

Abstract
The understanding of the fixation of mutations within human tissues and their subsequent clonal expansion is a considerable problem, of which little is known. We have previously shown that nononcogenic mutations in the mitochondrial genome occur in one of a number of morphologically normal colonic crypt stem cells, the progeny of which later occupy the whole crypt. We propose that these wholly mutated crypts then clonally expand by crypt fission, where each crypt divides into two mutated daughter crypts. Here we show that (i) mutated crypts in the process of fission share the same mutated mitochondrial genotype not present in neighboring cytochrome c oxidase-positive crypts (the odds of this being a random event are ≥2.48 × 109:1); (ii) neighboring mutated crypts have the same genotype, which is different from adjacent cytochrome c oxidase-positive crypts; (iii) mutated crypts are clustered together throughout the colon; and (iv) patches of cytochrome c oxidase-deficient crypts increase in size with age. We thus demonstrate definitively that crypt fission is the mechanism by which mutations spread in the normal human colon. This has important implications for the biology of the normal adult human colon and possibly for the growth and spread of colorectal neoplasms.
Keywords: fission, mitochondria
Colorectal cancer is considered a disease of colonic tissue stem cells. The high turnover of cells within the crypt (1, 2) means that stem cells are the only cell type that remain in the crypt long enough to acquire sufficient prooncogenic mutations. In addition, stem cells possess the ability to self renew, and if cancer is seen as a dysregulation of self renewal, fewer mutations may be required to activate this machinery. We have previously argued that colorectal adenomas begin as a mutated stem cell at the base of the crypt, whose progeny eventually colonize the crypt to become a monocryptal or unicryptal adenoma, which then expands by crypt fission (the “bottom-up” theory) (3). Although this proposal is not universally accepted (4), it does invoke a pivotal role for stem cells in carcinogenesis. At present, little is known of the mechanisms by which DNA mutations within morphologically normal colonic crypt stem cells are established and spread throughout the nondysplastic colon.
Mitochondria are the major generators of cellular ATP through oxidative phosphorylation. mtDNA is a small (16.6-kb) self-replicating molecule that encodes 13 essential proteins of the mitochondrial oxidative phosphorylation complexes, 2 rRNA and 22 tRNA genes. Within cells, there are thousands of mitochondria, each containing multiple copies of mtDNA. mtDNA mutations are random, increase with age (5, 6), and are thought to occur because of the presence of free-radical-generating enzymes, potentiated by poor DNA repair mechanisms and the lack of protective histones (7, 8). These mutations can affect all copies of the mitochondrial genome (homoplasmy) or a proportion thereof (heteroplasmy). For a mutated mitochondrial genotype to result in a mutated cellular phenotype, homoplasmy or high levels of heteroplasmy must be present (9). In some cells, there is a high level of mutation involving the mitochondrial cytochrome c oxidase genes and associated with cytochrome c oxidase deficiency. We have shown that these cells clonally expand to occupy first part and then the whole crypt (also known as monoclonal conversion) (10), where the crypt becomes entirely colonized by mutated cells. Such conversion has previously been shown in mice that have DNA mutations in nuclear housekeeping genes such as G6PD and metallothionein induced by ethylnitrosourea (11, 12). Colonic crypts in mice (13) and humans (14–16) are clonal populations; we have shown that one of a number of colonic stem cells can acquire a mutation in the mitochondrial genome, resulting in detectable cytochrome c oxidase deficiency only after the age of 40.
Thus, in humans, one stem cell has the potential to expand and replace the whole crypt stem cell population, either stochastically or by a selective advantage induced by the mutation, a phenomenon known as niche succession after Kim and Shibata (17). We and others have argued that these crypts then clonally expand by crypt fission, dividing to form two daughter crypts, and that this is the mechanism by which stem cell clones expand in the colon and indeed in the entire gastrointestinal tract (11, 14). This proposal would predict that neighboring colonic crypts are closely related to each other. We have previously shown that the X-linked patch in the adult human colon is large, ≈450 crypts, so colonic crypts can develop into large families (15). But this does not tell us anything about the development and spread of mutations occurring later in life. Indeed, salient studies by Kim and Shibata (18) have suggested that this simple proposal is more complex: detailed analysis of the methylation status of nonexpressed genes, such as MyoD, showed that adjacent crypts are no more related to each other than those 15 cm away; moreover, crypts in fission showed the same differences. This could mean either that crypts apparently in fission are fusing rather than dividing (17, 19, 20), or that the fission process is so protracted that niche succession occurs in the dividing partners.
In this study, we show that morphologically normal human colonic crypts are able to divide by crypt fission by demonstrating that stochastic mutations in mtDNA resulting in a phenotypic deficiency of cytochrome c oxidase are identical in both arms of a crypt that is bifurcating. Furthermore, we show that patches of neighboring crypts deficient in cytochrome c oxidase also share identical mitochondrial mutations, indicating that crypt fission is a mechanism by which mutations can spread within the colon. Also, we calculate the statistical likelihood that crypts deficient in cytochrome c oxidase are clustered and demonstrate that patches of mutated crypts increase in size with age, providing further evidence of the development of clonal patches in the human colon.
Results
Normal Human Colonic Crypts Divide and mtDNA Mutations Spread by Crypt Fission. We first determined that a crypt, in the process of fission, maintains mtDNA mutations in both daughter crypts. Single cytochrome c oxidase-deficient cells from both arms of a bifurcating wholly cytochrome c oxidase-deficient crypt, in addition to cells from cytochrome c oxidase-positive neighboring crypts, were microdissected (Fig. 1 a–i), and the sequence of the entire mitochondrial genome was determined in all of the cells. This identified an mtDNA mutation, 4733T>C in the MTND2 gene, in cells from both arms of the cytochrome c oxidase-deficient crypt in fission (Fig. 1k), whereas the cytochrome c oxidase-positive cells maintained their wild-type genotype (Fig. 1j). Whereas the 4733T>C mutation predicts a synonymous change and is probably not the reason for the observed cytochrome c oxidase deficiency, it does show that a single mutation is able to persist in newly forming crypts. Furthermore, this analysis provides evidence that crypts divide by fission.
Fig. 1.

Both arms of a cytochrome c oxidase-deficient crypt in fission share identical mutations. (a–i) Serial sections were made of a single crypt in fission, and single cells were isolated from each arm and from a single neighboring cytochrome c oxidase-positive crypt. Cells from both arms of the cytochrome c oxidase-deficient crypt in fission had a common 4733 T>C transition j is the neighboring cytochrome c oxidase-positive crypt, and k is from the deficient crypt in fission.
Crypt mtDNA Mutations Spread to Form Clonal Patches. We hypothesized that, if any given mtDNA mutation were able to establish itself in the stem cell population of newly formed crypts over time, the mutation would appear in other neighboring crypts as fission was repeated, resulting in the formation of a patch. To test this theory, we sequenced the entire mitochondrial genome from single colonocytes microdissected from three neighboring cytochrome c oxidase-deficient crypts (none of which was undergoing fission), in addition to colonocytes from three neighboring cytochrome c oxidase-positive crypts. A total of 17 recognized polymorphic variants were found in all six of the cells that were sequenced. However, all of the colonocytes from the three cytochrome c oxidase-deficient crypts harbored an additional mutation, 6277A>G, in the mitochondrial cytochrome c oxidase I gene, which predicts a Gly125Asp amino acid substitution in cytochrome c oxidase protein (Fig. 2a). Gly-125 is very highly conserved throughout evolution (21) and as such is highly likely to be the cause of the cytochrome c oxidase deficiency. The same mutation was not seen in any of the adjacent cytochrome c oxidase-positive crypts that were sequenced (Fig. 2b). In another patient, the same protocol revealed not one but two shared mutations found in both cytochrome c oxidase-deficient crypts [a 7275T>C transition mutation, which predicts a Ser458Pro amino acid substitution in the mitochondrial cytochrome c oxidase I gene (Fig. 2c) and a neutral polymorphic variant (C303ins) variant (Fig. 2e)]. Neither 7275T>C nor the C303ins was present in adjacent cytochrome c oxidase-positive crypts (Fig. 2 d and f).
Fig. 2.

Single cells were isolated from each of the cytochrome c oxidase-deficient crypts and from adjacent cytochrome c oxidase-positive crypts. In one patient, each cytochrome c oxidase-deficient cell had a 6277 A>G transition (a) which is not present in the cytochrome c oxidase-positive cells (b). In another patient, each cytochrome c oxidase-deficient cell had a 7275 T>C transition (c) and a C311 insertion (e) not present in the cytochrome c oxidase-positive cells (d and f).
Phenotypically Identical Crypts Are Clustered. Sequencing analysis gives a very strong indication of how mtDNA mutations spread within the human colon at the crypt level but not the overall picture of what is happening in the colon. We studied 14 patients and identified cytochrome c oxidase-deficient crypts by immunohistochemistry. We therefore labeled each crypt as either cytochrome c oxidase-positive or -deficient in an entire field (X no. crypts per field = 1,337 ± 94.83) and applied a Monte–Carlo simulation method to derive a control probability distribution of crypt cluster statistics, against which the same statistics measured on each sample is compared (Materials and Methods). To achieve this, two computer programs [biaqim and clan (cluster analysis), Tadrosoft] were used. biaqim was used to spatially map cytochrome c oxidase-deficient and -positive crypts throughout a low-power (×20) en face image of the colon. clan statistically determined the relative risk (RR) that a cytochrome c oxidase-deficient crypt also has at least one other cytochrome c oxidase-deficient crypt as a nearest neighbor, χ2, and the proportion (Nprop) of all cytochrome c oxidase-deficient crypts with at least one other as a nearest neighbor. To achieve significance, each observed data set was compared with 1,000 control probability distributions (random data sets), and significance was taken as >95% of random data sets being less than the observed data set. Fig. 3a shows the frequency of all images' RR. Only images taken from patient 11 had a RR <1 (indicating no statistical clustering within that patient). Observed RR of >1 indicate deficient crypts are clustered. When compared with 1,000 random assignations of cytochrome c oxidase-deficient labels, 91.18% (31/34) images achieved >95% RR significance, 82.35% (28/34) achieved >95% χ2 significance, and 88.24% (32/34) achieved >95% Nprop significance (Fig. 3b and Table 1, which is published as supporting information on the PNAS web site). On the rare occasion that an individual image did not reach the 95% level, there was at least one other image from that patient that did, or that patient was the younger of the patients in whom we would not expect to see statistical clustering, because so few mutated crypts were present. Analysis was performed by two independent observers. No intraobserver bias was found (Fig. 3c) on 15 randomly selected images from a total of 34. We have shown (10) that the number of cytochrome c oxidase-deficient crypts increases with age, and this patient data set shows a similar age-dependent increase (see Fig. 6, which is published as supporting information on the PNAS web site). Patient 11 (40 years old) did not show any statistical clustering of mutated crypts, and this would fit with age-dependent observations. Statistical analysis has confirmed that cytochrome c oxidase-deficient crypts are indeed clustered within the normal human colon and provides conclusive evidence in support of the development of the patch.
Fig. 3.

Analysis of clan-generated data. (a) The observed RR for any image was >1 in all patients except patient 11 (40 years of age) who had very few mutated crypts. (b) Observed versus randomly generated data for RR, χ2, and Nprop (91.18%, 82.35%, and 88.24%, respectively) for all images achieved significance at the 95% level. (c) All images were analyzed by two independent researchers in a blinded fashion, and no intraobserver differences were found [correlation coefficient (R2) of 0.85].
Patches of Phenotypically Identical Crypts Expand with Age. We have shown that phenotypically and genotypically identical crypts are able to divide by fission into a patch of at least two crypts or more. We have also shown that these crypts are statistically clustered together. A prediction could therefore be made that patch size would increase with age. To test this hypothesis, we calculated the mean patch size of cytochrome c oxidase-deficient crypts within colonic tissue sections of 34 patients. A patch was defined as at least two phenotypically identical crypts in juxtaposition to each other within the width of a single crypt. The mean patch size versus age is shown in Fig. 4, which shows that there is an age-dependent increase in patch size (R2 = 0.8741).
Fig. 4.

Mean patch size of cytochrome c oxidase-deficient crypts increases with age. Patients have been grouped according to decade (31–40, 41–50, 51–60, 61–70, 71–80, and 81–90). Results are ± SEM. Although individual deficient crypts were observed in patients under the age of 40, no patches of two or more deficient crypts were seen. R2 = 0.8741, and the equation of best-fit curve is y = 0.20090.03295x.
Discussion
We have demonstrated that the progeny of a morphologically normal human colonic crypt stem cell that contains an mtDNA mutation can expand to occupy a whole crypt, and that this crypt then further expands by fission to form a patch (illustrated in Fig. 5). This study demonstrates that crypt fission is an active process of clonal expansion of crypts in the normal human intestine. Previously, it had been shown that in the normal adult human colon, all crypts were monoclonal in nature (16), but this study demonstrates in humans that clonal patches develop by crypt fission.
Fig. 5.

A proposed model of how DNA mutations spread in the human colon. The initial mutation can occur anywhere within the epithelium but can persist only in stem cells. Occasionally, a stem cell carrying a DNA mutation can dominate the crypt, leading to all cells within that crypt also carrying the mutation. At some point, this crypt will divide by fission, and this process will carry on through the life of the host. If the mutation is prooncogenic, this may affect the rate at which this occurs, resulting in a rapid spread of potentially cancerous mutations.
Other researchers have argued that what was thought to be fission was actually fusion of two crypts colliding (17–19). We have shown here that both arms of a bifurcating crypt have identical mtDNA mutations; the odds of this occurring by chance are so small (at least 2.48 × 109:1, per Materials and Methods) as to refute the possibility of two identically mutated crypts merging. We have no reason to believe that the biology of cytochrome c oxidase-deficient crypts differs from cytochrome c oxidase-positive crypts (10).
The data of Kim and Shibata (17, 18) indicate, therefore, that the process of clonal expansion by crypt fission is slow, and that it would be useful to combine mtDNA genotyping and CpG methylation status in the same crypts. Our data would predict that, in conditions such as ulcerative colitis, where crypt fission is prominent, patches would be larger. This is a testable hypothesis ongoing in our laboratories, which also underlines the potential importance of crypt fission in the spread of dysplasias and neoplasms in the gut. In fact, in ulcerative colitis with dysplasia, the same aneuploid stem cell line can be found in large areas of mucosa (22). Our results would also predict that when a crypt becomes filled with dysplastic cells, it can then divide by fission, and it is by consecutive fission events that a dysplastic crypt expands into aberrant crypt foci. It is likely that such a mechanism is also responsible for the spread of mutated clones in the stomach and the esophagus in Barrett's mucosa; rapid expansion, for example of p16–/– clones, has been reported (23).
Our data could be extrapolated to conform to the “bottom-up” pattern of dysplastic spread (3). An adenomatous crypt would, therefore, begin to divide from the base of the crypt at the stem cell region and bifurcate into two adenomatous daughter crypts. Previous studies have shown there is a significant increase in the amount of crypt budding and bifurcation in patients with familial adenomatous polyposis (24, 25), and the data we have presented here have provided a likely mechanism by which the early development of aberrant crypt foci develop. As yet, the mechanism(s) initiating fission are unknown but may include the control of stem cell number by factors such as the bone morphogenic protein/SMAD4 pathway (26).
Taken together with the results of Kim and Shibata, who have shown that adjacent colonic crypts and even crypts in fission have quite dissimilar methylation patterns, it seems that crypt fission may take a long time in the human. In the mouse, the time for fission appears to take ≈12 h, with a possible crypt cycle time of ≈112 days (27). It has also been suggested that normal adult crypts in the human colon divide every 17 years (27, 28). A fission time of 27 months does not seem unreasonable, which could be the coefficient time for such divergence in methylation signatures to occur.
We have previously shown that the adult colon has large families of adjacent crypts (≈450) (15), which show the same X-linked gene, indicating that after lyonization, early stem cells from the colonic mucosal crypts expand, implicitly by crypt fission, to form large families of related crypts, so far as sharing the same X-chromosome is concerned. However, Kim and Shibata results (17, 18) show that the methylation patterns of adjacent crypts diverge markedly by stem cell niche succession, but every 8 years or so, a “bottleneck” is reached, where all stem cells in the crypt are closely related. Superimposed on this process, throughout life, is crypt fission, whereby a crypt expands to form a group or patch of closely related crypts, insofar as the genotype is concerned, even while their unique methylation patterns are diverging.
Materials and Methods
Patients. For cluster analysis, 1.0 × 1.5-cm blocks of morphologically normal colonic mucosa were taken from colectomy specimens of 14 patients with colorectal cancer (10 patients), diverticular disease (3 patients), or cecal stricture (1 patient). Blocks were selected from outside the resection margins of tumors and, in the case of diverticular disease, in noninflamed mucosa. Specimens were either fixed in 4% paraformaldehyde and embedded in paraffin or snap-frozen in liquid nitrogen-cooled isopentane. For patch-size analysis, the same 14 patients' blocks were used plus blocks taken from an additional 20 patients stored in tissue archives, reorientated en face. Reorientated blocks were not of sufficient size and continuity to perform cluster analysis. Ethical approval was sought and obtained from the Newcastle and North Tyneside Local Research Ethics Committees and the University College Hospital Research Ethics Committee.
Immunohistochemistry. Colonic sections were cut to a thickness of 4 μm. Antigen retrieval was performed by pressure cooking the slides for 3 min in boiling 0.1 M sodium citrate buffer, pH 6.0, after dewaxing them in xylene and rehydrating then through decreasing ethanol and finally with distilled water. Nonspecific staining was blocked by adding protein block serum-free ready-to-use buffer (DAKO) for 15 min then washed three times in Tris-buffered saline with 0.1% Triton X-100. A 1:100 dilution of mouse anti-human cytochrome c oxidase subunit I was made in Tris/Triton X-100 and applied to the sections for 30 min in a humid chamber. Slides were then washed three times in TBS/Triton. Sections were then incubated in neat CheMate (Envision) anti-rabbit/anti-mouse conjugated with horseradish peroxidase (DAKO) and developed in 4 mM diaminobenzidene tetrachloride (DAKO). The sections were lightly counterstained in haematoxylin, dehydrated through increasing ethanol washes, cleared with xylene, and mounted in DPX. Isotype-matched and without primary controls revealed no nonspecific staining.
Histochemistry. Frozen colon samples were mounted in OCT compound for sectioning. Sequential histochemical staining for cytochrome c oxidase and succinate dehydrogenase histochemistry (the presence of which was used to highlight the absence of cytochrome c oxidase activity) was carried out on 20-μm transverse sections as follows. Sections were first incubated in cytochrome c oxidase incubation medium (100 μM cytochrome c/4 mM diaminobenzidine tetrahydrochloride/20 μg/ml catalase in 0.2 M phosphate buffer, pH 7.0) for 50 min at 37°C. They were then washed in PBS, pH 7.4, for 3 × 5 min and incubated in SDH incubation medium (130 mM sodium succinate/200 μM phenazine methosulphate/1 mM sodium azide/1.5 mM nitroblue tetrazolium in 0.2 M phosphate buffer, pH 7.0) for 45 min at 37°C. Sections were washed in PBS for 3 × 5 min, dehydrated in a graded ethanol series (70%, 95%, and 100%) and left to air dry for 1 h.
Isolation of Total DNA from Individual Cells. Single cells from cytochrome c oxidase-deficient and cytochrome c oxidase-positive crypts were cut into sterile 0.5-ml PCR tubes by using the Leica (Deerfield, IL) Laser Microdissection (AS-LMD) System. After centrifugation (7,000 × g for 10 min), the cell was lysed in 14 μl of cell lysis buffer (50 mM Tris·HCl, pH 8.5/1 mM EDTA/0.5% Tween-20/200 ng/ml proteinase K) at 55°C for 2 h and then 95°C for 10 min to denature the proteinase K.
mtDNA Sequencing of Individual Colonocytes. The entire sequence of the mitochondrial genome from microdissected crypts was determined by using DNA extracted from single-cell lysates (see above). The mitochondrial genome was amplified in overlapping fragments by using a series of M13-tailed oligonucleotide primer pairs, as described (29). PCR products were sequenced by using BigDye Ver. 3.1 terminator cycle sequencing chemistries on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems) and compared directly with the revised Cambridge reference sequence (rCRS) by using sequence analysis and seqscape software (Applied Biosystems), as described (29).
Image Analysis for Spatial Clustering. Low-power (×20) digital images of tissue sections of normal colonic mucosa, cut en face, stained with anticytochrome c oxidase subunit 1, were captured by using an Optronics Magnafire digital microscope camera (Optronics International, Chelmsford, MA). A maximum of three individual images were taken from different sites on each section and up to two blocks used from the same patient taken from different regions of the colon. A total of 29 images (from a total of 10 patients) were of sufficient quality to use for analysis. Images with inominate grooves, inflammation, or areas with luminal spaces were discarded.
To determine the presence/strength of any spatial clustering of cytochrome c oxidase-deficient colonic crypts, we devised a method that calculates three separate statistical measures of spatial clustering, that takes into account the actual geometrical configuration of crypts in the colonic mucosal images under study, and that does not make assumptions about specific simplified geometries [e.g., we do not assume the crypts are arranged in an orderly hexagonal array, as in some previous methods of cluster analysis (30)]. The method, for each image, is divided into an interactive step and an automated step. The interactive step involves generating a simplified labeled image of crypt positions and types, which is then fed as input into the second step, a custom-written program (called clan for cluster analysis, Tadrosoft, London) that performs the statistical analysis. The simplified labeled image was made by marking the center point of all of the crypts in each field with a single pixel, and each of these pixels was labeled by the operator as either cytochrome c oxidase-deficient (called an index crypt) or -positive (a background crypt). This was performed by using the freely available biaqim image analysis software. The labeled image was then analyzed by clan, which measured the value of the three cluster statistics (defined below) and also generated control distributions for each of these statistics by measuring the value of that statistic on 1,000 random images. Each random image was generated by randomly assigning crypt pixels to either index or background status while keeping the absolute number of index and background pixels, as well as the positions of all pixels, the same as for the image under study (thus a control distribution is calculated for each image as well as for each of the three statistics). In this way, the original geometric configuration of colonic crypts is taken into account when generating control probability distributions rather than assumptions being made about idealized configurations.
Definition of Cluster Statistics. Nprop. The proportion of index crypts in an image with at least one adjacent crypt also of index (i.e., cytochrome c oxidase-deficient) type.
RR. The RR of an index crypt also having adjacent neighbors that are also of index type calculated as;
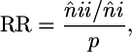 |
where n̂ii is the mean number of index crypt adjacent neighbors that are also index crypts, n̂i is the mean number of index crypt adjacent neighbors, and p is the proportion of all crypts in the image field that are of index type.
χ2. A χ2 statistic calculated as
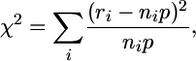 |
where the summation is over all index crypts, i.
In the above definitions, the definition of a crypt as being “adjacent” to any given index crypt was based on an exclusion circle of radius Ra, where all crypts closer than Ra pixels from a given index crypt are considered adjacent neighbors of that index crypt. The value of Ra was chosen as Ra = X + 5SD, where X is the mean of the minimum distance of each crypt to its closest neighbor calculated from all crypts in a given image, and SD is the standard deviation of these minimum distances. This choice of Ra was arrived at empirically, because it was found to agree well with an exclusion radius based on actual measurements of the distances from any given crypt to its immediately adjacent neighbors on a sample of index crypts. Because this is a statistical measure of adjacency, an heuristic constraint was enforced to ensure that Ra was a reasonable measure of crypt adjacency in that only those index crypts with five or six adjacent neighbors (defined by using the Ra exclusion radius) were eligible for calculation of the statistics. This heuristic is based on the observation that very few crypts have seven or more or four or less adjacent neighbors. The vast majority of crypts in each image studied were found to be eligible for analysis by these criteria.
Patch-Size Analysis. A patch was defined as at least two cytochrome c oxidase-deficient crypts in juxtaposition to one another within a distance of no more than the width of a single crypt. Paraffin-embedded blocks were taken from 34 patients (age range of 35–85 years), which were previously stored in tissue archives. The blocks were reorientated to provide an en face image. Tissue sections were subjected to immunohistochemistry (described above) for cytochrome c oxidase. The numbers of completely deficient crypts within each patch were counted manually at a magnification of ×200. Results were expressed as the mean patch size per patient ± SEM. The mean number of crypts per image was 489.4 ± 202.45.
Odds Ratios. The minimal odds of two random identical mtDNA mutations occurring at the same site in two separate crypts by chance were calculated as [16,600:1 (genome size) × 3 (possible transition mutations)] × [16,600:1 × 3] = 2.48 × 109:1. This does not take into consideration that other types of mutations are also found; therefore, it provides only minimal odds.
Supplementary Material
Acknowledgments
We thank Dr. Matthew Lovell, Histopathology Unit, Cancer Research UK, for art work. L.C.G. is a Medical Research Council Student. This work was supported in part by the Leicester University Hospital Research Trust (to S.A.C.M. and J.A.Z.J.), CORE (formerly the Digestive Disorders Foundation) (to S.L.P.), and the Wellcome Trust (to D.M.T. and R.W.T.).
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: clan, cluster analysis; RR, relative risk.
References
- 1.Karam, S. M. (1999) Front. Biosci. 4, D286–D298. [DOI] [PubMed] [Google Scholar]
- 2.Wright, N. A. & Alison, M. R. (1984) The Biology of Epithelial Cell Populations (Clarendon, Oxford, U.K.).
- 3.Preston, S. L., Wong, W. M., Chan, A. O., Poulsom, R., Jeffery, R., Goodlad, R. A., Mandir, N., Elia, G., Novelli, M., Bodmer, W. F., et al. (2003) Cancer Res. 63, 3819–3825. [PubMed] [Google Scholar]
- 4.Shih, I. M., Wang, T. L., Traverso, G., Romans, K., Hamilton, S. R., Ben-Sasson, S., Kinzler, K. W. & Vogelstein, B. (2001) Proc. Natl. Acad. Sci. USA 98, 2640–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brierley, E. J., Johnson, M. A., Lightowlers, R. N., James, O. F. & Turnbull, D. M. (1998) Ann. Neurol. 43, 217–223. [DOI] [PubMed] [Google Scholar]
- 6.Michikawa, Y., Mazzucchelli, F., Bresolin, N., Scarlato, G. & Attardi, G. (1999) Science 286, 774–779. [DOI] [PubMed] [Google Scholar]
- 7.Carew, J. S. & Huang, P. (2002) Mol. Cancer 1, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor, R. W. & Turnbull, D. M. (2005) Nat. Rev. Genet. 6, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sciacco, M., Bonilla, E., Schon, E. A., DiMauro, S. & Moraes, C. T. (1994) Hum. Mol. Genet. 3, 13–19. [DOI] [PubMed] [Google Scholar]
- 10.Taylor, R. W., Barron, M. J., Borthwick, G. M., Gospel, A., Chinnery, P. F., Samuels, D. C., Taylor, G. A., Plusa, S. M., Needham, S. J., Greaves, L. C., et al. (2003) J. Clin. Invest. 112, 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park, H. S., Goodlad, R. A. & Wright, N. A. (1995) Am. J. Pathol. 147, 1416–1427. [PMC free article] [PubMed] [Google Scholar]
- 12.Anne Cook, H., Williams, D. & Anne Thomas, G. (2000) J. Pathol. 191, 306–312. [DOI] [PubMed] [Google Scholar]
- 13.Winton, D. J., Blount, M. A. & Ponder, B. A. (1988) Nature 333, 463–466. [DOI] [PubMed] [Google Scholar]
- 14.Garcia, S. B., Park, H. S., Novelli, M. & Wright, N. A. (1999) J. Pathol. 187, 61–81. [DOI] [PubMed] [Google Scholar]
- 15.Novelli, M., Cossu, A., Oukrif, D., Quaglia, A., Lakhani, S., Poulsom, R., Sasieni, P., Carta, P., Contini, M., Pasca, A., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 3311–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novelli, M. R., Williamson, J. A., Tomlinson, I. P., Elia, G., Hodgson, S. V., Talbot, I. C., Bodmer, W. F. & Wright, N. A. (1996) Science 272, 1187–1190. [DOI] [PubMed] [Google Scholar]
- 17.Kim, K. M. & Shibata, D. (2002) Oncogene 21, 5441–5449. [DOI] [PubMed] [Google Scholar]
- 18.Kim, K. M. & Shibata, D. (2004) BMC Gastroenterol. 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slorach, E. M., Campbell, F. C. & Dorin, J. R. (1999) J. Cell Sci. 112, 3029–3038. [DOI] [PubMed] [Google Scholar]
- 20.Yatabe, Y., Tavare, S. & Shibata, D. (2001) Proc. Natl. Acad. Sci. USA 98, 10839–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu, C. H., Yeh, L. S., Huang, H., Arminski, L., Castro-Alvear, J., Chen, Y., Hu, Z., Kourtesis, P., Ledley, R. S., Suzek, B. E., et al. (2003) Nucleic Acids Res. 31, 345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brentnall, T. A., Crispin, D. A., Rabinovitch, P. S., Haggitt, R. C., Rubin, C. E., Stevens, A. C. & Burmer, G. C. (1994) Gastroenterology 107, 369–378. [DOI] [PubMed] [Google Scholar]
- 23.Wong, D. J., Paulson, T. G., Prevo, L. J., Galipeau, P. C., Longton, G., Blount, P. L. & Reid, B. J. (2001) Cancer Res. 61, 8284–8289. [PubMed] [Google Scholar]
- 24.Wasan, H. S., Park, H. S., Liu, K. C., Mandir, N. K., Winnett, A., Sasieni, P., Bodmer, W. F., Goodlad, R. A. & Wright, N. A. (1998) J. Pathol. 185, 246–255. [DOI] [PubMed] [Google Scholar]
- 25.Campbell, F., Geraghty, J. M., Appleton, M. A., Williams, E. D. & Williams, G. T. (1998) Hum. Pathol. 29, 1531–1535. [DOI] [PubMed] [Google Scholar]
- 26.He, X. C., Zhang, J., Tong, W. G., Tawfik, O., Ross, J., Scoville, D. H., Tian, Q., Zeng, X., He, X., Wiedemann, L. M., et al. (2004) Nat. Genet. 36, 1117–1121. [DOI] [PubMed] [Google Scholar]
- 27.Bjerknes, M. (1986) Biophys. J. 49, 1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Totafurno, J., Bjerknes, M. & Cheng, H. (1987) Biophys. J. 52, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor, R. W., Taylor, G. A., Durham, S. E. & Turnbull, D. M. (2001) Nucleic Acids Res. 29, E74–E84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howel, D. & Brunsdon, C. (1987) J. Neurol. Sci. 77, 49–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}