scholar]
- 34.Nishikawa, T., Edelstein, D., Du, X. L., Yamagishi, S., Matsumura, T., Kaneda, Y., Yorek, M. A., Beebe, D., Oates, P. J., Hammes, H. P., et al. (2000) Nature 404, 787–790. [DOI] [PubMed] [Google Scholar]
- 35.Du, X. L., Edelstein, D., Rossetti, L., Fantus, I. G., Goldberg, H., Ziyadeh, F., Wu, J. & Brownlee, M. (2000) Proc. Natl. Acad. Sci. USA 97, 12222–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pieper, G. M. (1999) Diabetologia 42, 204–213. [DOI] [PubMed] [Google Scholar]
- 37.Zavaroni, I., Piatti, P. M., Monti, L. D., Gasparini, P., Barilli, L. A., Massironi, P., Ardigo, D., Valsecchi, G., Delsignore, R. & Reaven, G. M. (2000) Metabolism 49, 959–961. [DOI] [PubMed] [Google Scholar]
- 38.Kamata, K., Miyata, N. & Kasuya, Y. (1998) Br. J. Pharmacol. 123, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez-Mañas, L., Angulo, J., Peiró, C., Liergo, J. L., Sanchez-Ferrer, A., Lopez-Doriga, P. & Sanchez-Ferrer, C. F. (1998) Br. J. Pharmacol. 123, 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattori, Y., Kawasaki, H., Abe, K. & Kanno, M. (1991) Am. J. Physiol. 261, H1086–H1094. [DOI] [PubMed] [Google Scholar]
- 41.Lash, J. M. & Bohlen, H. G. (1991) Circ. Res. 69, 1259–1268. [DOI] [PubMed] [Google Scholar]
- 42.Taylor, P. D., McCarthy, A. L., Thomas, C. R. & Poston, L. (1992) Br. J. Pharmacol. 107, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper, M. E., Rumble, J., Komers, R., Du, H. C., Jandeleit, K. & Chou, S. T. (1994) Diabetes 43, 1221–1228. [DOI] [PubMed] [Google Scholar]
- 44.Ohishi, K. & Carmines, P. K. (1995) J. Am. Soc. Nephrol. 5, 1559–1566. [DOI] [PubMed] [Google Scholar]
- 45.Schnackenberg, C. G. & Wilcox, C. S. (2001) Kidney Int. 59, 1859–1864. [DOI] [PubMed] [Google Scholar]
- 46.Mügge, A., Elwell, J. H., Peterson, T. E. & Harrison, D. G. (1991) Am. J. Physiol. 260, C219–C225. [DOI] [PubMed] [Google Scholar]
- 47.Huie, R. E. & Padmaja, S. (1993) Free Radical Res. Commun. 18, 195–199. [DOI] [PubMed] [Google Scholar]
- 48.Saenz de Tejada, I., Goldstein, I., Azadzoi, K., Krane, R. J. & Cohen, R. A. (1989) N. Engl. J. Med. 320, 1025–1030. [DOI] [PubMed] [Google Scholar]
- 49.Brautigan, D. L., Brown, M., Grindrod, S., Chinigo, G., Kruszewski, A., Lukasik, S. M., Bushweller, J. H., Horal, M., Keller, S., Tamura, S., et al. (2005) Biochemistry 44, 11067–11073. [DOI] [PubMed] [Google Scholar]
- 50.den Hartog, G. J., Haenen, G. R., Vegt, E., van der Vijgh, W. J. & Bast, A. (2003) Chem. Biol. Interact. 145, 33–39. [DOI] [PubMed] [Google Scholar]
- 51.Wohaieb, S. A. & Godin, D. V. (1987) Diabetes 36, 1014–1018. [DOI] [PubMed] [Google Scholar]
- 52.Baynes, J. W. (1991) Diabetes 40, 405–412. [DOI] [PubMed] [Google Scholar]
- 53.Giugliano, D., Ceriello, A. & Paolisso, G. (1996) Diabetes Care 19, 257–267. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi, T. & Kamata, K. (1999) Eur. J. Pharmacol. 367, 213–222. [DOI] [PubMed] [Google Scholar]
- 55.Rumble, J. R., Cooper, M. E., Soulis, T., Cox, A., Wu, L., Youssef, S., Jasik, M., Jerums, G. & Gilbert, R. E. (1997) J. Clin. Invest. 99, 1016–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schnackenberg, C. G. (2002) Curr. Opin. Pharmacol. 2, 121–125. [DOI] [PubMed] [Google Scholar]
- 57.Graf, E., Mahoney, J. R., Bryant, R. G. & Eaton, J. W. (1984) J. Biol. Chem. 259, 3620–3624. [PubMed] [Google Scholar]
- 58.Graf, E. & Empson, K. L. (1987) J. Biol. Chem. 262, 11647–11650. [PubMed] [Google Scholar]
- 59.Muraoka, S. & Miura, T. (2004) Life Sci. 74, 1691–1700. [DOI] [PubMed] [Google Scholar]
- 60.Wattanapitayakul, S. K., Weinstein, D. M., Holycross, B. J. & Bauer, J. (2000) FASEB J. 14, 271–278. [DOI] [PubMed] [Google Scholar]
- 61.Gow, A. J., Chen, Q., Hess, D. T., Day, B. J., Ischiropoulos, H. & Stamler, J. S. (2002) J. Biol. Chem. 277, 9637–9640. [DOI] [PubMed] [Google Scholar]
- 62.Bourgeois, F., Coady, M. J. & Lapointe, J. Y. (2005) J. Physiol. 563, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coady, M. J., Wallendorff, B., Gagnon, D. G. & Lapointe, J. Y. (2002) J. Biol. Chem. 277, 35219–35224. [DOI] [PubMed] [Google Scholar]
- 64.Sannomiya, P., Oliveira, M. A. & Fortes, Z. B. (1997) Br. J. Pharmacol. 122, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Du, X., Matsumura, T., Edelstein, D., Rossetti, L., Zsengeller, Z., Szabo, C. & Brownlee, M. (2003) J. Clin. Invest. 112, 986–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shinohara, M., Thornalley, P. J., Giardino, I., Beisswenger, P., Thorpe, S. R., Onorato, J. & Brownlee, M. (1998) J. Clin. Invest. 101, 1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hammes, H. P., Du, X., Edelstein, D., Taguchi, T., Matsumura, T., Ju, Q., Lin, J., Bierhaus, A., Nawroth, P., Hannak, D., et al. (2003) Nat. Med. 9, 294–299. [DOI] [PubMed] [Google Scholar]
- 68.Gow, A. J., McClelland, M., Garner, S. E., Malcolm, S. & Ischiropoulos, H. (1998) Methods Mol. Biol. 100, 291–299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}