

Abstract
Membranous nephropathy (MN) is a common cause of nephrotic syndrome in adults. Active and passive Heymann nephritis (HN) in rats are valuable experimental models because their features so closely resemble human MN. In HN, subepithelial immune deposits form in situ as a result of circulating antibodies. Complement activation leads to assembly of C5b-9 on glomerular epithelial cell (GEC) plasma membranes and is essential for sublethal GEC injury and the onset of proteinuria. This review revisits HN and focuses on areas of substantial progress in recent years. The response of the GEC to sublethal C5b-9 attack is not simply due to disruption of the plasma membrane but is due to the activation of specific signaling pathways. These include activation of protein kinases, phospholipases, cyclooxygenases, transcription factors, growth factors, NADPH oxidase, stress proteins, proteinases, and others. Ultimately, these signals impact on cell metabolic pathways and the structure/function of lipids and key proteins in the cytoskeleton and slit-diaphragm. Some signals affect GEC adversely. Thus C5b-9 induces partial dissolution of the actin cytoskeleton. There is a decline in nephrin expression, reduction in F-actin-bound nephrin, and loss of slit-diaphragm integrity. Other signals, such as endoplasmic reticulum stress, may limit complement-induced injury, or promote recovery. The extent of complement activation and GEC injury is dependent, in part, on complement-regulatory proteins, which act at early or late steps within the complement cascade. Identification of key steps in complement activation, the cellular signaling pathways, and the targets will facilitate therapeutic intervention in reversing GEC injury in human MN.
Keywords: passive Heymann nephritis, podocyte, complement, complement regulation, proteinuria, autoimmunity, cell signaling, cytoskeleton, slit-diaphragm
membranous nephropathy (MN) is an organ-specific autoimmune disease and a relatively common cause of nephrotic syndrome in adults. It is characterized by subepithelial immune deposits containing IgG and complement, expansion of the glomerular basement membrane (GBM), and diffuse effacement of podocyte [glomerular epithelial cell (GEC)] foot processes. Active and passive Heymann nephritis (HN) in rats have been widely used as models of human MN since 1965 (47). They remain valuable experimental tools because the functional and immunohistological features so closely resemble the human disease. In 1989, we reviewed the experimental literature on HN (118). Several key observations were documented at that time, most notably 1) that the immune deposits form in situ as a result of circulating antibodies, either injected as heterologous anti-Fx1A in passive HN (PHN) or endogenous autoantibodies after immunization with Fx1A, the antigenic preparation derived from proximal tubular brush border; that complement activation is essential for the onset of proteinuria; 2) that the effect of complement is mediated by assembly of C5b-9 and sublethal damage to podocytes; and 3) that sublethal GEC injury triggers a cascade of intracellular signaling events.
Since our review in 1989, the target antigen (gp330) was identified and shown to reside in coated pits on the soles of podocyte foot processes (35). Detailed immunohistological and cell culture studies showed that the subepithelial deposits form in situ as a result of antibody-mediated capping of the antigen on the podocyte surface and shedding of the antibody-antigen complexes into the GBM (7, 60). Subsequently, the cDNA for the HN antigen was cloned and shown to encode a large (~600 kDa) protein named megalin, a member of the LDL receptor family (35). Megalin and receptor-associated protein (RAP) appear to constitute the target antigen, and antibodies specific for an epitope within RAP promote cross-linking of the complexes to components of the GBM (63). Whereas targeting other podocyte surface antigens can induce proteinuria independently of complement (117), it appears that simply forming subepithelial immune deposits in the absence of complement fixation does not alter podocyte morphology or function (19). Identification of several podocyte proteins in inherited forms of nephrotic syndrome further substantiated the role of podocyte dysfunction in proteinuric kidney diseases and triggered much interest in the podocyte, podocyte-associated proteins, and signaling cascades in acquired forms of podocyte injury, including experimental and human MN. In addition, expanding knowledge of cell-surface and circulating complement regulatory proteins has opened new avenues for therapeutic intervention. The HN models have proved valuable in identifying the role of such proteins in protecting podocytes from complement-mediated damage and for testing proof of concept of anticomplementary molecules.
In this review, we have revisited the HN models with an update focused on three areas in which there has been substantial activity: complement-mediated cell signaling; regulation of complement; and alterations in podocyte structure and function.
COMPLEMENT AND THE ROLE OF THE C5B-9 MEMBRANE ATTACK COMPLEX IN EXPERIMENTAL MN
The complement system consists of the classical, alternative, and mannose-binding lectin pathways, altogether containing over 30 proteins involved in the activation and regulation of this system (143). Activation of the complement classical pathway was considered relevant to inflammatory glomerular diseases such as nephrotoxic serum nephritis (142), but its role in experimental MN was overlooked because of the lack of inflammation, considered the necessary product of complement activation in the glomerulus. With the appreciation that the complement C5b-9 membrane attack complex could have direct cellular effects, a series of studies performed in the 1980s showed a role for activation of C5b-9 on the podocyte in experimental MN (2, 18, 19). At the same time, it became clear that the intrinsic podocyte antigen now known as megalin was a target of antibodies in experimental MN (59, 76). Follow-up experiments showed that antibody-directed complement activation on the podocyte led to C5b-9 insertion in this cell, with subsequent shedding into the urine (62, 123). Additional proof for the relevance of complement in experimental MN came from the studies showing that complement inhibition with recombinant human complement receptor 1 (CR1; CD35) led to a reduction in proteinuria in this model (13). The exact effects of C5b-9 on podocytes have been the topic of significant efforts that are detailed below.
SIGNALING PATHWAYS ACTIVATED BY ASSEMBLY OF C5b-9
The consequences of C5b-9 assembly in the plasma membrane include formation of transmembrane channels or rearrangement of membrane lipids with loss of membrane integrity (Fig. 1). Nucleated cells require multiple C5b-9 lesions for lysis, whereas at lower doses C5b-9 induces sublethal (sublytic) injury (83, 94, 116). The response of a cell, including the GEC, to sublytic doses of C5b-9 attack is not simply due to disruption of the plasma membrane but rather to the activation of specific signaling pathways. Such signals include the activation of phospholipases, protein kinases, transcription factors, growth factors, proteinases, and others. Ultimately, these signals may impact on cell metabolic pathways and the structure or function of lipids and key cellular proteins in the cytoskeleton, slit-diaphragm, or other compartments. Some signals affect GEC adversely, whereas others appear to limit the extent of complement-induced injury, or promote recovery. In some cases, activation of a single pathway can lead to both cytotoxic and protective responses.
Fig. 1.

Signaling pathways activated by C5b-9 in cultured glomerular epithelial cells (GEC). [Ca2+]i, intracellular Ca2+ concentration; RTKs, receptor tyrosine kinases; PKC, protein kinase C; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; PLC, phospholipase C; PLA2, phospholipase A2; ER, endoplasmic reticulum; Hsp, heat shock protein; NF-κB, nuclear factor-κB; COX, cyclooxygenase; MMP, metalloproteinase; PDGF, platelet-derived growth factor; HB-EGF, heparin-binding epidermal growth factor-like factor; TGF, transforming growth factor; CDK, cyclin-dependant kinase; ECM, extracellular matrix.
Influx of Ca2+ and Transactivation of Receptor Tyrosine Kinases
Assembly of C5b-9 in cultured GECs leads to an increase in cytosolic free Ca2+ concentration ([Ca2+]) that is primarily due to calcium influx (20) (Fig. 2). Studies in GEC culture and in the glomeruli of rats with PHN showed that another early event that follows assembly of C5b-9 is the transactivation of receptor tyrosine kinases (9), including the epidermal growth factor receptor (EGFR), Neu, fibroblast growth factor receptor-2, and hepatocyte growth factor receptor (21, 23). Thus C5b-9 induced tyrosine phosphorylation of EGFR, and phosphorylated EGFR was able to bind the adaptor protein, Grb2 (127), and a peptide containing the SH2 and SH3 regions of phospholipase C-γ1 (PLC-γ1) (21, 23, 67). Furthermore, assembly of C5b-9 resulted in activation of the Ras-extracellular signal-regulated kinase (ERK) pathway (127), as well as PLC-γ1 and its downstream effectors, and these events were blocked with an EGFR inhibitor (17). Transactivated tyrosine kinases may serve as scaffolds for assembly and/or activation of proteins, which then lead to activation of downstream effector pathways, either independently, or in conjunction with increased cytosolic [Ca2+].
Fig. 2.
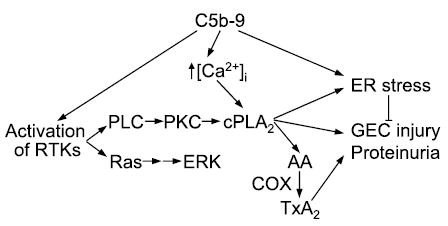
Activation of signaling pathways leading to GEC injury and proteinuria. Tx, thromboxane. See legend for Fig. 1 for other abbreviations.
Activation of Phospholipase A2
A key signaling pathway activated by C5b-9 involves release of arachidonic acid (AA) by phospholipase A2 (PLA2) and subsequent AA metabolism to prostanoids. In GEC in culture and in vivo, a major endogenous PLA2 isoform is cytosolic PLA2-α(cPLA2; group IV) (27, 49), and activation of cPLA2 has been linked with C5b-9 (16, 24, 98). As in other cells, cPLA2 activation in GEC is regulated by changes in cytosolic [Ca2+] and phosphorylation, the latter being dependent on PLC activation, production of 1,2-diacylglycerol, and activation of the protein kinase C (PKC) pathway (16, 98). In the GEC, cPLA2 localizes and hydrolyzes phospholipids at the plasma membrane, the membrane of the endoplasmic reticulum (ER), and the nuclear envelope, but not at mitochondria orGolgi (72). Thus the activation of cPLA2 and release of AA is compartmentalized to specific organelles. cPLA2 appears to be an important mediator of C5b-9-dependent GEC injury. First, the AA released by cPLA2 is metabolized in GECs via cyclooxygenases (COX)-1 and -2 to prostaglandin E2 and thromboxane A2, and inhibition of prostanoid production reduces proteinuria in PHN (see immediately below). Second, cPLA2 may mediate GEC injury more directly, by inducing cell membrane phospholipid hydrolysis and the ER stress response (see below).
Consequences of AA Metabolism
The AA released by C5b-9-induced activation of cPLA2 may be metabolized to prostanoids by COX enzymes (132). Resting GEC in culture primarily express COX-1. COX-2, but not COX-1, is upregulated by incubation with sublytic C5b-9 (135). The upregulation of COX-2 is dependent on activation of PKC, as well as the c-Jun NH2-terminal kinase (JNK) (66, 137). Release of AA results in increased prostaglandin E2 production, which was inhibited by 60% with the COX-2-selective inhibitor NS-398 (135). Glomeruli from rats with PHN express significantly more COX-1 and COX-2 than normal rat glomeruli. Prostaglandin E2 production in glomeruli of rats with PHN was about twofold greater than in control glomeruli. This increase was partially inhibited with NS-398 (135). Thus in GEC in culture C5b-9 increases the expression of COX-2, whereas COX-1 and -2 appear to be unregulated in vivo in PHN. Both isoforms of COX are involved in prostanoid production.
Earlier studies demonstrated that indomethacin, a nonselective COX inhibitor, reduced proteinuria in PHN (15, 148). Recently, the role of COX isoforms in the mediation of proteinuria in PHN was addressed by treating rats with either indomethacin or 5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methyl-sulfonyl)phenyl-2(5H)-furanone (DFU), a COX-2-selective inhibitor. Compared with vehicle-treated rats, treatment of PHN rats with DFU reduced proteinuria significantly (by ~33%), but to a lesser extent than indomethacin (56% reduction) (136). Therefore, inhibition of both COX-1 and -2 was required to achieve a maximum antiproteinuric effect. Both compounds reduced glomerular prostanoid generation significantly. Neither indomethacin nor DFU affected inulin clearance or glomerular expression of COX-1 and -2. In another study, the COX-2 inhibitor flosulide partially reduced proteinuria in PHN, but this drug also appeared to decrease expression of COX-1 and -2 (6). Other approaches to the blockade of prostanoid production have also been successful in reducing proteinuria in PHN. For example, treatment of rats with PHN with the thromboxane synthase inhibitor DP-1904 reduced proteinuria (87). Shifting production of dienoic prostanoids to inactive trienoic metabolites with a fish oil diet in PHN rats reduced production of glomerular thromboxane A2 and had both protective and therapeutic effects even after the onset of severe proteinuria (145). Thus a number of studies employing distinct experimental approaches support a role for prostanoids in exacerbating proteinuria in PHN.
The mechanism(s) by which prostanoids exacerbate proteinuria requires further study. One possibility is that prostanoids may contribute toward increasing glomerular capillary pressure, such that proteinuria is increased on a hemodynamic basis. Alternatively, in cultured GEC, COX inhibition reduced complement-induced cytotoxicity, and this reduction was reversed by the thromboxane A2 analog U-46619, suggesting that production of prostanoids may exacerbate GEC injury (136). On the other hand, effective substitution of GEC membrane phospholipids with omega-3 fatty acids substantially reduced thromboxane A2 production but did not protect the cells from C5b-9-mediated injury (43). Thus selective prostanoid inhibition may be an effective means of reducing proteinuria, perhaps independently of hemodynamic factors (77); however, the cellular mechanisms are still uncertain.
ER Injury and Stress Response
C5b-9-induced activation of cPLA2 results in damage to the membrane of the ER and induction of ER stress, i.e., the “unfolded protein response” (56). Brief incubation of GEC with complement perturbed the integrity of the ER membrane in a cPLA2-dependent manner (22). Exposure of GEC to chronic complement attack (4–24 h) resulted in increased expression of the ER stress proteins bip and grp94, and these increases were, at least in part, mediated via activation of cPLA2 (22). The increases in ER stress proteins were a direct result of cPLA2-mediated phospholipid hydrolysis (and, presumably, membrane injury) and were not due to products of phospholipid hydrolysis (e.g., AA, lysophospholipids) or their metabolites (i.e., prostanoids). Expression of bip antisense mRNA in GEC exacerbated complement-mediated GEC injury, thus confirming a functionally important protective role for ER stress proteins. Furthermore, “preconditioning” of GEC with other stimuli that can increase expression of ER stress proteins (e.g., puromycin aminonucleoside) limited the amount of injury during subsequent complement attack (22).
In vivo, expression of glomerular bip and grp94 was increased in rats with PHN (day 14) compared with control. Administration of a subnephritogenic dose of adriamycin or tunicamycin to rats also enhanced glomerular expression of bip and grp94. Based on these results, rats were treated with a subnephritogenic dose of adriamycin, or tunicamycin, and PHN was then induced in these pretreated rats and in untreated animals. Substantial proteinuria developed in untreated rats with PHN, whereas the amount of proteinuria was significantly lower in the rats with PHN that had been pretreated with the two compounds (days 7, 9, and 13) (22). Thus increasing ER stress protein expression can reduce C5b-9-mediated GEC injury in vivo. Adriamycin and tunicamycin did not decrease proteinuria by reducing the amount of glomerular antibody deposition, or complement activation, and serum creatinine was not significantly different among the three groups of rats, suggesting that there were no significant differences in renal function. This study provides a rationale for developing non-toxic methods to induce expression of ER stress proteins in vivo, which may eventually have applications to therapy of complement-mediated glomerular disease.
Reactive Oxygen Species
Phagocytes generate reactive oxygen species (ROS) in response to a variety of stimuli. This process is catalyzed by a multicomponent enzyme complex, the NADPH oxidase, which, in the presence of NADPH, reduces molecular oxygen to the superoxide anion (138). Superoxide can then be further metabolized to other ROS. Although the NADPH oxidase system is best characterized in phagocytic cells (138), GEC in culture and in vivo express components of the NADPH oxidase (44, 93). Incubation of cultured GEC with complement stimulated superoxide production via the NADPH oxidase. The mechanism involves activation of cPLA2 and release of AA (100).
Classically, production of ROS is believed to be associated with cell or tissue damage, but more recently, ROS (particularly at low concentrations) have been shown to function in signal transduction (36, 138). In GEC in culture and in vivo, complement-induced superoxide production led to activation of JNK, and in culture, JNK activation was associated with cytoprotection (100). In contrast, earlier studies showed that high amounts of ROS are produced in PHN and that ROS are associated with lipid peroxidation and modification of GEC membrane proteins and glomerular basement membrane components by malondialdehyde adducts (92, 93). Inhibition of ROS and lipid peroxidation with probucol (a cholesterol-lowering drug with antioxidant properties) reduced urine protein excretion in PHN, compared with a control group of rats that were treated with simvastatin (a cholesterol-lowering drug without antioxidant properties), suggesting that ROS exacerbate proteinuria by damaging the glomerular capillary wall (92). Thus in PHN, ROS may potentially play a dual role, both facilitating signal transduction and contributing to cell injury. The relative importance of these functions to the pathogenesis of PHN will require further study.
p38 Kinase
In addition to ERK and JNK, C5b-9 can activate the p38 kinase pathway in cultured GEC (1). Treatment of GEC with p38 inhibitors significantly augmented complement-mediated cytotoxicity (1). In contrast, cytotoxicity was reduced by the expression of a constitutively active mutant of transforming growth factor-activated kinase-1 (a kinase upstream of p38) (66). By analogy to cultured cells, p38 activity was also increased in glomeruli from rats with PHN, and treatment of these rats with a p38 inhibitor exacerbated proteinuria. In cultured GEC, complement induced phosphorylation of mitogen-activated protein kinase-associated protein kinase-2 (MAPKAPK-2), a kinase downstream of p38 (66). Heat shock protein-27 (HSP27) is a cytoskeleton-interacting substrate of MAPKAPK-2. Overexpression of wild-type HSP27, but not a phosphorylation-defective mutant, markedly reduced complement-mediated GEC injury (1). Therefore, activation of p38 appears to be protective against complement attack, and cytoprotection may involve HSP27.
Nuclear Factor-κB
Activation of the transcription factor nuclear factor κB (NF-κB) is central to inflammatory processes (79). In cultured GEC, complement can activate NF-κB and NF-κB-dependent gene transcription (137). In vivo, glomerular nuclear extracts from rats with PHN showed increased NF-κB binding activity compared with control rats (86). By immunohistochemistry, the p50 subunit demonstrated nuclear translocation, predominantly within podocytes. Expression of the NF-κB-dependent gene interleukin-1βwas increased in GEC of rats with PHN (at days 10 and 20), although expression of another NF-κB-dependent gene, matrix metalloproteinase-9 (MMP-9), did not change. NF-κB was inhibited in vivo by administration of pyrrolidone dithiocarbamate (PDTC). In mild PHN (i.e., PHN induced by a low dose of nephritogenic antibody, where rats developed modest proteinuria), PDTC produced a significant decline in proteinuria (on days 15 and 20), but PDTC had no effect in PHN with high levels of urine protein excretion. PDTC also induced a decline in MMP-9 expression but had no significant effect on the level of interleukin-1β (86). These results suggest that NF-κB is activated, and may, in part, contribute to proteinuria in PHN. The genes transcriptionally regulated by NF-κB require further elucidation.
Growth Factors and Signals for Proliferation
Responses to cell injury may include proliferation and/or apoptosis and may involve induction of growth factors, growth factor receptors, and proteins that regulate the cell cycle (128). Platelet-derived growth factor (PDGF) B-chain was upregulated on day 3 of PHN in both visceral and parietal GEC (38). The increase was evident before the onset of significant proteinuria. In contrast, PDGF-C expression was not demonstrated (33). Heparin-binding epidermal growth factor-like factor (HB-EGF) mRNA and protein were upregulated within 3 days following induction of PHN, before the onset of proteinuria (97). At this time point, HB-EGF was localized in the cytoplasm of GEC, whereas on day 21 HB-EGF showed a nodular pattern within GEC and along the GBM, suggesting secretion and binding to the GBM. The functional relevance of these growth factors will require further elucidation. Receptors for PDGF are not present in GEC; consequently, PDGF might act in a paracrine fashion. In contrast, HB-EGF could be involved in EGFR activation in GEC.
In vivo, the GEC is believed to be a terminally differentiated cell with a low capacity for proliferation under normal circumstances and after injury (128). In PHN, GEC do not proliferate, although an increase in proliferating cell nuclear antigen expression has been reported (38). Various changes in cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors have also been reported in PHN. There is pronounced upregulation of the CDK inhibitors p21 and p27 in association with decreased CDK2 kinase activity (128). The CDK inhibitor p57 was decreased (50). Cdc2, cyclins B1, B2, and D1 and phosphorylated histone 3 were increased (32, 101–104). Administration of exogenous basic fibroblast growth factor to rats with PHN attenuated the increase in p21 levels and increased GEC mitosis and ploidy (128, 129). Thus the low proliferative capacity of GEC in vivo in response to injury may be due, at least in part, to an increase in expression of specific cyclin kinase inhibitors, including p21 and p27.
In cultured GEC and in PHN, C5b-9 induced an increase in DNA damage (103, 104). This damage was associated with increases in p53, the p21 CDK inhibitor, the growth-arrest DNA damage-45 (GADD45) gene, and the checkpoint kinases-1 and -2. C5b-9-induced activation of ERK (17) appeared to protect GEC from DNA damage, as inhibition of the ERK pathway reduced the increases in p21 and GADD45 and augmented DNA damage (104). Induction of DNA damage by C5b-9 may explain, in part, why GEC proliferation is limited following injury.
Extracellular Matrix
The glomerular capillary wall is a size- and charge-selective filter that is critical for ultrafiltration and restricting passage of proteins (140). Several studies indicate that C5b-9 may induce changes in the expression of GBM components or enzymes involved in extracellular matrix metabolism. Steady-state mRNA levels of α3-α5 type IV collagen chains were reported to be increased in glomeruli of rats with PHN (on day 14), and complement depletion blunted the increase in α4 chain (81). In addition, there was de novo synthesis of type I collagen. Expression of agrin, the main heparan sulfate proteoglycan of the GBM, was decreased in active HN, and the reduction correlated inversely with the amount of proteinuria (111). MMP-9 mRNA and protein were upregulated in GEC in rats with PHN (days 5 and 15) (78). GEC heparanase expression was increased in PHN on days 5, 14, and 28, and the change on day 5 was abrogated by complement depletion (54). In addition, glomerular and urinary heparanase activities were increased on days 5 and 21. Administration of polyclonal anti-heparanase antibody significantly reduced proteinuria on day 5 (54, 68). Finally, transforming growth factor (TGF)-β family members may mediate changes in the metabolism of extracellular matrix components. Expression of TGF-β2 and TGF-β3 (but not TGF-β1), as well as TGF-β type I and type II receptors, was increased in PHN in a complement-dependent manner (130). Together, the results of these studies indicate that in PHN, assembly of C5b-9 in GEC may promote formation of an expanded and disorganized GBM. In addition, secreted proteolytic enzymes may be induced/upregulated in GEC and may play a role in the breakdown or turnover of glomerular capillary wall components, as well as in the pathogenesis of proteinuria.
PODOCYTE CYTOSKELETAL CHANGES INDUCED BY ANTIBODY AND COMPLEMENT
Condensation of the actin cytoskeleton at the base of effaced podocyte foot processes is a prominent feature of both human and experimental MN. This is accompanied by various alterations in the intervening filtration slits, including widening, formation of occluding-type junctions, and displacement and disruption of slit-diaphragms (31, 122). In an in vitro model of anti-Fx1A- and complement-mediated sublethal injury of rat GEC, C5b-9 assembly induced the dissolution of F-actin microfilaments and loss of the focal adhesion complexes that anchor the cytoskeleton to integrins (139). The in vitro changes were found to be reversible as the cells recovered when the antibodies and complement were removed (139). The disassembly of focal adhesions was associated with substantial ATP depletion and dephosphorylation of paxillin and other proteins but was not prevented by tyrosine phosphatase inhibition, nor was reassembly retarded by tyrosine kinase inhibition. There was also no evidence of proteolysis of the focal adhesion or cytoskeletal proteins, which argues for a dynamic disassembly and reassembly process perhaps mediated by Rho and downstream members of the Ras family of small GTPases. Interestingly, the α3β1-integrins, which attach podocytes to the underlying matrix, remained intact. This finding corresponds to in vivo immunoelectron microscopy observations showing normal distribution of β1-integrins on the basal aspect of effaced foot processes in proteinuric human and experimental diseases, including MN (61). It is also noteworthy that an increase has been reported in the expression of the antiadhesive protein SPARC, together with upregulation of the intermediate filament desmin in HN and other models of podocyte injury (37). Together, these findings of collapse of the actin cytoskeleton and altered adhesion to the GBM may account for loss of the column-like architecture and effacement of the foot processes in MN (Fig. 3).
Fig. 3.
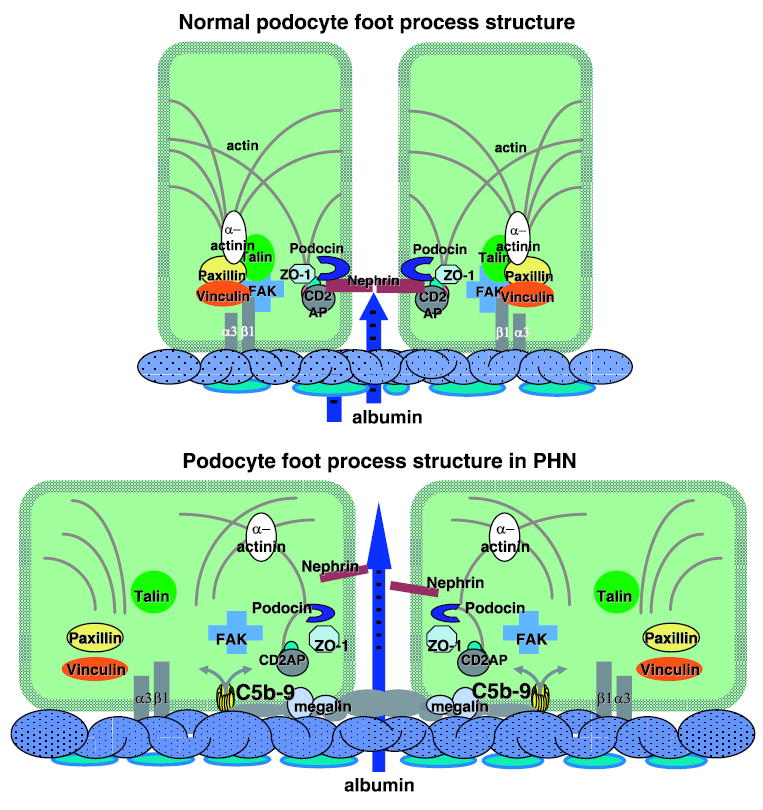
Normal podocyte foot process structure and alterations induced by antibody and complement C5b-9 in passive Heymann nephritis (PHN). CD2AP, CD2-associated protein; FAK, focal adhesion kinase; α3β1-integrin.
Alterations in the Slit-Diaphragm Complex
An impressive body of work over the past decade has produced substantial insight into the structural biology of the podocyte and its role in regulating glomerular permeability. This includes the identification of genes responsible for congenital forms of nephrotic syndrome in humans and mice, analysis of pathogenic mechanisms of podocyte injury in experimental models, and cell biological studies. This progress has been extensively reviewed in several recent articles (4, 74, 80, 140). These studies have established that 1) the podocyte is the primary target in most, if not all forms of inherited and acquired glomerular diseases in which the initial and major functional abnormality is proteinuria; 2) the slit-diaphragm that bridges the filtration slits between adjacent foot processes is the final filtration barrier; 3) the slit-diaphragm is a multicomponent structure including heterophilic and homophilic interactions between nephrin, and neph 1, and possibly cadherins; 4) the slit-diaphragm is linked to the cytoskeleton and anchored in a cholesterol-rich lipid-raft domain of the podocyte plasma membrane by a complex of proteins that includes podocin, CD2AP, and ZO-1; and 5) the slit-diaphragm complex has signaling properties mediated in part by Src family kinases,
Morphological alterations in the filtration slits have been well described in human and experimental MN (31, 122). In addition, several investigators have examined the integrity of the slit-diaphragm in acquired proteinuric diseases using nephrin as a marker. Thus it was reported that nephrin mRNA levels are diminished and that immunostaining for nephrin is reduced in amount and altered in distribution in human MN (30, 42, 144). Moreover, it was documented that the glomeruli of rats with PHN exhibit a progressive decline in nephrin mRNA and protein content from 1 wk to 8 mo after disease induction and that this decline is preventable by angiotensin II blockade (3). Interestingly, recent observations of angiotensin II receptor-mediated calcium flux in podocytes and the presence of a local angiotensin system on podocytes exposed to mechanical stress raise the possibility that the antiproteinuric action of angiotensin II blockade and preservation of nephrin may be due to cellular rather than purely hemodynamic effects (32, 34, 95).
Because the above observations on nephrin in MN were made after the onset of proteinuria, it is impossible to determine whether they were the cause of proteinuria or the result of podocyte injury. To examine this question, studies were undertaken at the onset of proteinuria in rats with PHN (147). These studies showed disruption or dislocation of slit-diaphragms and formation of occluding-type junctions before or at the onset of proteinuria. These changes were accompanied by a reduction in the amount and alteration in the distribution of nephrin on immunofluorescence and immunoelectron microscopy. In addition, sequential extraction of isolated glomeruli and Western blot analysis from individual rats at the onset of proteinuria on day 4 and 3 days later when proteinuria was well established showed a progressive decline in total nephrin, as well as a reduction in the fraction of nephrin that is bound to actin. In contrast, the amount and actin association of CD2AP and ZO-1 were not affected. In addition, complement depletion preserved slit-diaphragm morphology and the distribution of nephrin as well as inhibiting the development of proteinuria (120). Whereas complement-replete animals showed a significant reduction in the total amount of nephrin and the fraction of actin-associated nephrin, the amounts in the complement-depleted rats were similar to normal controls. Based on these findings, it was concluded that the onset of proteinuria in experimental MN is due to complement-dependent dissociation of nephrin from the actin cytoskeleton and loss of slit-diaphragm integrity. Considering that podocin is also essential for slit-diaphragm integrity, associates with the cytoplasmic tail of nephrin, and likely stabilizes nephrin and the slit-diaphragm in a lipid raft domain in the podocyte plasma membrane (126), a study was undertaken to examine the distribution and amount of podocin at the onset of proteinuria in PHN (88). Using a combination of dual-labeling immunofluorescence and Western blotting of sequentially extracted glomeruli, it was found that the locations of both nephrin and podocin are altered before the onset of proteinuria and that the two molecules dissociate from each other. Interestingly, although the total amounts of both proteins were reduced in PHN rats, the detergent-insoluble fraction of nephrin (lipid raft and/or actin associated) was reduced but not that of podocin. Moreover, a fraction of nephrin (but not podocin) could be recovered from the urine of PHN rats. These findings suggest that as nephrin dissociates from podocin and actin, it loses its tethers to the cytoskeleton and plasma membrane (Fig. 3), which allows dislocation of the slit-diaphragm and shedding into the urine. The appearance of nephrin in the urine, as well as studies that showed shedding of nephrin from complement injured cells in vitro (30), may explain the consistent finding that total nephrin is reduced in experimental and human MN. On the other hand, shedding of podocytes might also account for the appearance of nephrin in the urine of rats with PHN (102). In addition, these findings do not exclude the possibility that there is also reduced production or accelerated degradation of nephrin. In fact, reduced steady-state mRNA levels have been demonstrated as well (3, 42).
INFLUENCE OF COMPLEMENT REGULATORY PROTEINS IN EXPERIMENTAL MN
The alternative pathway of complement is spontaneously active, and there is the potential for bystander injury when appropriate complement activation occurs through any of the pathways. Hence, complement regulators are placed throughout the complement activation cascades (71, 84, 141), and a number of these are relevant to glomerular diseases (89, 107). The human podocyte bears CR1 (57), decay-accelerating factor (DAF; CD55) (110), and CD59 (114). CR1 and DAF are regulators of complement activation that limit activation of C3 and C5, whereas CD59 is an inhibitor of C5b-9 formation. In the rodent, CR1 has a more limited distribution and Crry appears to supplant its complement regulatory role on the podocyte (108).
The proximal tubular brush-border antigens (Fx1A) used to create the HN models of MN contain both Crry and CD59 (41). Thus neutralizing antibodies to these complement regulators may be present in the heterologous antisera injected into rats to induce PHN (108), as well as in the antibody repertoire generated by rats when actively immunized to develop MN (121). Such antibodies can bind to and inhibit intrinsic podocyte complement regulators and lead to unrestricted complement activation through the alternative pathway, thereby amplifying the activation of complement induced by other cell-bound antibodies via the classical pathway (108). Thus in the case of active HN, the presence of antibodies that inhibit podocyte Crry is necessary for anti-megalin antibodies to result in sufficient complement activation to induce podocyte injury (121). However, in PHN, sufficient antibody reactive with megalin and other podocyte antigens (134) may be present to overwhelm this intrinsic complement regulation, and disease proceeds irrespective of whether podocytes have functional complement regulators (14). Interestingly, intravenous administration of immunoglobulin (IVIG) to rats with PHN ameliorated proteinuria and deposition of C3c and C5b-9 without altering systemic complement levels or glomerular immune deposits (91). These findings suggest a possible therapeutic mechanism for IVIG involving complement regulation in autoimmune diseases. The relevance of podocyte complement regulators is also illustrated in another model of antibody-mediated podocyte injury, in which a hierarchical protection occurs with DAF and CD59 (70).
OTHER EFFECTS OF COMPLEMENT IN EXPERIMENTAL MN
There are other less direct roles for the complement system in experimental MN. Complement activation is intimately involved in the metabolism of immune complexes by limiting their size and interaction with receptors responsible for their clearance (46, 106). By virtue of its physicochemical characteristics, actively administered cationized bovine serum albumin becomes incorporated into subepithelial immune complexes, leading to a model of MN in which disease expression is dependent on generation of C5b-9 on the podocyte (45). In this model, the passage of immune complexes from subendothelial to subepithelial locations is partly dependent on C3 activation (131). Similarly, in chronic serum sickness, classical pathway complement activation appears to be required for immune deposits to migrate to the subepithelial space (109). Abnormalities or absence of early complement proteins can lead to altered tolerance, resulting in the development of autoantibodies and, in some cases, production of a lupus-like disease (10, 143) and, occasionally, development of a MN-like disease (69). Finally, it appears that complement proteins present in the glomerular ultrafiltrate in proteinuric diseases such as MN can be activated via the alternative pathway and lead to C5b-9 generation on tubular epithelium (8, 90). As reviewed in detail elsewhere, it is conceivable that such tubular damage from complement activation is responsible for progressive interstitial inflammation and fibrosis (52).
IMPLICATIONS FOR HUMAN MN
Whereas the target antigen (megalin) responsible for in situ immune deposit formation in HN has been identified on the soles of podocyte foot processes in rats (35), megalin is not expressed on human podocytes (58, 73) and the pathogenesis of human MN remains unknown (12). Nonetheless, given the remarkable similarities between human and experimental MN, it is likely that circulating autoantibodies gain access to an antigen on the podocyte foot processes in human MN, although the identity of the human antigen(s) remains unknown in the majority of cases. Perhaps the most exciting observation related to the pathogenesis of MN is a recent report of a case of antenatal MN, an exceedingly uncommon condition (25). The mother of the child was found to be homozygous deficient in a neutral endopeptidase (NEP) and developed circulating alloantibodies at the time of a previous miscarriage. These crossed the placental barrier and caused the formation of subepithelial immune deposits in the fetus, and the clinical and pathological manifestations of MN were found after birth. Serum from the mother and antiserum to NEP colocalized on podocytes of normal kidney sections, and the mother’s serum formed membranous-type glomerular deposits of IgG when injected into rabbits. In follow-up studies, three individuals from two other families were identified with a similar pathogenesis (26). Interestingly, a NEP-deficient female from one of these families bore four children, none of whom developed MN; while she had measurable anti-NEP antibodies, these were IgG4 and not the complement-fixing IgG1 subclass, which the authors suggested could explain their lack of pathogenicity (26). Whereas these reports support the concept of in situ immune deposit formation by circulating antibodies to a podocyte antigen in human MN, it is unlikely that NEP is the target antigen in the majority of cases of idiopathic MN. Discovering the human MN antigen is of much more than academic interest. It will aid the development of a diagnostic immunoassay and allow speciation of the autoantibodies. This will allow rational therapy with anti-B cell therapy in those cases with high titers of circulating antibodies (115) and targeted application of complement inhibitors and regulators (55, 113, 133, 146) in those cases with antibodies belonging to a complement-fixing subclass (IgG1 or IgG3).
One might question the role of complement activation in human MN because the non-complement-fixing IgG4 isotype predominates in the immune deposits (29, 53), and the C1qr2s2 complex is quite large (Mr ~ 800 kDa), which undoubtedly impedes its access to the subepithelial space. Still, there is a good deal of circumstantial evidence that complement is activated in subepithelial immune deposits, including the presence of C3 and C5b-9 in many cases (48, 99) and C3d in all cases of human MN (28); C3d is the stable C3 product generated from iC3b by factor I, presumably relying on podocyte CR1 as a cofactor. Despite these apparent contradictions, it does seem likely that in human MN complement activation is initiated through the classical pathway with possible amplification by the alternative pathway, a scenario that may be more common than previously appreciated (51). One possible explanation for how classical pathway activation is initiated is that IgG antibodies, with and without in vitro C1q binding properties, are able to cooperate and synergize in activating both the classical and alternative pathways to produce a lytic response greater than that expected from the C1q binding antibodies alone (5). This is particularly true when the different antibodies identify different antigen epitopes (119) as is likely to be the case in a polyclonal disorder like human MN. Alternatively, the non-complement-fixing antibodies may render the target cells more susceptible to subthreshold amounts of complement-fixing antibodies (112).
As noted above, the complement regulators DAF, CR1, and CD59 are present on human podocytes. Although these have not been described as the target of antibodies, it is interesting that CR1 is reduced on podocytes in human MN, as is also the case in lupus nephritis (82). Such acquired CR1 deficiency could increase the susceptibility of podocytes to complement activation by antibodies in subepithelial immune deposits (82). As in experimental MN, C5b-9 is assembled on the podocytes of patients with MN, which is evident histologically (26, 99) and by examining urine for C5b-9 (65, 125). Complicating the utility of urinary C5b-9 measurement is alternative complement pathway activation on proximal tubular cells noted previously, which also can result in the appearance of C5b-9 in the urine of proteinuric patients, including those with MN (85, 96). Thus the utility of C5b-9 in stratifying patients with MN is unclear (11). Based on the clear pathogenesis of experimental MN in which C5 activation leads to C5b-9 generation on the podocyte, a multicenter, double-blind controlled study of an inhibitory humanized anti-C5 monoclonal antibody (eculizumab) was performed in 130 patients with idiopathic MN. While there was no effect on the primary end point of proteinuria after 16 wk of therapy, patients given up to 1 yr of this agent in an open-label extension had impressive responses, which have led to optimism with this approach, although its fate is uncertain (Quigg RJ, unpublished observations). Better characterization of the circulating autoantibodies once the antigen is identified may allow selection of patients most likely to benefit from such therapy.
MN derives its name from the progressive GBM expansion that surrounds and encases the subepithelial immune deposits. This is due to increased deposition of the GBM type IV collagen isoforms α3(IV), α4(IV), and α5(IV), as well as laminin and nidogen (64), most likely derived from increased synthesis by the injured podocytes as in experimental MN (39, 81). Subsequent accumulation of the α1(IV) and α2(IV) type IV collagen isoforms in the subendothelial GBM appears to encroach on, and narrow, the capillary lumen (64). These changes in the GBM and findings in experimental MN may help explain the observation that only a fraction of patients with MN respond to anti-B cell therapy with rituximab (anti-CD20) or eculizumab (anti-C5). Thus proteinuric kidneys transplanted from rats with active HN into normal syngeneic rats had persistent proteinuria, albeit at a lower level, despite resolution of glomerular complement and the absence of new antibody deposition (75). This is likely due to the extensive remodeling and disorganization of the GBM (40, 81), which prevent the reconstitution of the normal cell-matrix interface and cause persistent abnormalities in podocyte architecture. As a corollary, it might be expected that therapies targeting B cells and complement activation in MN are most likely to be effective when there are high titers of autoantibody, active immune deposition, and complement activation (105, 124, 125) before extensive GBM remodeling has occurred.
Identification of the cellular signaling pathways and their products may also point the way for therapeutic intervention. For example, if one were able to achieve the same level of membrane phospholipid substitution with omega-3 fatty acids as achieved in rats (145), a substantial reduction in thromboxane A2 and proteinuria might be seen. Other promising therapeutic avenues arising from the signaling work described above include cytoprotection against complement attack by inducing ER stress proteins, inhibition of intracellular oxidant injury, and selective activation of p38 MAP kinase or HSP27. A better understanding of the cellular mechanisms of cytoskeleton and slit-diaphragm disassembly and the advent of small molecule kinase inhibitors and activators provides hope of reversing the damage to podocytes after immune injury has already occurred. In the final analysis, effective therapy will likely involve a combination of approaches, specifically targeting autoantibody-producing B cells, inhibiting complement activation, and providing cytoprotection by angiotensin blockade and the intracellular modalities noted above.
Acknowledgments
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-30932 (D. J. Salant), DK-067658 (D. J. Salant ), and DK-41873 (R. J. Quigg), the Canadian Institutes of Health Research (A. V. Cybulsky), the Kidney Foundation of Canada (A. V. Cybulsky), and Fonds de la Recherche en Santé du Québec (A. V. Cybulsky).
References
- 1.Aoudjit L, Stanciu M, Li H, Lemay S, Takano T. p38 Mitogen-activated protein kinase protects glomerular epithelial cells from complement-mediated cell injury. Am J Physiol Renal Physiol. 2003;285:F765–F774. doi: 10.1152/ajprenal.00100.2003. [DOI] [PubMed] [Google Scholar]
- 2.Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol. 1989;135:185–194. [PMC free article] [PubMed] [Google Scholar]
- 3.Benigni A, Tomasoni S, Gagliardini E, Zoja C, Grunkemeyer JA, Kalluri R, Remuzzi G. Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol. 2001;12:941–948. doi: 10.1681/ASN.V125941. [DOI] [PubMed] [Google Scholar]
- 4.Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol. 2004;15:1382–1391. doi: 10.1097/01.asn.0000130167.30769.55. [DOI] [PubMed] [Google Scholar]
- 5.Bindon CI, Hale G, Waldmann H. Importance of antigen specificity for complement-mediated lysis by monoclonal antibodies. Eur J Immunol. 1988;18:1507–1514. doi: 10.1002/eji.1830181006. [DOI] [PubMed] [Google Scholar]
- 6.Blume C, Heise G, Muhlfeld A, Bach D, Schror K, Gerhardz CD, Grabensee B, Heering P. Effect of flosulide, a selective cyclooxygenase 2 inhibitor, on passive Heymann nephritis in the rat. Kidney Int. 1999;56:1770–1778. doi: 10.1046/j.1523-1755.1999.00742.x. [DOI] [PubMed] [Google Scholar]
- 7.Camussi G, Brentjens JR, Noble B, Kerjaschki D, Malavasi F, Roholt OA, Farquhar MG, Andres G. Antibody-induced redistribution of Heymann nephritis on the surface of cultured glomerular visceral epithelial cells: possible role in the pathogenesis of Heymann glomerulonephritis. J Immunol. 1985;135:2409–2416. [PubMed] [Google Scholar]
- 8.Camussi G, Rotunno M, Segoloni G, Brentjens JR, Andres GA. In vitro alternative pathway activation of complement by the brush border of proximal tubules of normal rat kidney. J Immunol. 1982;128:1659–1663. [PubMed] [Google Scholar]
- 9.Carpenter G. Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J Cell Biol. 1999;146:697–702. doi: 10.1083/jcb.146.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 11.Cattran DC, Wald R, Brenchley PE, Coupes B. Clinical correlates of serial urinary membrane attack complex estimates in patients with idiopathic membranous nephropathy. Clin Nephrol. 2003;60:7–12. [PubMed] [Google Scholar]
- 12.Couser WG and Alpers CE. Membranous nephropathy. In: Immunologic Renal Diseases (2nd ed.), edited by Neilson EG and Couser WG. Philadelphia, PA: Lippincott Williams and Wilkins, 2001, p. 1029–1055.
- 13.Couser WG, Johnson RJ, Young BA, Yeh CG, Toth CA, Rudolph AR. The effects of soluble recombinant complement receptor 1 on complement-mediated experimental glomerulonephritis. J Am Soc Nephrol. 1995;5:1888–1894. doi: 10.1681/ASN.V5111888. [DOI] [PubMed] [Google Scholar]
- 14.Cunningham PN, Hack BK, Ren G, Minto AW, Morgan BP, Quigg RJ. Glomerular complement regulation is overwhelmed in passive Heymann nephritis. Kidney Int. 2001;60:900–909. doi: 10.1046/j.1523-1755.2001.060003900.x. [DOI] [PubMed] [Google Scholar]
- 15.Cybulsky AV, Lieberthal W, Quigg RJ, Rennke HG, Salant DJ. A role for thromboxane in complement-mediated glomerular injury. Am J Pathol. 1987;128:45–51. [PMC free article] [PubMed] [Google Scholar]
- 16.Cybulsky AV, Monge JC, Papillon J, McTavish AJ. Complement C5b-9 activates cytosolic phospholipase A2 in glomerular epithelial cells. Am J Physiol Renal Fluid Electrolyte Physiol. 1995;269:F739–F749. doi: 10.1152/ajprenal.1995.269.5.F739. [DOI] [PubMed] [Google Scholar]
- 17.Cybulsky AV, Papillon J, McTavish AJ. Complement activates phospholipases and protein kinases in glomerular epithelial cells. Kidney Int. 1998;54:360–372. doi: 10.1046/j.1523-1755.1998.00013.x. [DOI] [PubMed] [Google Scholar]
- 18.Cybulsky AV, Quigg RJ, Salant DJ. The membrane attack complex in complement-mediated glomerular epithelial cell injury: formation and stability of C5b-9 and C5b-7 in rat membranous nephropathy. J Immunol. 1986;137:1511–1516. [PubMed] [Google Scholar]
- 19.Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement-induced glomerular epithelial cell injury: the role of the membrane attack complex in rat membranous nephropathy. J Clin Invest. 1986;77:1096–1107. doi: 10.1172/JCI112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cybulsky AV, Salant DJ, Quigg RJ, Badalamenti J, Bonventre JV. Complement C5b-9 complex activates phospholipases in glomerular epithelial cells. Am J Physiol Renal Fluid Electrolyte Physiol. 1989;257:F826–F836. doi: 10.1152/ajprenal.1989.257.5.F826. [DOI] [PubMed] [Google Scholar]
- 21.Cybulsky AV, Takano T, Papillon J, Khadir A, Bijian K, Le Berre L. The actin cytoskeleton facilitates complement-mediated activation of cytosolic phospholipase A2. Am J Physiol Renal Physiol. 2004;286:F466–F476. doi: 10.1152/ajprenal.00260.2003. [DOI] [PubMed] [Google Scholar]
- 22.Cybulsky AV, Takano T, Papillon J, Khadir A, Liu J, Peng H. Complement C5b-9 membrane attack complex increases expression of endoplasmic reticulum stress proteins in glomerular epithelial cells. J Biol Chem. 2002;277:41342–41351. doi: 10.1074/jbc.M204694200. [DOI] [PubMed] [Google Scholar]
- 23.Cybulsky AV, Takano T, Papillon J, McTavish AJ. Complement C5b-9 induces receptor tyrosine kinase transactivation in glomerular epithelial cells. Am J Pathol. 1999;155:1701–1711. doi: 10.1016/S0002-9440(10)65485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cybulsky AV, Takano T, Papillon J, McTavish AJ. Complement-induced phospholipase A2 activation in experimental membranous nephropathy. Kidney Int. 2000;57:1052–1062. doi: 10.1046/j.1523-1755.2000.00932.x. [DOI] [PubMed] [Google Scholar]
- 25.Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, Deschenes G, Ronco PM. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346:2053–2060. doi: 10.1056/NEJMoa012895. [DOI] [PubMed] [Google Scholar]
- 26.Debiec H, Nauta J, Coulet F, van der Burg M, Guigonis V, Schurmans T, de Heer E, Soubrier F, Janssen F, Ronco P. Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet. 2004;364:1252–1259. doi: 10.1016/S0140-6736(04)17142-0. [DOI] [PubMed] [Google Scholar]
- 27.Dessen A. Structure and mechanism of human cytosolic phospholipase A2. Biochim Biophys Acta. 2000;1488:40–47. doi: 10.1016/s1388-1981(00)00108-6. [DOI] [PubMed] [Google Scholar]
- 28.Doi T, Kanatsu K, Nagai H, Suehiro F, Kuwahara T, Hamashima Y. Demonstration of C3d deposits in membranous nephropathy. Nephron. 1984;37:232–235. doi: 10.1159/000183255. [DOI] [PubMed] [Google Scholar]
- 29.Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y. Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol. 1984;58:57–62. [PMC free article] [PubMed] [Google Scholar]
- 30.Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, Reponen P, Tryggvason K, Camussi G. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol. 2001;158:1723–1731. doi: 10.1016/S0002-9440(10)64128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drumond MC, Kristal B, Myers BD, Deen WM. Structural basis for reduced glomerular filtration capacity in nephrotic humans. J Clin Invest. 1994;94:1187–1195. doi: 10.1172/JCI117435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 33.Eitner F, Ostendorf T, Van Roeyen C, Kitahara M, Li X, Aase K, Grone HJ, Eriksson U, Floege J. Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J Am Soc Nephrol. 2002;13:910–917. doi: 10.1681/ASN.V134910. [DOI] [PubMed] [Google Scholar]
- 34.Endlich N, Kress KR, Reiser J, Uttenweiler D, Kriz W, Mundel P, Endlich K. Podocytes respond to mechanical stress in vitro. J Am Soc Nephrol. 2001;12:413–422. doi: 10.1681/ASN.V123413. [DOI] [PubMed] [Google Scholar]
- 35.Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. 1995;6:35–47. doi: 10.1681/ASN.V6135. [DOI] [PubMed] [Google Scholar]
- 36.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 37.Floege J, Alpers CE, Sage EH, Pritzl P, Gordon K, Johnson RJ, Couser WG. Markers of complement-dependent and complement-independent glomerular visceral epithelial cell injury in vivo. Expression of antiadhesive proteins and cytoskeletal changes. Lab Invest. 1992;67:486–497. [PubMed] [Google Scholar]
- 38.Floege J, Johnson RJ, Alpers CE, Fatemi-Nainie S, Richardson CA, Gordon K, Couser WG. Visceral glomerular epithelial cells can proliferate in vivo and synthesize platelet-derived growth factor B-chain. Am J Pathol. 1993;142:637–650. [PMC free article] [PubMed] [Google Scholar]
- 39.Floege J, Johnson RJ, Gordon K, Yoshimura A, Campbell C, Iruela-Arispe L, Alpers CE, Couser WG. Altered glomerular extracellular matrix synthesis in experimental membranous nephropathy. Kidney Int. 1992;42:573–585. doi: 10.1038/ki.1992.321. [DOI] [PubMed] [Google Scholar]
- 40.Fogel MA, Boyd CD, Leardkamolkarn V, Abrahamson DR, Minto AW, Salant DJ. Glomerular basement membrane expansion in passive Heymann nephritis. Absence of increased synthesis of type IV collagen, laminin, or fibronectin. Am J Pathol. 1991;138:465–475. [PMC free article] [PubMed] [Google Scholar]
- 41.Funabashi K, Okada N, Matsuo S, Yamamoto T, Morgan BP, Okada H. Tissue distribution of complement regulatory membrane proteins in rats. Immunology. 1994;81:444–451. [PMC free article] [PubMed] [Google Scholar]
- 42.Furness PN, Hall LL, Shaw JA, Pringle JH. Glomerular expression of nephrin is decreased in acquired human nephrotic syndrome. Nephrol Dial Transplant. 1999;14:1234–1237. doi: 10.1093/ndt/14.5.1234. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein DJ, Wheeler DC, Salant DJ. Effects of omega-3 fatty acids on complement-mediated glomerular epithelial cell injury. Kidney Int. 1996;50:1863–1871. doi: 10.1038/ki.1996.507. [DOI] [PubMed] [Google Scholar]
- 44.Greiber S, Munzel T, Kastner S, Muller B, Schollmeyer P, Pavenstadt H. NAD(P)H oxidase activity in cultured human podocytes: effects of adenosine triphosphate. Kidney Int. 1998;53:654–663. doi: 10.1046/j.1523-1755.1998.00796.x. [DOI] [PubMed] [Google Scholar]
- 45.Groggel GC, Adler S, Rennke HG, Couser WG, Salant DJ. Role of the terminal complement pathway in experimental membranous nephropathy in the rabbit. J Clin Invest. 1983;72:1948–1957. doi: 10.1172/JCI111159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hebert LA. The clearance of immune complexes from the circulation of man and other primates. Am J Kidney Dis. 1991;17:352–361. doi: 10.1016/s0272-6386(12)80488-4. [DOI] [PubMed] [Google Scholar]
- 47.Heymann W, Kmetec EP, Wilson SG, Hunter JL, Hackel DB, Okuda R, Cuppage F. Experimental autoimmune renal disease in rats. Ann NY Acad Sci. 1965;124:310–322. doi: 10.1111/j.1749-6632.1965.tb18966.x. [DOI] [PubMed] [Google Scholar]
- 48.Hinglais N, Kazatchkine MD, Bhakdi S, Appay MD, Mandet C, Grossetete J, Bariety J. Immunohistochemical study of the C5b-9 complex of complement in human kidneys. Kidney Int. 1986;30:399–410. doi: 10.1038/ki.1986.198. [DOI] [PubMed] [Google Scholar]
- 49.Hirabayashi T, Shimizu T. Localization and regulation of cytosolic phospholipase A2. Biochim Biophys Acta. 2000;1488:124–138. doi: 10.1016/s1388-1981(00)00115-3. [DOI] [PubMed] [Google Scholar]
- 50.Hiromura K, Haseley LA, Zhang P, Monkawa T, Durvasula R, Petermann AT, Alpers CE, Mundel P, Shankland SJ. Podocyte expression of the CDK-inhibitor p57 during development and disease. Kidney Int. 2001;60:2235–2246. doi: 10.1046/j.1523-1755.2001.00057.x. [DOI] [PubMed] [Google Scholar]
- 51.Holers VM, Thurman JM. The alternative pathway of complement in disease: opportunities for therapeutic targeting. Mol Immunol. 2004;41:147–152. doi: 10.1016/j.molimm.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 52.Hsu SI, Couser WG. Chronic progression of tubulointerstitial damage in proteinuric renal disease is mediated by complement activation: a therapeutic role for complement inhibitors? J Am Soc Nephrol. 2003;14:S186–S191. doi: 10.1097/01.asn.0000070032.58017.20. [DOI] [PubMed] [Google Scholar]
- 53.Imai H, Hamai K, Komatsuda A, Ohtani H, Miura AB. IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy, and lupus nephritis. Kidney Int. 1997;51:270–276. doi: 10.1038/ki.1997.32. [DOI] [PubMed] [Google Scholar]
- 54.Kanellis J, Levidiotis V, Khong T, Cox AJ, Stacker SA, Gilbert RE, Cooper ME, Power DA. A study of VEGF and its receptors in two rat models of proteinuria. Nephron Physiol. 2004;96:P26–P36. doi: 10.1159/000075577. [DOI] [PubMed] [Google Scholar]
- 55.Kaplan M. Eculizumab (Alexion) Curr Opin Investig Drugs. 2002;3:1017–1023. [PubMed] [Google Scholar]
- 56.Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, Arnold SM. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3:411–421. doi: 10.1038/nrm829. [DOI] [PubMed] [Google Scholar]
- 57.Kazatchkine MD, Fearon DT, Appay MD, Mandet C, Bariety J. Immunohistochemical study of the human glomerular C3b receptor in normal kidney and in seventy-five cases of renal diseases: loss of C3b receptor antigen in focal hyalinosis and in proliferative nephritis of systemic lupus erythematosus. J Clin Invest. 1982;69:900–912. doi: 10.1172/JCI110529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerjaschki D. Pathogenetic concepts of membranous glomerulopathy (MGN) J Nephrol. 2000;13(Suppl 3):S96–S100. [PubMed] [Google Scholar]
- 59.Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci USA. 1982;79:5557–5581. doi: 10.1073/pnas.79.18.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerjaschki D, Miettinen A, Farquhar MG. Initial events in the formation of immune deposits in passive Heymann nephritis. J Exp Med. 1987;166:109–128. doi: 10.1084/jem.166.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kerjaschki D, Ojha PP, Susani M, Horvat R, Binder S, Hovorka A, Hillemanns P, Pytela R. A β1-integrin receptor for fibronectin in human kidney glomeruli. Am J Pathol. 1989;134:481–489. [PMC free article] [PubMed] [Google Scholar]
- 62.Kerjaschki D, Schulze M, Binder S, Kain R, Ojha PP, Susani M, Horvat R, Baker PJ, Couser WG. Transcellular transport and membrane insertion of the C5b-9 membrane attack complex of complement by glomerular epithelial cells in experimental membranous nephropathy. J Immunol. 1989;143:546–552. [PubMed] [Google Scholar]
- 63.Kerjaschki D, Ullrich R, Exner M, Orlando RA, Farquhar MG. Induction of passive Heymann nephritis with antibodies specific for a synthetic peptide derived from the receptor-associated protein. J Exp Med. 1996;183:2007–2015. doi: 10.1084/jem.183.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim Y, Butkowski R, Burke B, Kleppel MM, Crosson J, Katz A, Michael AF. Differential expression of basement membrane collagen in membranous nephropathy. Am J Pathol. 1992;139:1381–1388. [PMC free article] [PubMed] [Google Scholar]
- 65.Kon SP, Coupes B, Short CD, Solomon LR, Raftery MJ, Mallick NP, Brenchley PE. Urinary C5b-9 excretion and clinical course in idiopathic human membranous nephropathy. Kidney Int. 1995;48:1953–1958. doi: 10.1038/ki.1995.496. [DOI] [PubMed] [Google Scholar]
- 66.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 67.Lee SB, Rhee SG. Significance of PIP2 hydrolysis and regulation of phospholipase C isozymes. Curr Opin Cell Biol. 1995;7:183–189. doi: 10.1016/0955-0674(95)80026-3. [DOI] [PubMed] [Google Scholar]
- 68.Levidiotis V, Freeman C, Tikellis C, Cooper ME, Power DA. Heparanase is involved in the pathogenesis of proteinuria as a result of glomerulonephritis. J Am Soc Nephrol. 2004;15:68–78. doi: 10.1097/01.asn.0000103229.25389.40. [DOI] [PubMed] [Google Scholar]
- 69.Lhotta K, Wurzner R, Rumpelt HJ, Eder P, Mayer G. Membranous nephropathy in a patient with hereditary complete complement C4 deficiency. Nephrol Dial Transplant. 2004;19:990–993. doi: 10.1093/ndt/gfh008. [DOI] [PubMed] [Google Scholar]
- 70.Lin F, Salant DJ, Meyerson H, Emancipator S, Morgan BP, Medof ME. Respective roles of decay-accelerating factor and CD59 in circumventing glomerular injury in acute nephrotoxic serum nephritis. J Immunol. 2004;172:2636–2642. doi: 10.4049/jimmunol.172.4.2636. [DOI] [PubMed] [Google Scholar]
- 71.Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. Control of the complement system. Adv Immunol. 1996;61:201–283. doi: 10.1016/s0065-2776(08)60868-8. [DOI] [PubMed] [Google Scholar]
- 72.Liu J, Takano T, Papillon J, Khadir A, Cybulsky AV. Cytosolic phospholipase A2α associates with plasma membrane, endoplasmic reticulum and nuclear membrane in glomerular epithelial cells. Biochem J. 2001;353:79–90. [PMC free article] [PubMed] [Google Scholar]
- 73.Lundgren S, Carling T, Hjalm G, Juhlin C, Rastad J, Pihlgren U, Rask L, Akerstrom G, Hellman P. Tissue distribution of human gp330/megalin, a putative Ca2+-sensing protein. J Histochem Cytochem. 1997;45:383–392. doi: 10.1177/002215549704500306. [DOI] [PubMed] [Google Scholar]
- 74.Ly J, Alexander M, Quaggin SE. A podocentric view of nephrology. Curr Opin Nephrol Hypertens. 2004;13:299–305. doi: 10.1097/00041552-200405000-00006. [DOI] [PubMed] [Google Scholar]
- 75.Makker SP, Kanalas JJ. Course of transplanted Heymann nephritis kidney in normal host. Implications for mechanism of proteinuria in membranous glomerulonephropathy. J Immunol. 1989;142:3406–3410. [PubMed] [Google Scholar]
- 76.Makker SP, Singh AK. Characterization of the antigen (gp600) of Heymann nephritis. Lab Invest. 1984;50:287–293. [PubMed] [Google Scholar]
- 77.McCarthy ET, Sharma M. Indomethacin protects permeability barrier from focal segmental glomerulosclerosis serum. Kidney Int. 2002;61:534–541. doi: 10.1046/j.1523-1755.2002.00172.x. [DOI] [PubMed] [Google Scholar]
- 78.McMillan JI, Riordan JW, Couser WG, Pollock AS, Lovett DH. Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J Clin Invest. 1996;97:1094–1101. doi: 10.1172/JCI118502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mercurio F, Manning AM. Multiple signals converging on NF-κB. Curr Opin Cell Biol. 1999;11:226–232. doi: 10.1016/s0955-0674(99)80030-1. [DOI] [PubMed] [Google Scholar]
- 80.Miner JH. Focusing on the glomerular slit diaphragm: podocin enters the picture. Am J Pathol. 2002;160:3–5. doi: 10.1016/S0002-9440(10)64341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Minto AW, Kalluri R, Togawa M, Bergijk EC, Killen PD, Salant DJ. Augmented expression of glomerular basement membrane specific type IV collagen isoforms (α3-α5) in experimental membranous nephropathy. Proc Assoc Am Physicians. 1998;110:207–217. [PubMed] [Google Scholar]
- 82.Moll S, Miot S, Sadallah S, Gudat F, Mihatsch MJ, Schifferli JA. No complement receptor 1 stumps on podocytes in human glomerulopathies. Kidney Int. 2001;59:160–168. doi: 10.1046/j.1523-1755.2001.00476.x. [DOI] [PubMed] [Google Scholar]
- 83.Morgan BP. Effects of the membrane attack complex of complement on nucleated cells. Curr Top Microbiol Immunol. 1992;178:115–140. doi: 10.1007/978-3-642-77014-2_8. [DOI] [PubMed] [Google Scholar]
- 84.Morgan BP and Harris CL. Regulation in the activation pathways. In: Complement Regulatory Proteins San Diego, CA: Academic, 1999, p. 41–136.
- 85.Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol. 2000;11:700–707. doi: 10.1681/ASN.V114700. [DOI] [PubMed] [Google Scholar]
- 86.Mudge SJ, Paizis K, Auwardt RB, Thomas RJ, Power DA. Activation of nuclear factor-κB by podocytes in the autologous phase of passive Heymann nephritis. Kidney Int. 2001;59:923–931. doi: 10.1046/j.1523-1755.2001.059003923.x. [DOI] [PubMed] [Google Scholar]
- 87.Nagao T, Nagamatsu T, Suzuki Y. Effect of DP-1904, a thromboxane A2 synthase inhibitor, on passive Heymann nephritis in rats. Eur J Pharmacol. 1996;316:73–80. doi: 10.1016/s0014-2999(96)00662-0. [DOI] [PubMed] [Google Scholar]
- 88.Nakatsue T, Koike H, Han GD, Suzuki K, Yuan H, Salant DJ, Gejyo F, Shimizu F, Kawachi H. Nephrin and podocin dissociate at the onset of proteinuria in experimental membranous nephropathy. Kidney Int. 2005;67:2239–2253. doi: 10.1111/j.1523-1755.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- 89.Nangaku M. Complement regulatory proteins in glomerular diseases. Kidney Int. 1998;54:1419–1428. doi: 10.1046/j.1523-1755.1998.00130.x. [DOI] [PubMed] [Google Scholar]
- 90.Nangaku M, Pippin J, Couser WG. Complement membrane attack complex (C5b-9) mediates interstitial disease in experimental nephrotic syndrome. J Am Soc Nephrol. 1999;10:2323–2331. doi: 10.1681/ASN.V10112323. [DOI] [PubMed] [Google Scholar]
- 91.Nangaku M, Pippin J, Richardson CA, Schulze M, Young BA, Alpers CE, Gordon KL, Johnson RJ, Couser WG. Beneficial effects of systemic immunoglobulin in experimental membranous nephropathy. Kidney Int. 1996;50:2054–2062. doi: 10.1038/ki.1996.529. [DOI] [PubMed] [Google Scholar]
- 92.Neale TJ, Ojha PP, Exner M, Poczewski H, Ruger B, Witztum JL, Davis P, Kerjaschki D. Proteinuria in passive Heymann nephritis is associated with lipid peroxidation and formation of adducts on type IV collagen. J Clin Invest. 1994;94:1577–1584. doi: 10.1172/JCI117499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neale TJ, Ullrich R, Ojha P, Poczewski H, Verhoeven AJ, Kerjaschki D. Reactive oxygen species and neutrophil respiratory burst cytochrome b558 are produced by kidney glomerular cells in passive Heymann nephritis. Proc Natl Acad Sci USA. 1993;90:3645–3649. doi: 10.1073/pnas.90.8.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicholson-Weller A, Halperin JA. Membrane signaling by complement C5b-9, the membrane attack complex. Immunol Res. 1993;12:244–257. doi: 10.1007/BF02918256. [DOI] [PubMed] [Google Scholar]
- 95.Nitschke R, Henger A, Ricken S, Gloy J, Muller V, Greger R, Pavenstadt H. Angiotensin II increases the intracellular calcium activity in podocytes of the intact glomerulus. Kidney Int. 2000;57:41–49. doi: 10.1046/j.1523-1755.2000.00810.x. [DOI] [PubMed] [Google Scholar]
- 96.Ogrodowski JL, Hebert LA, Sedmak D, Cosio FG, Tamerius J, Kolb W. Measurement of SC5b-9 in urine in patients with the nephrotic syndrome. Kidney Int. 1991;40:1141–1147. doi: 10.1038/ki.1991.326. [DOI] [PubMed] [Google Scholar]
- 97.Paizis K, Kirkland G, Polihronis M, Katerelos M, Kanellis J, Power DA. Heparin-binding epidermal growth factor-like growth factor in experimental models of membranous and minimal change nephropathy. Kidney Int. 1998;53:1162–1171. doi: 10.1046/j.1523-1755.1998.00846.x. [DOI] [PubMed] [Google Scholar]
- 98.Panesar M, Papillon J, McTavish AJ, Cybulsky AV. Activation of phospholipase A2 by complement C5b-9 in glomerular epithelial cells. J Immunol. 1997;159:3584–3594. [PubMed] [Google Scholar]
- 99.Papagianni AA, Alexopoulos E, Leontsini M, Papadimitriou M. C5b-9 and adhesion molecules in human idiopathic membranous nephropathy. Nephrol Dial Transplant. 2002;17:57–63. doi: 10.1093/ndt/17.1.57. [DOI] [PubMed] [Google Scholar]
- 100.Peng H, Takano T, Papillon J, Bijian K, Khadir A, Cybulsky AV. Complement activates the c-Jun N-terminal kinase/stress-activated protein kinase in glomerular epithelial cells. J Immunol. 2002;169:2594–2601. doi: 10.4049/jimmunol.169.5.2594. [DOI] [PubMed] [Google Scholar]
- 101.Petermann A, Hiromura K, Pippin J, Blonski M, Couser WG, Kopp J, Mundel P, Shankland SJ. Differential expression of d-type cyclins in podocytes in vitro and in vivo. Am J Pathol. 2004;164:1417–1424. doi: 10.1016/S0002-9440(10)63228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Petermann AT, Krofft R, Blonski M, Hiromura K, Vaughn M, Pichler R, Griffin S, Wada T, Pippin J, Durvasula R, Shankland SJ. Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 2003;64:1222–1231. doi: 10.1046/j.1523-1755.2003.00217.x. [DOI] [PubMed] [Google Scholar]
- 103.Petermann AT, Pippin J, Hiromura K, Monkawa T, Durvasula R, Couser WG, Kopp J, Shankland SJ. Mitotic cell cycle proteins increase in podocytes despite lack of proliferation. Kidney Int. 2003;63:113–122. doi: 10.1046/j.1523-1755.2003.00723.x. [DOI] [PubMed] [Google Scholar]
- 104.Pippin JW, Durvasula R, Petermann A, Hiromura K, Couser WG, Shankland SJ. DNA damage is a novel response to sublytic complement C5b-9-induced injury in podocytes. J Clin Invest. 2003;111:877–885. doi: 10.1172/JCI15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pruchno CJ, Burns MW, Schulze M, Johnson RJ, Baker PJ, Couser WG. Urinary excretion of C5b-9 reflects disease activity in passive Heymann nephritis. Kidney Int. 1989;36:65–71. doi: 10.1038/ki.1989.162. [DOI] [PubMed] [Google Scholar]
- 106.Quigg RJ. Complement and the kidney. J Immunol. 2003;171:3319–3324. doi: 10.4049/jimmunol.171.7.3319. [DOI] [PubMed] [Google Scholar]
- 107.Quigg RJ. Role of complement and complement regulatory proteins in glomerulonephritis. Springer Semin Immunopathol. 2003;24:395–410. doi: 10.1007/s00281-002-0116-9. [DOI] [PubMed] [Google Scholar]
- 108.Quigg RJ, Holers VM, Morgan BP, Sneed AE., 3rd Crry and CD59 regulate complement in rat glomerular epithelial cells and are inhibited by the nephritogenic antibody of passive Heymann nephritis. J Immunol. 1995;154:3437–3443. [PubMed] [Google Scholar]
- 109.Quigg RJ, Lim A, Haas M, Alexander JJ, He C, Carroll MC. Immune complex glomerulonephritis in C4- and C3-deficient mice. Kidney Int. 1998;53:320–330. doi: 10.1046/j.1523-1755.1998.00723.x. [DOI] [PubMed] [Google Scholar]
- 110.Quigg RJ, Nicholson-Weller A, Cybulsky AV, Badalamenti J, Salant DJ. Decay accelerating factor regulates complement activation on glomerular epithelial cells. J Immunol. 1989;142:877–882. [PubMed] [Google Scholar]
- 111.Raats CJ, Luca ME, Bakker MA, Van Der Wal A, Heeringa P, Van Goor H, Van Den Born J, De Heer E, Berden JH. Reduction in glomerular heparan sulfate correlates with complement deposition and albuminuria in active Heymann nephritis. J Am Soc Nephrol. 1999;10:1689–1699. doi: 10.1681/ASN.V1081689. [DOI] [PubMed] [Google Scholar]
- 112.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004;4:326–334. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 113.Rioux P. TP-10 (AVANT Immunotherapeutics) Curr Opin Invest Drugs. 2001;2:364–371. [PubMed] [Google Scholar]
- 114.Rooney IA, Davies A, Griffiths D, Williams JD, Davies M, Meri S, Lachmann PJ, Morgan BP. The complement-inhibiting protein, protectin (CD59 antigen), is present and functionally active on glomerular epithelial cells. Clin Exp Immunol. 1991;83:251–256. doi: 10.1111/j.1365-2249.1991.tb05623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ruggenenti P, Chiurchiu C, Brusegan V, Abbate M, Perna A, Filippi C, Remuzzi G. Rituximab in idiopathic membranous nephropathy: a one-year prospective study. J Am Soc Nephrol. 2003;14:1851–1857. doi: 10.1097/01.asn.0000071511.35221.b3. [DOI] [PubMed] [Google Scholar]
- 116.Rus HG, Niculescu FI, Shin ML. Role of the C5b-9 complement complex in cell cycle and apoptosis. Immunol Rev. 2001;180:49–55. doi: 10.1034/j.1600-065x.2001.1800104.x. [DOI] [PubMed] [Google Scholar]
- 117.Salant DJ, Natori Y, and Kawachi H. Glomerular injury due to antibody alone. In: Immunologic Renal Diseases (2nd ed.), edited by Neilson EG and Couser WG. Philadelphia, PA: Lippincott-Raven, 2001, p. 347–365.
- 118.Salant DJ, Quigg RJ, Cybulsky AV. Heymann nephritis: mechanisms of renal injury. Kidney Int. 1989;35:976–984. doi: 10.1038/ki.1989.81. [DOI] [PubMed] [Google Scholar]
- 119.Sandberg AL, Cisar JO, Siraganian RP, Mudrick LL, Armstrong MW. Complement activation by individual and combinations of monoclonal antibodies to Actinomyces viscosus T14V fimbriae: a probe for epitope distribution on these polymeric proteins. Mol Immunol. 1990;27:787–794. doi: 10.1016/0161-5890(90)90088-h. [DOI] [PubMed] [Google Scholar]
- 120.Saran AM, Yuan H, Takeuchi E, McLaughlin M, Salant DJ. Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int. 2003;64:2072–2078. doi: 10.1046/j.1523-1755.2003.00305.x. [DOI] [PubMed] [Google Scholar]
- 121.Schiller B, He C, Salant DJ, Lim A, Alexander JJ, Quigg RJ. Inhibition of complement regulation is key to the pathogenesis of active Heymann nephritis. J Exp Med. 1998;188:1353–1358. doi: 10.1084/jem.188.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schneeberger EE, Grupe WE. The ultrastructure of the glomerular slit diaphragm in autologous immune complex nephritis. Lab Invest. 1976;34:298–305. [PubMed] [Google Scholar]
- 123.Schulze M, Baker PJ, Perkinson DT, Johnson RJ, Ochi RF, Stahl RA, Couser WG. Increased urinary excretion of C5b-9 distinguishes passive Heymann nephritis in the rat. Kidney Int. 1989;35:60–68. doi: 10.1038/ki.1989.8. [DOI] [PubMed] [Google Scholar]
- 124.Schulze M, Pruchno CJ, Burns M, Baker PJ, Johnson RJ, Couser WG. Glomerular C3c localization indicates ongoing immune deposit formation and complement activation in experimental glomerulonephritis. Am J Pathol. 1993;142:179–187. [PMC free article] [PubMed] [Google Scholar]
- 125.Schulze MS, Donadio JV, Pruchno CJ, Baker PJ, Johnson RJ, Stahl RAK, Watkins S, Martin DC, Wurzner R, Gotze O, Couser WG. Elevated urinary excretion of the C5b-9 complex in membranous nephropathy. Kidney Int. 1991;40:533–538. doi: 10.1038/ki.1991.242. [DOI] [PubMed] [Google Scholar]
- 126.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 128.Shankland SJ. Cell-cycle control and renal disease. Kidney Int. 1997;52:294–308. doi: 10.1038/ki.1997.335. [DOI] [PubMed] [Google Scholar]
- 129.Shankland SJ, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Henne K, Hockenberry DM, Johnson RJ, Couser WG. Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int. 1997;52:404–413. doi: 10.1038/ki.1997.347. [DOI] [PubMed] [Google Scholar]
- 130.Shankland SJ, Pippin J, Pichler RH, Gordon KL, Friedman S, Gold LI, Johnson RJ, Couser WG. Differential expression of transforming growth factor-beta isoforms and receptors in experimental membranous nephropathy. Kidney Int. 1996;50:116–124. doi: 10.1038/ki.1996.294. [DOI] [PubMed] [Google Scholar]
- 131.Sheerin NS, Springall T, Carroll M, Sacks SH. Altered distribution of intraglomerular immune complexes in C3-deficient mice. Immunology. 1999;97:393–399. doi: 10.1046/j.1365-2567.1999.00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 133.Song H, He C, Knaak C, Guthridge JM, Holers VM, Tomlinson S. Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activation. J Clin Invest. 2003;111:1875–1885. doi: 10.1172/JCI17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Susani M, Schulze M, Exner M, Kerjaschki D. Antibodies to glycolipids activate complement and promote proteinuria in passive Heymann nephritis. Am J Pathol. 1994;144:807–819. [PMC free article] [PubMed] [Google Scholar]
- 135.Takano T, Cybulsky AV. Complement C5b-9-mediated arachidonic acid metabolism in glomerular epithelial cells: role of cyclooxygenase-1 and -2. Am J Pathol. 2000;156:2091–2101. doi: 10.1016/S0002-9440(10)65080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takano T, Cybulsky AV, Cupples WA, Ajikobi DO, Papillon J, Aoudjit L. Inhibition of cyclooxygenases reduces complement-induced glomerular epithelial cell injury and proteinuria in passive Heymann nephritis. J Pharmacol Exp Ther. 2003;305:240–249. doi: 10.1124/jpet.102.043604. [DOI] [PubMed] [Google Scholar]
- 137.Takano T, Cybulsky AV, Yang X, Aoudjit L. Complement C5b-9 induces cyclooxygenase-2 gene transcription in glomerular epithelial cells. Am J Physiol Renal Physiol. 2001;281:F841–F850. doi: 10.1152/ajprenal.2001.281.5.F841. [DOI] [PubMed] [Google Scholar]
- 138.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 139.Topham PS, Haydar SA, Kuphal R, Lightfoot JD, Salant DJ. Complement-mediated injury reversibly disrupts glomerular epithelial cell actin microfilaments and focal adhesions. Kidney Int. 1999;55:1763–1775. doi: 10.1046/j.1523-1755.1999.00407.x. [DOI] [PubMed] [Google Scholar]
- 140.Tryggvason K, Wartiovaara J. Molecular basis of glomerular permselectivity. Curr Opin Nephrol Hypertens. 2001;10:543–549. doi: 10.1097/00041552-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 141.Turnberg D, Botto M. The regulation of the complement system: insights from genetically-engineered mice. Mol Immunol. 2003;40:145–153. doi: 10.1016/s0161-5890(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 142.Unanue E, Dixon FJ. Experimental glomerulonephritis. IV. Participation of complement in nephrotoxic nephritis. J Exp Med. 1964;119:965–982. doi: 10.1084/jem.119.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 144.Wang SX, Rastaldi MP, Patari A, Ahola H, Heikkila E, Holthofer H. Patterns of nephrin and a new proteinuria-associated protein expression in human renal diseases. Kidney Int. 2002;61:141–147. doi: 10.1046/j.1523-1755.2002.00114.x. [DOI] [PubMed] [Google Scholar]
- 145.Weise WJ, Natori Y, Levine JS, O’Meara YM, Minto AW, Manning EC, Goldstein DJ, Abrahamson DR, Salant DJ. Fish oil has protective and therapeutic effects on proteinuria in passive Heymann nephritis. Kidney Int. 1993;43:359–368. doi: 10.1038/ki.1993.54. [DOI] [PubMed] [Google Scholar]
- 146.Whiss PA. Pexelizumab Alexion. Curr Opin Invest Drugs. 2002;3:870–877. [PubMed] [Google Scholar]
- 147.Yuan H, Takeuchi E, Taylor GA, McLaughlin M, Brown D, Salant DJ. Nephrin dissociates from actin, and its expression is reduced in early experimental membranous nephropathy. J Am Soc Nephrol. 2002;13:946–956. doi: 10.1681/ASN.V134946. [DOI] [PubMed] [Google Scholar]
- 148.Zoja C, Benigni A, Verroust P, Ronco P, Bertani T, Remuzzi G. Indomethacin reduces proteinuria in passive Heymann nephritis in rats. Kidney Int. 1987;31:1335–1343. doi: 10.1038/ki.1987.147. [DOI] [PubMed] [Google Scholar]