

Abstract
Recent decades have seen tremendous growth in our understanding of the cognitive dysfunctions observed in Huntington's disease (HD). Advances in neuroimaging have contributed greatly to this growth. We reviewed the role that structural and functional neuroimaging techniques have played in elucidating the cerebral bases of the cognitive deficits associated with HD. We conducted a computer-based search using PubMed and PsycINFO databases to retrieve studies of patients with HD published between 1965 and December 2004 that reported measures on cognitive tasks and used neuroimaging techniques. Structural neuroimaging has provided important evidence of morphological brain changes in HD. Striatal and cortical atrophy are the most common findings, and they correlate with cognitive deficits in attention, working memory and executive functions. Functional studies have also demonstrated correlations between striatal dysfunction and cognitive performance. Striatal hypoperfusion and decreased glucose utilization correlate with executive dysfunction. Hypometabolism also occurs throughout the cerebral cortex and correlates with performance on recognition memory, language and perceptual tests. Measures of presynaptic and postsynaptic dopamine biochemistry have also correlated with measurements of episodic memory, speed of processing and executive functioning. Aided by the results of numerous neuroimaging studies, it is becoming increasingly clear that cognitive deficits in HD involve abnormal connectivity between the basal ganglia and cortical areas. In the future, neuroimaging techniques may shed the most light on the pathophysiology of HD by defining neurodegenerative disease phenotypes as a valuable tool for knowing when patients become “symptomatic,” having been in a gene-positive presymptomatic state, and as a biomarker in following the disease, thereby providing a prospect for improved patient care.
Medical subject headings: brain imaging, cognition, functional neuroimaging, Huntington disease, structural neuroimaging
Abstract
Au cours des dernières décennies, notre compréhension des dysfonctionnements cognitifs observés dans les cas de chorée de Huntington (CH) a beaucoup progressé. Les progrès de la neuro-imagerie ont contribué énormément à cette évolution. Nous avons étudié le rôle que les techniques de neuro-imagerie structurelle et fonctionnelle ont joué dans l'explication des bases cérébrales des déficits cognitifs associés à la CH. Nous avons effectué une recherche informatique dans les bases de données PubMed et PsycINFO pour en extraire des études portant sur des patients atteints de CH, qui ont été publiées entre 1965 et décembre 2004 et où l'on a signalé des mesures portant sur des tâches cognitives et utilisé des techniques de neuro-imagerie. La neuro-imagerie structurelle a produit des données importantes sur les changements morphologiques du cerveau provoqués par la CH. Les constatations les plus courantes sont une atrophie des corps striés et du cortex, qui est associée aux déficits cognitifs de l'attention, de la mémoire de travail et des fonctions d'exécution. Des études de fonction ont aussi démontré des liens entre le dysfonctionnement striatal et le rendement cognitif. L'hypoperfusion striatale et la diminution de l'utilisation du glucose sont reliés au dysfonctionnement de l'exécution. Il y a aussi hypométabolisme dans tout le cortex cérébral, ce qui est relié aux résultats de tests de mémoire de reconnaissance, de langue et de perception. On a aussi constaté des liens entre les mesures biochimiques de la dopamine présynaptique et postsynaptique et les mesures de la mémoire épisodique, de la rapidité de traitement et du fonctionnement d'exécution. Compte tenu des résultats de nombreuses études de neuro-imagerie, il devient de plus en plus clair que les déficits cognitifs des patients atteints de CH mettent en cause une connectivité anormale entre les noyaux gris centraux et les régions corticales. À l'avenir, les techniques de neuro-imagerie pourront sans doute expliquer le mieux la pathophysiologie de la CH en définissant les phénotypes des maladies neurodégénératives comme outil valable qui aide à savoir quand les patients deviennent « symptomatiques » après avoir été dans un état présymptomatique positif sur le plan génétique, et comme marqueur biologique permettant de suivre la maladie, ce qui permet d'envisager une amélioration du soin des patients.
Introduction
Huntington's disease (HD) is a monogenic neurodegenerative disorder clinically characterized by progressive involuntary movements, neuropsychiatric disturbances and cognitive impairments. At a molecular level, the disease is caused by an extended trinucleotide (CAG) repeat on chromosome 4,1 which results in widespread neuronal degeneration preferentially within the striatum.2 As the disease progresses, the pathological process eventually involves other brain regions, such as the frontal and temporal lobes,3 and at death the brain can have lost 25% of its volume.4 Because the primary pathology in HD lies in subcortical regions, the cognitive deficits in HD have been classically characterized, despite some recent controversy, as a subcortical dementia syndrome, that is, a pattern of cognitive deficits that suggest dysfunction of the frontal–subcortical neuronal circuitry.
Within the last 4 decades, great strides have been made that have furthered our understanding of the neural bases of HD. Advances in neuroimaging have played a prominent role in this effort. The demonstration of basal ganglia and cortical atrophy by structural brain imaging studies has provided strong in-vivo evidence of structural cerebral changes in patients with HD.5,6 The synergy between neuropsychology and neuroimaging techniques7 has been used to identify the neural correlates of cognitive deficits in patients with HD.
In this article, we review the neuroimaging studies that have explored the processes underlying the cognitive deficits in HD. After a brief description of the cognitive dysfunctions and morphological brain changes in HD, we will present a synopsis of the relevant neuroimaging literature. We conducted a computer-based search using PubMed and PsycINFO databases to retrieve studies of patients with HD published between 1965 and December 2004 that reported measures on cognitive tasks and used neuroimaging techniques. We have purposely focused our review on functional and structural neuroimaging and cognitive dysfunction of adult-onset HD. Although cognitive dysfunction is believed to be an important aspect in the early onset variant and in presymptomatic gene carriers, the neuroimaging literature concerning these areas is beyond the intended scope of this review.
Cognitive dysfunction in HD
Cognitive dysfunction has always been considered an intrinsic feature of HD; however, the natural history of HD-related cognitive impairment is still not completely understood mainly because of the heterogeneous nature of the disorder and wide variation in the design of the longitudinal studies conducted to date.
Although there is evidence for preclinical cognitive dysfunction in HD,8,9 studies suggest that there may be variation in the nature and time course of the earliest changes, as well as variation in the evolution of such cognitive deficits over time.
Longitudinal neuropsychological studies of asymptomatic mutation carriers have found subtle cognitive deficits in psychomotor, attentional and executive functions,10 as well as deficits in semantic verbal fluency and visual working memory,8 before the onset of motor symptoms.9
Some cognitive deficits appear to have a gradual onset with an insidious progression, whereas others appear to have a relatively acute onset, occurring in a stepwise fashion in the immediate period around clinical onset. For example, Snowden and colleagues11 observed subtle changes in psychomotor performance in individuals with the HD mutation that were apparent years before the onset of HD and progressed insidiously as the disease progressed, whereas other changes, particularly those in memory, appeared around the time of clinical onset and had a more pronounced decline.
In the early stages of the disorder, cognitive dysfunctions in attention, concentration,12,13 visuospatial abilities, emotional processing14 and memory15 become more apparent, but again vary in their evolution over time. One of the largest cohorts of patients with mild-to-moderate HD in whom cognitive decline8 was examined showed a progressive impairment in attention, executive function and immediate memory. In addition, tasks of psychomotor functioning (Trail-Making Test A [TMT A], the reading and colour naming subsets of the Stroop Test, Symbol Digit Modality Test [SDMT]) and executive function and planning (Trail-Making Test B [TMT-B] and Tower of London Test) showed the greatest decline over time.8 On the other hand, patients also exhibited an overall deficit in recognizing emotion, with particularly severe problems regarding recognition of expressions of disgust from both faces and voices.14
Regardless of the variation in the progression of the dysfunction, cognitive changes become more severe as the disorder evolves. At later stages of the disease, patients exhibit deficits in executive functioning especially on tests that require planning, problem solving, cognitive flexibility and freedom from distraction, such as the Wisconsin Card Sorting Test16 and the Tower of London Test.17 Deficits are particularly evident on tests in which patients must inhibit inappropriate responses or where sufficient cognitive and behavioural flexibility is demanded (i.e., shifting cognitive or behavioural sets).18
Visuospatial abilities also become more affected throughout the development of HD. Patients have shown deficits on tasks that require visuospatial processing skills, such as the Block Design subtest of the Wechsler Adult Intelligence Scale (WAIS), the Clock Drawing Test, the Porteus Maze Test, the Benton Visual Retention Test and the Halsted–Reitan Neuropsychological Test Battery.19,20 More specific visuospatial abilities have also been shown to be impaired in patients with HD. Compared with patients with Alzheimer's disease, patients with HD performed worse on tests that involved personal and extra-personal visuospatial orientation, such as the Money Road Map Test.20 Compared with control subjects, patients with HD have shown impairments in overall processing capacity and spatial manipulation domains.16,18
As time progresses, memory deficits become more manifest. Impairments reported in patients with HD range from deficits in tasks involving short-term memory21 to long-term memory tests, including deficits in declarative memory (i.e., episodic memory, working memory)22 and procedural memory (i.e., implicit memory).23 In a meta-analysis used to estimate the consistency, strength and selectivity of neuropsychological deficits in HD, Zakzanis24 indicated that patients with HD are most deficient on tests of delayed recall (more specifically, verbal and visual delayed recall) followed by tests of memory acquisition, cognitive flexibility and abstraction, manual dexterity, attention and concentration, performance skill and verbal skill. The neuropsychological pattern, as demonstrated by Zakzanis, of poor performance on delayed recall and memory acquisition tests together with relatively good performance on recognition tests suggests a retrieval-based memory deficit due to frontostriatal dysfunction. However, a recent study showed that patients with HD exhibited similarly impaired performance in both recall and recognition memory.24
Ultimately, cognitive impairments will gradually worsen to a dementia characterized by a slowing of information processing, decreased motivation, depression, apathy and personality changes, although language is relatively spared. This pattern of deficits has been called a subcortical dementia syndrome16 and is thought to be distinct from the frank amnesia, aphasia, apraxia and agnosia that embodies the cortical dementia syndrome associated with disorders such as Alzheimer's disease.25
Although the many neuropsychological deficits and their patterns of deterioration over time appear quite disparate on the surface, many researchers suggest that most of these deficits derive from impairments of function of the striatum and frontostriatal circuits. A neurodegenerative disorder such as HD usually involves multiple neuronal systems and, as HD evolves, the cognitive deficit becomes more extensive paralleling the pathological involvement of extra-striatal regions (the neocortex). In this context, it seems that functions mediated by the striatum are affected first and further deficits slowly develop over years following, in many cases, the proposed dorsal–ventral progression of the neuropathology through the striatum.2 Other deficits, however, may depend on cortical changes arising as a secondary result of loss of striatal projections to the cerebral cortex and may evolve once a threshold of striatal pathology has been reached.11
Structural imaging studies in HD
The morphological brain changes that occur during the course of HD are observable with in-vivo brain imaging techniques. Many studies have used volumetric analysis of computed tomography (CT) scans and magnetic resonance images (MRIs) to demonstrate progressive bilateral atrophy of the striatum,26,27 which may occur gradually even years before motor symptoms appear.28 Studies have suggested that the earliest changes in HD occur in the caudate nucleus.29 Other regions of the striatum such as the putamen and globus pallidus are also affected, particularly following disease progression,29,30 with additional evidence of thalamic changes.31 Recent evidence from neuroimaging studies suggests that the neurodegenerative changes in HD extend to cortical grey-matter and cerebral white-matter regions,3,32,33 although the onset and progression of these changes are only beginning to be understood. Reduced volume of the frontal5,28,34 and temporal lobes35 in patients with HD has also been observed in some studies, although this may primarily reflect white-matter loss.36 Other studies have reported reduced hippocampal, entorhinal cortex and brainstem volumes,3 as well as reductions in cerebellum32 and total brain volume.3
Such structural changes, in the striatum, for example, have been associated with specific cognitive deficits including attention, working memory and executive functions (Table 1). Harris et al37 obtained volumetric measures of the basal ganglia in 15 patients with HD and 19 controls. They found significant atrophy in the putamen of patients with HD. Volumetric measures of the putamen significantly correlated with neurologic examination scores, whereas caudate volumes correlated with measures of general cognition (the Mini-Mental State Examination [MMSE]) supporting the idea that a variety of motor circuits involve the putamen but not the caudate, whereas cognitive circuits are thought to involve the caudate more than the putamen.37 Similar findings have been observed in other studies. Bamford and colleagues27 found that performance of patients with HD in tests of complex psychomotor skills (i.e., the Stroop Test, Trail-Making Test) and tests of visuospatial processing (i.e., Porteus Mazes, immediate and delayed recall of figures, a figure-copying test [Rey–Osterrieth Complex Figure copy], the WAIS-Revised Block Design subset, Raven's Progressive Matrices) was significantly related to the degree of caudate atrophy.
Table 1
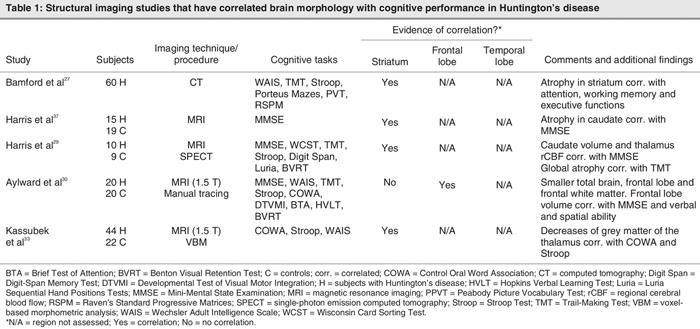
In a study using MRI and single-photon emission computed tomography (SPECT), Harris et al29 found that the putamen was the region that showed the greatest atrophy, whereas the caudate was the region with the greatest reduction in cerebral blood flow in patients with HD when compared with controls. Caudate volumes correlated with scores on the MMSE, and global atrophy correlated with error scores on the Trail-Making Test,29 a measure of visual scanning, sequencing, set shifting and motor skills.
Finally, Aylward et al30 found that moderately affected patients with HD had significantly smaller total brain volumes and decreased frontal lobe white matter. Frontal lobe volumes correlated with MMSE scores and measures of verbal and spatial ability.30
These studies suggest that structural changes co-occur with a decrease in general and specific cognitive abilities in patients with HD. Structural changes are relatively specific in the early stages of the illness, affecting primarily the caudate and putamen. Deterioration in psychomotor and executive functions, as well as visuospatial processing and memory, appear to derive primarily from basal ganglia pathology that disrupts normal cognitive processes mediated by frontostriatal circuits.
Functional imaging studies in HD
Functional brain imaging is based on the premise that focal neuronal activity is related to either regional cerebral blood flow (rCBF), the local degree of glucose metabolism or regional changes in receptor binding. There are 2 major classes of functional imaging studies: one examines the resting pattern of activity (resting-state studies) and the other assesses changes in cerebral activity during the performance of a task (neurocognitive-activation studies).
It has been hypothesized in HD that neurons may be at risk for some time before they die, and symptoms and cognitive difficulties may reflect neuronal dysfunction rather than neuronal loss. Thus, it is possible that functional brain imaging modalities may be more sensitive to the early changes that occur in disease than are structural studies.
Resting-state studies in HD
Despite some discrepant findings, studies of resting cerebral metabolism using SPECT or positron emission tomography (PET) have revealed metabolic abnormalities in the striatal and extra-striatal regions in HD,38–40 reinforcing the mainstream theory of a causative association between pathology in the basal ganglia, abnormal perfusion in the cerebral cortex and cognitive dysfunctions (Table 2). For example, SPECT studies have revealed that there is a reduction in rCBF in the striatum (caudate and putamen) and the cortex (prefrontal cortex) of patients with HD.40–42 Reduced rCBF in frontotemporal regions that are known to be involved in memory and cognition in general40–42 correlated with performance on the MMSE and episodic memory tests,43–42 whereas decreased rCBF in the caudate nucleus correlated with performance on tasks of executive function in some studies.39
Table 2
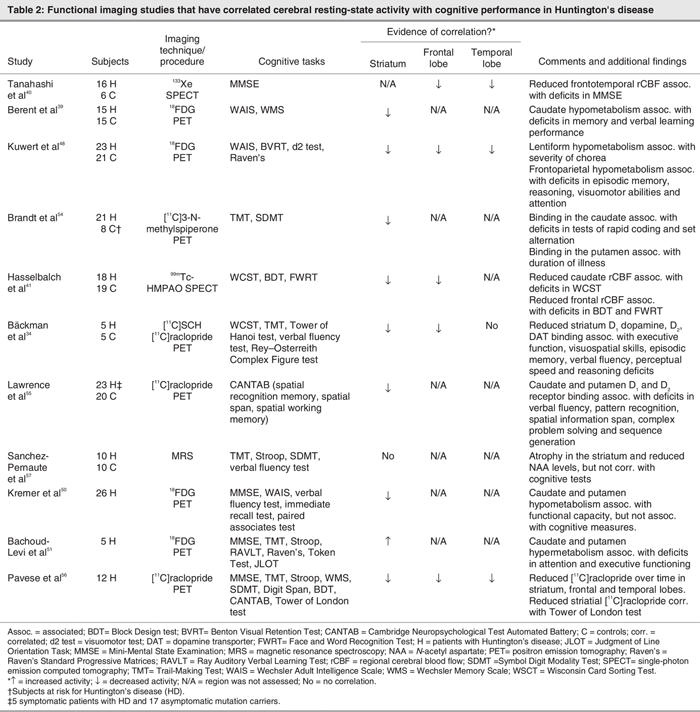
A reduction in glucose utilization in the striatum of patients with HD was initially discovered in the first PET study of HD.46 These findings have been confirmed by subsequent studies.39,47–49 Caudate hypometabolism was shown to correlate with performance on tasks of immediate recall memory, verbal associative learning and executive functions.39
Kuwert and colleagues48 found that in addition to more pronounced decreases of striatal (caudate and lentiform nucleus) glucose metabolism in patients with HD, a reduction in glucose utilization, another marker of neural activity, was also observed in cortical regions (frontoparietal and temporo-occipital). Further analyses of the relation between glucose metabolism and clinical symptoms showed that the severity of chorea was significantly correlated with the metabolic reductions within the lentiform nucleus, whereas the severity of cognitive deficits (as assessed by tests of episodic memory, reasoning, attention and visuomotor abilities) significantly correlated with metabolic reductions in frontoparietal, as well as temporo-occipital, cortices.48 Again, these studies reaffirmed the direct association between cognitive and clinical symptoms, as well as subcortical and cortical changes in neural activity in a widely distributed set of brain regions.
Reductions of cerebral glucose metabolism become more severe as the disease progresses and correlates with deterioration in function. In a large follow-up study of PET measurements in patients with HD, Kremer et al50 studied patients with HD who took lamotrigine over the course of 5 years. Resting-state studies showed hypometabolism in cortical areas (frontal and temporal), the thalamus and the basal ganglia relative to controls. Over the 5-year period, deterioration of cerebral glucose metabolism was observed. The most profound decrease in metabolic rates occurred in the basal ganglia, with an average yearly decline of 7%, with the most important decrease occurring in the last 6 months of this 5-year period. The authors found a positive correlation between basal ganglia hypometabolism and measures of functional capacity over time. However, they could not find any significant correlations between such abnormalities in cerebral metabolism and measures of global cognitive performance, such as performance on the MMSE, WAIS, and word fluency and immediate recall tests. Conversely, these parameters of cognitive function improved over time in both the placebo and the lamotrigine group.50 Because this effect was found in both groups, it is likely to reflect the effect of practice or familiarization with the assessment process.
Indeed, certain therapeutic procedures may slow or even reverse the degenerative process of HD. Bachoud-Levi and colleagues51 studied 5 patients with HD who received neural fetal transplantation (intrastriatal grafts bilaterally) in order to explore the therapeutic potential for the functional, motor and cognitive changes in patients with HD. Motor and cognitive functions were periodically evaluated, and repeated PET scanning was done in order to assess changes in cerebral metabolic activity over time. They found that grafts in the striatum were metabolically active even after 2 years. In 3 of the 5 patients, increased metabolic activity in the striatum correlated with improved cognitive performance in tasks of attention and executive functioning.51
Correlations between striatum dysfunction and cognitive performance have also been demonstrated by functional studies of neurotransmission systems. Using dopamine neurotransmission parameters (measures of presynaptic and postsynaptic dopamine biochemistry), PET studies have found marked reductions in dopamine binding in the striatum34,39,52,53 and in cortical regions (frontal and temporal cortex)53 of patients with HD. Brandt and colleagues54 observed an abnormal reduction in D2 dopamine receptor binding in the striatum of patients with HD and found that the severity of impairment exhibited by patients on cognitive tests requiring rapid coordination and set alternation (i.e., Trail-Making Test) correlated with D2 receptor binding in the caudate.54 Bäckman and colleagues34 also found reductions in all dopamine neurotransmission parameters (such as D1 and D2 receptor density and dopamine transporter density) in the striatum (and to a lesser extent in the temporal cortex), which showed a strong relation with severity of impairment on tests of executive function, visuospatial skill, episodic memory, verbal fluency, perceptual speed and reasoning.34
These correlations between striatal dopamine markers and cognitive deficits have also been found in individuals whose disease is at a preclinical stage55 and progress linearly as HD evolves. Lawrence and colleagues55 found that striatum D1 and D2 receptor binding were reduced in both symptomatic and asymptomatic individuals with the HD gene mutation and that the reduction was significantly related to the severity of impairment on tests of verbal fluency, spatial memory span, planning and sequence generation.55 In a longitudinal assessment of the loss of D2 receptor binding in the striatum, Pavese et al56 evaluated 12 patients with HD with 2 serial PET scans 1 year apart; 6 of the patients had a third PET scan 4 months later. They found a progressive striatal and cortical loss of dopamine receptors. The performance on all neuropsychological measures deteriorated over time, but only scores on the Tower of London Test correlated with striatal dopamine neurotransmission parameters.56
Finally, in a study using magnetic resonance spectroscopy (MRS), a noninvasive technique that provides information about biochemical and metabolic processes occurring within the brain, Sanchez-Pernaute and colleagues57 found reduced metabolic signals of markers of neuronal health and energy metabolism (such as creatinine, a marker for cellular energy, and N-acetyl aspartate, a marker for neuronal/axonal density) in the striatum of patients with HD. Reduced creatinine levels were associated with impaired performance on tasks of attention, verbal fluency and visuomotor skills.57
Neurocognitive-activation studies in HD
To date, just a few neuroimaging studies have used cognitive-activation paradigms in HD. Table 3 lists functional imaging studies that assessed cerebral activity during cognitive performance in HD. In a study that compared patients with HD and patients with schizophrenia (both disorders with a pathophysiology implicating abnormalities in the dopamine system), Goldberg and colleagues58 observed rCBF changes during the performance of a test of prefrontal cortical functioning (Wisconsin Card Sorting Test [WCST]). They found increased frontal blood flow in patients with HD during the performance of the WCST, in contrast to the diminished prefrontal blood flow shown in the group with schizophrenia. The increased cortical rCBF observed in patients with HD correlated positively with caudate atrophy (i.e., the greater the atrophy, the greater the increase in rCBF during task performance), but no significant correlation between the other cognitive score (WAIS) and rCBF was found.58
Table 3
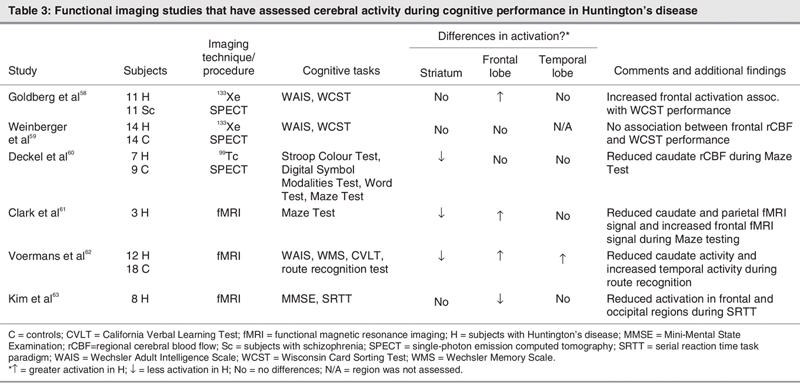
These results differ from the findings of Weinberger and colleagues,59 who examined the relation between cortical rCBF and cognitive function in HD. They found that even when patients with HD manifested overt cognitive impairment on the computerized version of the WCST, they had normal cortical rCBF during the WCST performance.59
Deckel and colleagues60 used SPECT to study rCBF in patients with HD at rest and during the performance of a test that activates frontal brain regions (maze testing). During the resting condition, they found increased rCBF in the cortex and striatum (with the exception of the caudate nucleus) in patients with HD. On the other hand, during the test condition, a positive correlation was found between executive functioning performance (as measured by the Porteus Maze Test) and reduced rCBF in the striatum and orbital cortex.60
Using functional MRI (fMRI), Clark et al61 examined neural activity in patients with HD during performance of the Porteus Maze Task. They observed reduced fMRI signal in patients with HD relative to controls in occipital, parietal and sensorimotor cortices and in the caudate, whereas increased signal was found in the left postcentral and right middle frontal gyri.61
In an fMRI study that investigated the interaction between the hippocampus and the basal ganglia in navigational memory, Voermans and colleagues62 studied 12 patients with HD and 18 controls using a route recognition navigational test. They found that the hippocampus in patients with HD compensates for caudate dysfunction during route recognition. Control subjects showed greater activity in the right caudate, whereas the patients with HD showed greater activity in the right hippocampus during performance of the route recognition test.
Finally, in another fMRI study, Kim et al63 used a procedural learning task in order to unmask functional striatal abnormalities in the brains of individuals in the early stages of clinical HD. Although the basal ganglia structures were not differentially activated between groups during the performance of a serial reaction-time task, they found reduced activation in the HD group relative to healthy controls in the right middle frontal, left middle occipital, left precuneus and left middle frontal gyri.
Taken together, these studies show that some levels of cortical function in HD correlate with performance on behavioural tasks, suggesting that dysfunction in both striatal and cortical regions contributes to cognitive status in these patients.
Discussion
By combining neuroimaging techniques with improved knowledge of human cognitive processes, the last decades have seen tremendous advances in our understanding of cognitive abnormalities in neurodegenerative disorders such as HD. The goal of the current paper was to review the literature related to neuroimaging and cognitive dysfunction in HD so as to uncover the most consistent findings linking cerebral abnormalities and cognitive deficits.
Structural neuroimaging has provided important evidence of morphological brain changes in HD3,37 extending beyond the basal ganglia, and some correlations with neuropsychological measures have been reported.3,35 Striatal29,30 and cortical atrophy5,30,34 are the most common findings throughout the literature, and some associations with cognitive deficits in attention, working memory and executive functions have been shown. Further work is needed to determine the precise relation between cognitive and regional volume changes and may provide an important surrogate marker of disease onset or disease progression, or both.
Functional brain imaging has also enhanced our understanding of cognitive deficits in HD. Complementing the evidence of structural cerebral changes in patients with HD, some functional studies have also demonstrated correlations between striatal dysfunction and cognitive performance. Significant metabolic40–42 and neurotransmission changes34 occur in the striatal region in the brains of patients with HD. These changes include hypoperfusion, decreased glucose utilization and reductions in striatal neurotransmitter parameters, all correlating with psychomotor and executive functions, episodic memory and visuospatial processing.40,41
Further evidence that impairment of the cerebral cortex may account for some of the cognitive deficits observed in patients with HD has been provided by functional studies. Significant declines in glucose metabolic rate occur throughout the cerebral cortex.48,50 This hypometabolism is more prominent in frontal regions and correlates with performance on recognition memory, language and perceptual tests.58,60 At present, functional neuroimaging studies using cognitive paradigms have been few and the findings equivocal.59,61–63 Ambiguous frontal activation patterns may be related to differences in functional imaging techniques or to differences in the sensitivity of the cognitive paradigm employed. In addition, differences in performance levels between patient and control groups could also account for these contradictory findings.
Together, the studies reviewed here support the specific role of the striatum in the cognitive dysfunction of patients with HD as a part of a complex neuronal architecture interlinking cortical regions and the basal ganglia. The particular structural and functional changes in frontal regions, shown by neuroimaging and neuropsychological tests, are in concordance with the existence of several anatomically and functionally distinct circuits that link the striatum with regions of the frontal cortex.64 These findings reinforce the general theories suggesting a causative association between frontostriatal circuitry pathology and cognitive dysfunction in HD.65
Findings from the studies described here suggest that the basal ganglia should be viewed as components of circuits organized in parallel, remaining largely segregated from one another,66 that are thought to be involved in the performance of higher-order cognitive processes such as attention, memory and executive functions. Functions mediated by the striatum may be affected first and deficits may evolve insidiously over many years, whereas other functional deficits may depend on cortical changes as a secondary result of loss of striatal projections to the cerebral cortex.11
A few caveats about drawing strong conclusions from brain imaging studies in HD seem relevant here. First, there are differences in patient selection (i.e., population size, differences in severity of disease, etc.) in the studies reviewed. For instance, some patient groups had severe movement disorders, making it difficult to obtain brain images. In addition, many patients with HD take different types of medications, which may have an effect on the results. These constraints are the cause for even greater concern in patients with both a movement disorder and cognitive problems. Movement artifacts and interference in performance by anxiety, lack of concentration or motivation during imaging can also influence the results. Furthermore, some studies were conducted before the identification of the HD gene in 1993;1 thus, earlier studies can be problematic in terms of the HD diagnosis (because there was no assurance of gene status in their samples at that time).
Second, the correlational design used in most of these studies has been criticized, because such an approach can be misleading given the progressive nature of this disorder. Indeed, as the disease advances, regional levels of brain function and pathology might worsen in tandem, thus, a correlation between 2 such indices could reflect collinearity instead of a cause–effect relation. Furthermore, neuropsychological tests vary in the distributed cognitive networks required and, thus, may highlight different cortical and subcortical deficits.4 In addition, cognitive testing can be influenced through practice: whereas a single cognitive assessment may be misleading, repeated ones may produce a problem in practice effects. It is also important to know what aspects of cognition the test is tapping into and to be conscious of the fact that the cognitive strategies employed by subjects with HD and other subjects may vary. For example, common tests of general cognition employed in numerous HD studies, such as MMSE, are poor at looking at frontostriatal circuits and thus correlations between this measure and changes in the basal ganglia are not necessary helpful.
The inherent limitations of brain imaging techniques should also be taken into account for an accurate interpretation of the findings. For instance, imaging at any given time cannot differentiate between local neurodegenerative consequences of the underlying disease versus the indirect effect of damaged tissue on other brain regions. The ideal way to address this issue would be a histological comparison with the neuroimaging studies; however, an alternative approach would be to compare, briefly, the brain involvement expected from postmortem studies of HD with neuroimaging findings.
In addition to these considerations, we must also take into account that functional neuroimaging studies (PET, fMRI) depend on the integrity of the vascular and metabolic response to neuronal activity. This neurovascular coupling may not always be intact in HD; thus, changes in vascular parameters may either reflect functional (neuronal) changes or simply a disturbance in neurovascular coupling.61,67
Despite these limitations, further advances in neuroimaging development and imaging analysis software will nevertheless give us greater spatial and temporal resolution, enhancing our understanding of the cognitive dysfunction associated with HD. Morphometric markers may provide a sensitive and reliable method to determine whether a drug is effective in symptomatic patients. In the future, functional neuroimaging's greatest contribution toward uncovering the pathophysiology of HD could be by defining neurodegenerative disease phenotypes both during preclinical and early clinical disease periods. Functional brain imaging tests can be done repeatedly and can assess temporal changes in the neuronal degeneration both before and after the development of clinical symptoms. They can also provide a window to the neuronal dysfunction and data complementary to clinical and cognitive evaluation, as well as a tangible assessment of the efficacy of novel treatment strategies. Finally, the application of several imaging techniques in parallel may give us the opportunity to gather even more revealing information about the metabolic, functional and neuroanatomical alterations that take place during the various stages of the disease and is an exciting and promising development for future research on this complex neurodegenerative disorder.
Acknowledgments
This study was supported by the Huntington Society of Canada. We thank Hazel Sutton for her assistance with editing the manuscript.
Footnotes
Contributors: All authors contributed substantially to drafting and revising the article, and each gave final approval for the article to be published.
Competing interests: None declared.
Correspondence to: Dr. Martin Lepage, Brain Imaging Group, Douglas Hospital Research Centre, 6875 LaSalle Blvd., Montréal QC H4H 1R3; martin.lepage@mcgill.ca
References
- 1.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993;72:971-83. [DOI] [PubMed]
- 2.Vonsattel JP, Myers RH, Stevens TJ, et al. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 1985; 44:559-77. [DOI] [PubMed]
- 3.Rosas HD, Koroshetz WJ, Chen YI, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology 2003;60:1615-20. [DOI] [PubMed]
- 4.Andrews TC, Brooks DJ. Advances in the understanding of early Huntington's disease using the functional imaging techniques of PET and SPET. Mol Med Today 1998;4:532-9. [DOI] [PubMed]
- 5.Starkstein SE, Brandt J, Bylsma F, et al. Neuropsychological correlates of brain atrophy in Huntington's disease: a magnetic resonance imaging study. Neuroradiology 1992;34:487-9. [DOI] [PubMed]
- 6.Simmons JT, Pastakia B, Chase TN, et al. Magnetic resonance imaging in Huntington disease. AJNR Am J Neuroradiol 1986;7:25-8. [PMC free article] [PubMed]
- 7.Oscar-Berman M, Zola-Morgan SM, Oberg RG, et al. Comparative neuropsychology and Korsakoff's syndrome. III–Delayed response, delayed alternation and DRL performance. Neuropsychologia 1982;20:187-202. [DOI] [PubMed]
- 8.Ho AK, Sahakian BJ, Brown RG, et al. Profile of cognitive progression in early Huntington's disease. Neurology 2003;61:1702-6. [DOI] [PubMed]
- 9.Lemiere J, Decruyenaere M, Evers-Kiebooms G, et al. Longitudinal study evaluating neuropsychological changes in so-called asymptomatic carriers of the Huntington's disease mutation after 1 year. Acta Neurol Scand 2002;106:131-41. [DOI] [PubMed]
- 10.Kirkwood SC, Siemers E, Hodes ME, et al. Subtle changes among presymptomatic carriers of the Huntington's disease gene. J Neurol Neurosurg Psychiatry 2000;69:773-9. [DOI] [PMC free article] [PubMed]
- 11.Snowden JS, Craufurd D, Thompson J, et al. Psychomotor, executive, and memory function in preclinical Huntington's disease. J Clin Exp Neuropsychol 2002;24:133-45. [DOI] [PubMed]
- 12.Butters N, Wolfe J, Granholm E, et al. An assessment of verbal recall, recognition and fluency abilities in patients with Huntington's disease. Cortex 1986;22:11-32. [DOI] [PubMed]
- 13.Claus JJ, Mohr E. Attentional deficits in Alzheimer's, Parkinson's, and Huntington's diseases. Acta Neurol Scand 1996;93:346-51. [DOI] [PubMed]
- 14.Sprengelmeyer R, Young AW, Calder AJ, et al. Loss of disgust. Perception of faces and emotions in Huntington's disease. Brain 1996;119:1647-65. [DOI] [PubMed]
- 15.Brandt J, Bylsma FW, Gross R, et al. Trinucleotide repeat length and clinical progression in Huntington's disease. Neurology 1996;46:527-31. [DOI] [PubMed]
- 16.Paulsen JS, Butters N, Sadek JR, et al. Distinct cognitive profiles of cortical and subcortical dementia in advanced illness. Neurology 1995;45:951-6. [DOI] [PubMed]
- 17.Lange KW, Sahakian BJ, Quinn NP, et al. Comparison of executive and visuospatial memory function in Huntington's disease and dementia of Alzheimer type matched for degree of dementia. J Neurol Neurosurg Psychiatry 1995;58:598-606. [DOI] [PMC free article] [PubMed]
- 18.Lawrence AD, Watkins LH, Sahakian BJ, et al. Visual object and visuospatial cognition in Huntington's disease: implications for information processing in corticostriatal circuits. Brain 2000;123:1349-64. [DOI] [PubMed]
- 19.Brandt J, Butters N. The neuropsychology of Huntington's disease. Trends Neurosci 1986;9:118-20.
- 20.Brouwers P, Cox C, Martin A, et al. Differential perceptual-spatial impairment in Huntington's and Alzheimer's dementias. Arch Neurol 1984;41:1073-6. [DOI] [PubMed]
- 21.Kirkwood SC, Su JL, Conneally P, et al. Progression of symptoms in the early and middle stages of Huntington disease. Arch Neurol 2001;58:273-8. [DOI] [PubMed]
- 22.Sprengelmeyer R, Canavan AG, Lange HW, et al. Associative learning in degenerative neostriatal disorders: contrasts in explicit and implicit remembering between Parkinson's and Huntington's diseases. Mov Disord 1995;10:51-65. [DOI] [PubMed]
- 23.Redondo-Verge L. [Cognitive deterioration in Huntington disease.] Rev Neurol 2001;32:82-5. [PubMed]
- 24.Zakzanis KK. The subcortical dementia of Huntington's disease. J Clin Exp Neuropsychol 1998;20:565-78. [DOI] [PubMed]
- 25.Cummings JL. Behavioral and psychiatric symptoms associated with Huntington's disease. Adv Neurol 1995;65:179-86. [PubMed]
- 26.Bamford KA, Caine ED, Kido DK, et al. Clinical-pathologic correlation in Huntington's disease: a neuropsychological and computed tomography study. Neurology 1989;39:796-801. [DOI] [PubMed]
- 27.Bamford KA, Caine ED, Kido DK, et al. A prospective evaluation of cognitive decline in early Huntington's disease: functional and radiographic correlates. Neurology 1995;45:1867-73. [DOI] [PubMed]
- 28.Aylward EH, Brandt J, Codori AM, et al. Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at-risk persons. Neurology 1994;44:823-8. [DOI] [PubMed]
- 29.Harris GJ, Aylward EH, Peyser CE, et al. Single photon emission computed tomographic blood flow and magnetic resonance volume imaging of basal ganglia in Huntington's disease. Arch Neurol 1996;53:316-24. [DOI] [PubMed]
- 30.Aylward EH, Li Q, Stine OC, et al. Longitudinal change in basal ganglia volume in patients with Huntington's disease. Neurology 1997;48:394-9. [DOI] [PubMed]
- 31.Jernigan TL, Salmon DP, Butters N, et al. Cerebral structure on MRI, part II: specific changes in Alzheimer's and Huntington's diseases. Biol Psychiatry 1991;29:68-81. [DOI] [PubMed]
- 32.Fennema-Notestine C, Archibald SL, Jacobson MW, et al. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology 2004;63:989-95. [DOI] [PubMed]
- 33.Kassubek J, Juengling FD, Kioschies T, et al. Topography of cerebral atrophy in early Huntingdon's disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry 2004;75:213-20. [PMC free article] [PubMed]
- 34.Bäckman L, Robins-Wahlin TB, Lundin A, et al. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain 1997;120:2207-17. [DOI] [PubMed]
- 35.Dierks T, Linden DE, Hertel A, et al. Multimodal imaging of residual function and compensatory resource allocation in cortical atrophy: a case study of parietal lobe function in a patient with Huntington's disease. Psychiatry Res 1999;90:67-75. [PubMed]
- 36.Aylward EH, Anderson NB, Bylsma FW, et al. Frontal lobe volume in patients with Huntington's disease. Neurology 1998;50:252-8. [DOI] [PubMed]
- 37.Harris GJ, Pearlson GD, Peyser CE, et al. Putamen volume reduction on magnetic resonance imaging exceeds caudate changes in mild Huntington's disease. Ann Neurol 1992;31:69-75. [DOI] [PubMed]
- 38.Bartenstein P, Weindl A, Spiegel S, et al. Central motor processing in Huntington's disease. A PET study. Brain 1997;120:1553-67. [DOI] [PubMed]
- 39.Berent S, Giordani B, Lehtinen S, et al. Positron emission tomographic scan investigations of Huntington's disease: cerebral metabolic correlates of cognitive function. Ann Neurol 1988;23:541-6. [DOI] [PubMed]
- 40.Tanahashi N, Meyer JS, Ishikawa Y, et al. Cerebral blood flow and cognitive testing correlate in Huntington's disease. Arch Neurol 1985;42:1169-75. [DOI] [PubMed]
- 41.Hasselbalch SG, Oberg G, Sorensen SA, et al. Reduced regional cerebral blood flow in Huntington's disease studied by SPECT. J Neurol Neurosurg Psychiatry 1992;55:1018-23. [DOI] [PMC free article] [PubMed]
- 42.Sax DS, Powsner R, Kim A, et al. Evidence of cortical metabolic dysfunction in early Huntington's disease by single-photon-emission computed tomography. Mov Disord 1996;11:671-7. [DOI] [PubMed]
- 43.Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 1992;99:195-231. [DOI] [PubMed]
- 44.Tulving E, Markowitsch HJ. Memory beyond the hippocampus. Curr Opin Neurobiol 1997;7:209-16. [DOI] [PubMed]
- 45.Cabeza R, Nyberg L. Imaging cognition II: an empirical review of 275 PET and fMRI studies. J Cogn Neurosci 2000;12:1-47. [DOI] [PubMed]
- 46.Kuhl DE, Phelps ME, Markham CH, et al. Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann Neurol 1982;12:425-34. [DOI] [PubMed]
- 47.Hayden MR, Martin WR, Stoessl AJ, et al. Positron emission tomography in the early diagnosis of Huntington's disease. Neurology 1986;36:888-94. [DOI] [PubMed]
- 48.Kuwert T, Lange HW, Langen KJ, et al. Cortical and subcortical glucose consumption measured by PET in patients with Huntington's disease. Brain 1990;113:1405-23. [DOI] [PubMed]
- 49.Young AB, Penney JB, Starosta-Rubinstein S, et al. PET scan investigations of Huntington's disease: cerebral metabolic correlates of neurological features and functional decline. Ann Neurol 1986; 20:296-303. [DOI] [PubMed]
- 50.Kremer B, Clark CM, Almqvist EW, et al. Influence of lamotrigine on progression of early Huntington disease: a randomized clinical trial. Neurology 1999;53:1000-11. [DOI] [PubMed]
- 51.Bachoud-Levi AC, Remy P, Nguyen JP, et al. Motor and cognitive improvements in patients with Huntington's disease after neural transplantation. Lancet 2000;356:1975-9. [DOI] [PubMed]
- 52.Bohnen NI, Koeppe RA, Meyer P, et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology 2000; 54:1753-9. [DOI] [PubMed]
- 53.Ginovart N, Lundin A, Farde L, et al. PET study of the pre-and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain 1997;120:503-14. [DOI] [PubMed]
- 54.Brandt J, Folstein SE, Wong DF, et al. D2 receptors in Huntington's disease: positron emission tomography findings and clinical correlates. J Neuropsychiatry Clin Neurosci 1990;2:20-7. [DOI] [PubMed]
- 55.Lawrence A, Sahakian B, Robbins T. Cognitive functions and corticostriatal circuits: insights from Huntington's disease. Trends Cogn Sci 1998;2:379-88. [DOI] [PubMed]
- 56.Pavese N, Andrews TC, Brooks DJ, et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington's disease: a PET study. Brain 2003;126:1127-35. [DOI] [PubMed]
- 57.Sanchez-Pernaute R, Garcia-Segura JM, del Barrio Alba A, et al. Clinical correlation of striatal 1H MRS changes in Huntington's disease. Neurology 1999;53:806-12. [DOI] [PubMed]
- 58.Goldberg TE, Berman KF, Mohr E, et al. Regional cerebral blood flow and cognitive function in Huntington's disease and schizophrenia. A comparison of patients matched for performance on a prefrontal-type task. Arch Neurol 1990;47:418-22. [DOI] [PubMed]
- 59.Weinberger DR, Berman KF, Iadarola M, et al. Prefrontal cortical blood flow and cognitive function in Huntington's disease. J Neurol Neurosurg Psychiatry 1988;51:94-104. [DOI] [PMC free article] [PubMed]
- 60.Deckel AW, Weiner R, Szigeti D, et al. Altered patterns of regional cerebral blood flow in patients with Huntington's disease: a SPECT study during rest and cognitive or motor activation. J Nucl Med 2000;41:773-80. [PubMed]
- 61.Clark VP, Lai S, Deckel AW. Altered functional MRI responses in Huntington's disease. Neuroreport 2002;13:703-6. [DOI] [PubMed]
- 62.Voermans NC, Petersson KM, Daudey L, et al. Interaction between the human hippocampus and the caudate nucleus during route recognition. Neuron 2004;43:427-35. [DOI] [PubMed]
- 63.Kim JS, Reading SA, Brashers-Krug T, et al. Functional MRI study of a serial reaction time task in Huntington's disease. Psychiatry Res 2004;131:23-30. [DOI] [PubMed]
- 64.Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev 1995;20:91-127. [DOI] [PubMed]
- 65.Ring HA, Serra-Mestres J. Neuropsychiatry of the basal ganglia. J Neurol Neurosurg Psychiatry 2002;72:12-21. [DOI] [PMC free article] [PubMed]
- 66.Strick P, Dum R, Mushiake H. Basal ganglia “loops” with the cerebral cortex. In: Kimura M, Graybiel A, editors. Functions of the cortico-basal ganglia loop. Tokyo: Springer-Verlag; 1995. p. 106-24.
- 67.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci 2004;5:347-60. [DOI] [PubMed]