

Abstract
Clostridium difficile colitis is a major complication of antibiotic therapy. Antibiotics cause a reduction in bacteria that normally reside in the colon. If an antibiotic-treated patient ingests C. difficile bacteria, this organism may proliferate in the colon because it is resistant to most antibiotics and because it does not have to compete with the normal bacteria for nutrients. If the C. difficile organism has the gene for toxin production, the toxin can produce a colitis. In addition to antibiotics, other proposed risk factors for development of C. difficile colitis include advanced age, contact with infected patients and with their health care providers, impaired immune function, suppression of gastric acid secretion by a proton pump inhibitor, and postpyloric tube feeding. Many of the risk factors become simultaneously focused on patients admitted to the hospital. The incidence of C. difficile disease has been rising, and strains have become more virulent. In some forms of the disease, the patient doesn't have diarrhea, and in such patients C. difficile can be deadly but difficult to diagnose. The standard treatment, with metronidazole or vancomycin, fails to work in up to 25% of patients with the fulminant form of colitis. Since C. difficile causes only 20% of cases of antibiotic-associated diarrhea, a specific test is needed to diagnose this organism. Toxigenic cultureis highly specific but not available at most institutions. The tests that are available—enzyme-linked immunosorbent assay and fecal cytotoxicity assay—have high false-negative rates, even in patients with severe clinical disease, creating a diagnostic dilemma. The only proven way to reduce the risk of C. difficile disease is implementation of an antibiotic management program in conjunction with enhanced infection control procedures.
My experience with antibiotic-associated colitis began in the early 1970s—although at that time the Clostridium difficile etiology was not known. A Dallas internist told me that numerous patients who were taking clindamycin were getting diarrhea and colitis. At that time, clindamycin had been on the market for only a few years. Larry Hoberman, Dick Norgaard, and Lannie Hughes, who were gastroenterology fellows, gathered data on 16 cases from Parkland Hospital, the Veterans Affairs Hospital, Methodist Hospital, and Baylor University Medical Center (BUMC). No pathogenic organisms could be cultured in any of these patients, and there was no evidence of staphylococcal overgrowth. All 16 patients had severe colitis, and 7 had pseudomembranous colitis. None had small bowel disease. The average age of the patients was 53, and 13 of the 16 were women. Four patients—25%—died (1).
Several possible etiologies were considered, including direct toxicity from or an allergy to clindamycin, bowel ischemia induced by the antibiotic, an altered bacterial flora, emergence of a new bacterial pathogen, and a virus. Since the physicians did not know the cause of the disease, they treated the patients as if they had ulcerative colitis. Fourteen patients received systemic steroids; 7, steroid enemas; 6, Lomotil; and 2, colectomies (1). None of the patients received vancomycin because they were not thought to have staphylococcal enterocolitis.
Within just a few years, C. difficile was discovered as the etiologic agent of antibiotic colitis (Table 1). Ironically, C. difficile turned out to be sensitive to vancomycin, which might have been used in our patients had we not done tests to rule out staphylococcal enterocolitis (6). C. difficile bacteria had been discovered years earlier, in 1935 (7). In their attempt to understand the development of normal bacterial flora in neonates, Hall and O'Toole found a new anaerobe—which they called Bacillus difficilis because of the difficulty involved in its isolation and study. It wasn't pathogenic in newborns infants, but it was pathogenic in guinea pigs via a toxin. The bacillus has since been placed in the genus Clostridium. The organism is depicted microscopically in (Figure 1), which shows both the vegetative bacteria and spores. Although the spores are inactive, they can rapidly change into vegetative forms, which can then induce disease under the right circumstances.
Table 1.
Some important steps in the discovery of Clostridium difficile as the major etiologic agent in antibiotic colitis
| Author | Published | Findings |
| Larson et al (2) | May 1977 | Fecal cytotoxin was found in patients with clindamycin colitis. |
| Rifkin et al (3) | November 1977 | Fecal cytotoxin was neutralized by antisera to Clostridium sordellii. |
| Bartlett et al (4) | November 1977 | Clostridial toxins were the cause of clindamycin colitis in hamsters. |
| C. difficile was cultured from patients with pseudomembranous colitis. | ||
| George et al (5) | March 1978 | Culture broth had a cytotoxic effect that was neutralized by antitoxin. The organism was sensitive to vancomycin. |
Figure 1.

Gram-stained smear of Clostridium difficile prepared from a broth culture. The rod-shaped vegetative cells are shorter and narrower than those of C. perfringens; abundant subterminal and “free” spores are present (× 500). Reprinted from reference 8 with permission from McGraw Hill.
PATHOGENESIS
C. difficile is resistant to most antibiotics. When a person takes an antibiotic, the normal colonic bacteria are reduced. Under these conditions, C. difficile is uninhibited by the normal bacterial products that tend to suppress it, and it has access to additional colonic nutrients. The C. difficile then proliferates. If it lacks the gene for toxin production, no disease develops. However, if it produces toxins A and B, it may cause colitis. The mechanism by which C. difficile toxins damage colonocytes is shown in Figure 2. The result of this damage is a loss of polarity of colonocytes, impaired cell migration, and cell death (9), which lead to colonic inflammation and in some cases pseudomembranous colitis (Figure 3).
Figure 2.
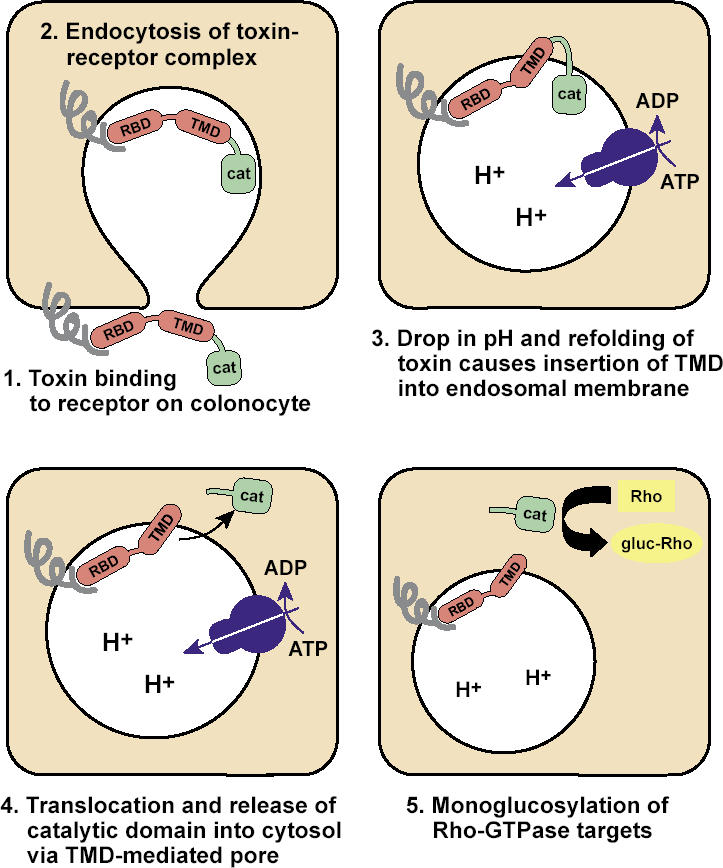
Mechanisms of Clostridium difficile toxin injury to colonocytes, as modified from reference 9. RBD indicates receptor-binding domain of the toxin; TMD, the transmembrane domain of the toxin; CAT, the catalytic domain of the toxin. Five steps are depicted. (1) Toxin RBD binds to a receptor located on the luminal membrane of a colonocyte. (2) The toxin-receptor complex is endocytosed. (3) Acid is secreted into the endocytosed vesicle by a metabolic process; the resulting drop in pH and refolding of the toxin causes insertion of the TMD portion of the toxin into the endosomal membrane. (4) There is a translocation and release of the CAT portion of the toxin into the cytosol of the colonocyte. (5) The CAT portion of the toxin causes monoglycosylation of Rho-GTPases. Not shown: Monoglycosylation of Rho-ATPases by C. difficile toxins destroys the actin cytoskeleton of the colonic epithelial cell, resulting in a loss of cell polarity, impaired cell migration, and cell death. Note: A lack of development of toxin-binding receptor on the colonocytes of newborn infants may explain the absence of colitis and diarrhea in neonates with C. difficile infections.
Figure 3.
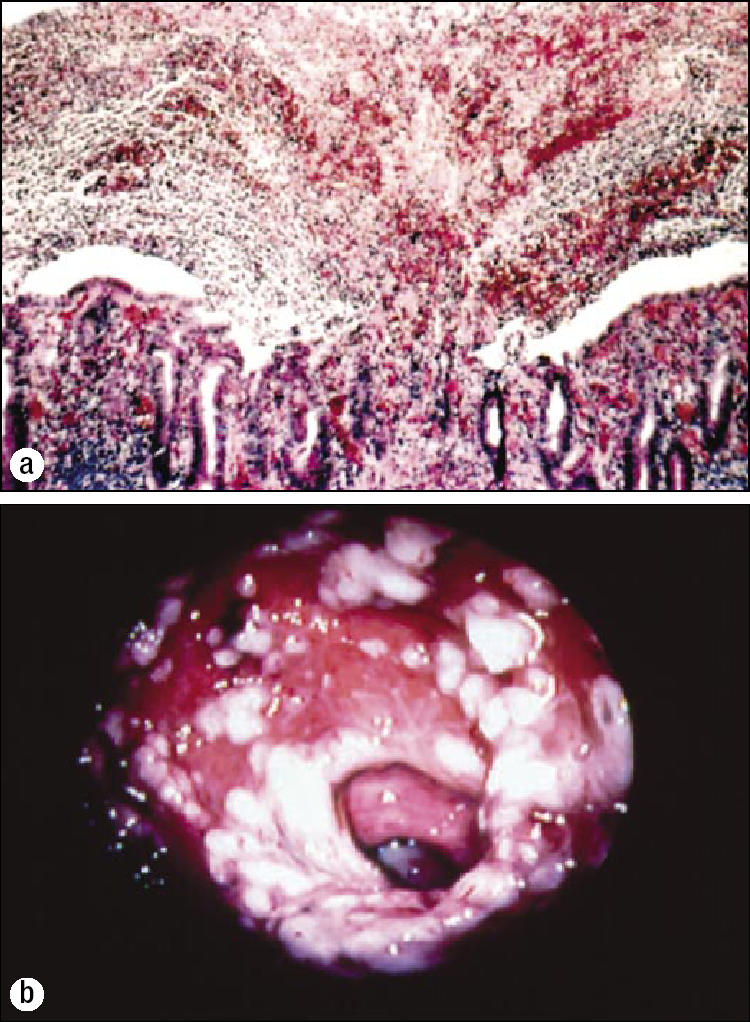
Pseudomembranous colitis. In contrast to antibiotic-associated diarrhea, which has several causes, C. difficile is considered the only cause of antibiotic-associated pseudomembranous colitis. (a) Microscopic appearance of a “volcanic eruption.” The exudate contains fibrin, albumin, and polymorphonuclear cells. Vascular permeability is increased via tumor necrosis factor alpha and inflammatory interleukins. Photo: James Williams, MD, Department of Pathology, Mayo Clinic Scottsdale; reproduced from reference 10 with permission from the American Medical Association. (b) Endoscopic appearance: raised, white or yellowish nodules, usually 2 to 10 mm in diameter, often with skip areas of inflamed or normal-looking mucosa. They may coalesce to form a membrane that covers a large segment ofinflamed mucosa. The pseudomembranes can be easily stripped off with forceps. Reprinted from reference 11 with permission from The European Society of Clinical Microbiology and Infectious Diseases.
C. difficile does not invade the colonic mucosa, and it does not cause disease if no toxin is produced. Even when a toxin is produced, some people become carriers or develop a mild, self-limited diarrhea while others develop severe colitis and may have multiple relapses of the disease. The clinical result depends on the briskness of the serum antibody response to the toxin. Aboudola et al showed that after infection with C. difficile, symptom-free carriers of the infection had a median of 3 enzyme-linked immunosorbent assay (ELISA) units per mL of serum IgG antitoxin A, and patients with a single episode of C. difficile–associated disease had 6 ELISA units. On the other hand, those patients with recurrent disease had only 0.7 units (12). Recurrent C. difficile colitis, then, appears to be associated with a lack of protective immunity to C. difficile toxins.
The serum antibody level to C. difficile can be increased with a vaccine, which is still experimental. In 2005, Sougioultzis et al tested the vaccine in three patients with serious, recurrent disease that was unresponsive to treatment. After vaccination, all three subjects had no further recurrence of diarrhea and were able to discontinue treatment with vancomycin. Further, in two of the three, the increases in serum IgG antibodies were striking: 3-fold and 4-fold increases in antitoxin A antibodies and 52-fold and 20-fold increases in antitoxin B antibodies(13). In 1997, researchers showed that passive immunotherapy with intravenous immunoglobulin was also effective in patients with impaired serum antitoxin response and refractory C. difficile colitis (14). The antibodies are believed to block the effects of the toxins until the normal flora can recover; the antibody effect is on the toxins and not on the organism itself.
Almost any antibiotic can lead to C. difficile intestinal disease. Diarrhea usually begins 4 to 9 days after the patient starts an antibiotic, but it can also develop up to 8 weeks after an antibiotic is discontinued (15). Generally, C. difficile disease is caused by genotypes and strains that are resistant to the precipitating antibiotic. Penicillins, the cephalosporins, and clindamycin are most apt to precipitate the disease (16). However, even vancomycin—which is effective in treating C. difficile disease—can cause the disease. This probably occurs because vancomycin suppresses both normal flora and the vegetative forms of C. difficile, but not its spores. When vancomycin is discontinued, the spores germinate and the new vegetative forms flourish in the altered bacterial environment.
NOSOCOMIAL TRANSMISSION
C. difficile intestinal disease occurs primarily in the elderly who are hospitalized or have recently been hospitalized. Johal et al's 2004 study of 136 patients showed that C. difficile diarrhea (which followed antibiotic exposure in 96%) started in the hospital in 98 patients (72%) and in the community in 38 patients (28%). In addition, 87% of those in whom the disease began in the community had been hospitalized during the preceding 12 months. The average age of the patients was 79 in the inpatient group and 77 in the community group (17).
McFarland et al reported that of 399 patients who were shown not to have C. difficile upon admission to the hospital, 83 (21%) tested positive for the organism during their hospitalization. Fifty-two of the infected patients (63%) remained asymptomatic, but 31 (37%) developed diarrhea (18). Thus, almost 8% of the admitted patients developed a symptomatic infection with C. difficile during their hospitalization.
When the researchers used immunoblot typing to differentiate individual strains of C. difficile, they found that C. difficile is spread by other patients (especially those with diarrhea) and by the hands of health care workers (18). The hands of physicians and medical students were contaminated 75% of the time after they had been in contact with a patient with C. difficile; those of dialysis technicians, 66%; those of nurses, 56%; and those of physical therapists, 50%. Among contaminated health care workers, the undersides of the fingernails were affected in 43% of cases; the fingertips or palms, 37%; and the underside of rings, 20%. Wearing gloves and washing with chlorhexidine markedly reduce hand carriage of the bacteria, but alcohol is not effective (19).
The routine infection control measures used in most hospitals are no match for C. difficile. The spores survive desiccation for months and are resistant to conventional disinfectants. Environmental contamination occurs in 34% to 58% of sites in hospitals, even after detergent cleaning (20). These sites include carpets, clothing, blood pressure cuffs, thermometers, telephones, call buttons, scales, commodes, and tube feeding equipment. Intrahospital transfer of patients from one unit to another is common and creates an infection control nightmare. In addition, compliance with infection control measures is poor, and there are financial pressures to reduce spending on cleaning, infection control, and surveillance cultures to monitor cleaning standards.
Patients, then, come into a hospital that is teeming with C. difficile, an organism difficult to destroy with standard infection control measures. These patients are prescribed an antibiotic, which they presumably need, which suppresses their normal colonic flora. They are probably immunosuppressed—because of either their advanced age or their underlying illness. If that were not enough, other proposed risks are often superimposed.
PROTON PUMP INHIBITORS, TUBE FEEDINGS, AND CHEMOTHERAPY
Acid gastric juice is “nature's disinfectant” for the gastrointestinal tract. It kills the vegetative forms of C. difficile (21), just as it kills cholera, salmonella, and shigella. It is possible that acid gastric juice also protects against viral and prion infections (22).
Secretion of gastric acid is markedly reduced in patients with atrophic gastritis (which is common in elderly people)and by treatment with proton pump inhibitors (PPIs) and histamine H2 receptor antagonists (HRAs).When gastric acid secretion is inhibited, the volume of fluid secreted into the stomach falls markedly, the acid concentration in stomach fluid falls, and the gastric pH rises, as shown in Table 2. The marked reduction in gastric juice volume and the reduced concentration of acid allow ingested acid-sensitive the small bowel and colon.
Table 2.
Correlation of pH and acid concentration in gastric juice*
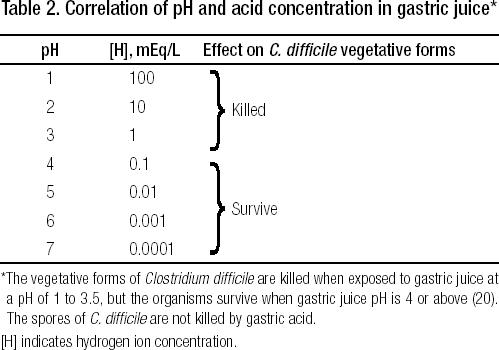
Hospitalized patients are often started on a PPI in order to prevent stomach and/or duodenal bleeding due to stress ulcers. If a patient is admitted to a BUMC intensive care unit (ICU) and a PPI is not prescribed, the physician will be notified: he or she will be asked to consider starting a PPI for stress ulcer prophylaxis, and almost always a PPI is started. Although PPIs are clearly indicated in ICU patients who are especially prone to develop gastroduodenal ulcers due to gastric hypersecretion of acid, such as in patients with acute head injuries (Cushing's ulcer), it seems surprising that PPIs have become the preferred agent for stress ulcer prophylaxis for virtually all patients admitted to ICUs. Most study groups developing practice guidelines have not endorsed PPIs as first-line therapy for this purpose and instead recommend either HRAs or sucralfate. Moreover, recent studies have shown that the incidence of serious stress ulcer bleeding in medical ICUs is low, even when no drug is prescribed for prophylaxis against gastroduodenal ulceration.
Although PPIs, HRAs, antacids, and sucralfate reduce the already low rate of bleeding from gastroduodenal ulceration, there is no proof that these drugs reduce mortality rate. Stress ulcer prophylaxis with two drugs that reduce gastric acidity (HRAs and antacids) led to higher, not lower, mortality rates in a meta-analysis (23) (Figure 4). Reliable mortality rates comparing PPIs with other anti–stress ulcer drugs are apparently not available. Unlike PPIs/HRAs/antacids, sucralfate reduces stress ulceration with very little reduction of gastric acidity (24). Although sucralfate was associated with reduced mortality in this metaanalysis when compared with HRAs and antacids (23), it showed no difference when compared with placebo, presumably reflecting the small effect of stress ulcer bleeding on mortality in modern ICUs. In general, these statistics suggest that suppression of gastric acidity increases the mortality rate in ICUs.
Figure 4.

Common odds ratio and 95% confidence interval (CI) for the risk of mortality determined by a metaanalysis of studies of stress ulcer prophylaxis. Reprinted from reference 23 with permission from Adis International.
Of course, many patients come to the hospital already taking PPIs. The drug companies have spent millions of dollars on direct-to-consumer advertising about PPIs as a treatment for gastroesophageal reflux disease, sometimes offering free samples for starters. The companies have provided assistance to professional gastroenterological associations, academic physicians, and practicing physicians. In my opinion, such marketing has resulted in overutilization of PPIs.
One of the results of PPI overutilization is that almost 50% of hospitalized patients receiving an antibiotic also receive a PPI, at least in Montreal (25). Moreover,there is no legitimate medical reason to markedly suppress gastric acid secretion in most patients who are treated with a PPI in the hospital (25), especially since the preponderance of published evidence suggests that PPIs increase the likelihood of getting C. difficile disease (Table 3). One study suggests that HRAs also increase the odds ratio of contracting this infection (28). The association between treatment with a PPI and development of C. difficile disease has been attributed to PPI suppression of gastric secretion to such a large extent that stomach acidity can no longer serve its natural disinfectant function. This explanation assumes that ingested vegetative forms of C. difficile, in addition to spores (which are resistant to gastric acid), are important in the transmission of C. difficile colitis. Alternatively, or in addition, PPIs may inhibit immune function (29), and this could also predispose to C. difficile disease.
Table 3.
Studies evaluating proton pump inhibitors as a risk factor for Clostridium difficile diarrhea
| Number of patients who received a PPI | |||||
| Reference | Type of study | n | C. difficile group | Control group | Comparison results |
| Shah, 2000 (26)* | Case control | 126 in each group | 46 (37%) | 49 (39%) | OR: no association |
| Cunningham, 2003 (27)* | Case control | 160 in each group | 58 (36%) | 31 (19%) | OR: 2.5 (CI, 1.5–4.2) |
| Preceding 8 weeks: | Preceding 8 weeks: | ||||
| Dial, 2004 (25) | Case control | 94 in each group | 60 (64%) | 34 (36%) | OR: 3.1 (CI, 1.7–5.6) |
| >6 months: 22 (23%) | >6 months: 4 (4%) | OR: 6.9 (CI, 2.3–20.8) | |||
| Univariate OR: 1.8 (CI, 1.2–2.9) | |||||
| Muto, 2005 (28) | Case control | 203 in each group | 78 (38%) | 54 (27%) | Multivariable logistic regression |
| OR: 2.4 (CI, 1.3–4.4) | |||||
| C. diff, taking PPI: 55/591 (9%) | Unadjusted RR: 2.1 (CI, 1.4–3.4) | ||||
| Dial, 2004 (25) | Cohort | 1187 given antibiotic | C. diff, not taking PPI: 26/596 (4%) | Adjusted RR: 2.1 (CI, 1.2–3.5) | |
Patients who start treatment with PPIs often continue taking them indefinitely, and that can result in added risks for other serious diseases (22). Since PPIs are started and continued without knowledge of baseline gastric acid secretion, patients with atrophic gastritis who are treated with a PPI are subjected to side effects from these drugs with little possibility of benefit. At the very least, physicians should specify an automatic termination date rather than allow ongoing renewal that in some cases can mean unnecessary suppression of gastric acidity for many years.
Postpyloric tube feedings increase the risk of C. difficile disease (30). The tube feeding materials may be contaminated with C. difficile, and protective gastric acid is bypassed because such feedings are infused beyond the stomach, directly into the small intestine.
The risk of C. difficile disease is also increased by chemotherapy. In their retrospective case-control study, Cunningham et al found that an antibiotic plus cytotoxic chemotherapy increased the odds ratio for developing C. difficile diarrhea to 17.3. When PPIs were added to the antibiotic and chemotherapy, the odds ratio was 43.2 (27). Chemotherapy suppresses the normal colonic flora to some extent, may impair immune responses, and retards repair function of colonic epithelial cells.
MEASURES TO REDUCE C. DIFFICILE INTESTINAL DISEASE
Enhanced infection control measures reduce the development of C. difficile disease (20). For example, researchers at the University of Pittsburgh Medical Center performed a case-control study with multivariate analysis and molecular subtyping. They identified 419 cases of C. difficile in 2000–2001, with 18 deaths (4.3% mortality rate) and 26 colectomies. One of their conclusions was that enhanced infection control and an antibiotic management program are both required to stem an outbreak (28).
Antibiotic management programs have been shown to reduce the incidence of C. difficile disease in the hospital by more than twofold. In 1994, a joint working group in the UK National Health Service developed the following recommendations for such a program (31):
Written guidelines on correct antimicrobial use
Restriction of susceptibility reports by the microbiology laboratory
Continuous education for health care workers, patients, and their families
Control and restriction of antimicrobial prescriptions
Automatic dates for termination of therapy
Avoidance of unnecessary use of antimicrobial agents
Avoidance of wide-spectrum antimicrobial agents when feasible
Avoidance of antimicrobial agents especially linked to a high risk of C. difficile–associated disease
Conservative use of antibiotics for prophylaxis against infections after surgery.
Two other pharmacologic methods are promising but currently unproven: probiotics, repopulating the colon with nontoxic organisms as antibiotics are given; and using PPIs only when there are specific indications for marked suppression of gastric acid secretion.
INCIDENCE AND VIRULENCE
The incidence of C. difficile disease is increasing. In Quebec, 3262 cases were reported in 2000, 3675 in 2001, 4097 in 2002, and 7004 in 2003 (32). The mortality rate increased from 12% in 2000 to 18% in 2003, and about half of these deaths were directly related to C. difficile intestinal disease. This death rate is approaching what it was in the 1970s, before physicians knew what caused the disease and how to treat it. The newer strains of C. difficile were shown to produce about 20 times more toxins A and B than previously studied strains (33, 34).
Other studies have confirmed the increasing virulence. Dial et al reported a study from the Davis Jewish Hospital in Montreal. Among 94 consecutive patients with C. difficile diarrhea, 18 (19%) required admission to the ICU because of fulminant disease; 12 (13%) developed acute renal failure requiring dialysis; 8 (9%) required colectomy; 21 (22%) had relapses; and 21 (22%) died (25).
CLINICAL SYMPTOMS
Patients with C. difficile intestinal disease usually have mild or severe diarrhea and abdominal pain, low-grade to high-grade fever, and leukocytosis; they may have hypovolemia, shock, and hypoalbuminemia (16). The latter occurs when exudate from colonic ulceration oozes albumin from the blood plasma into the colonic lumen; this albumin is digested or excreted in stool. Acute diarrhea with hypoalbuminemia is a good indicator of C. difficile intestinal disease.
Stools are usually not grossly bloody, but they can be. Occult blood is present in 30% of patients. Fecal leukocytosis is also present in one third of the patients. Colitis, when present, is usually most severe in the rectum and distal colon, but it can be present on the right side of the colon.
Some patients develop fulminant colitis associated with a colonic ileus, in which case the patient may not have diarrhea (Figure 5). Physicians rarely suspect C. difficile disease in the absence of diarrhea. A sigmoidoscopy to look for pseudomembranes and to obtain a stool sample for toxin assay may help reveal the diagnosis. A computed tomography scan is often diagnostic of C. difficile colitis, provided physicians think of this disease in the absence of diarrhea.
Figure 5.

Acute toxic megacolon in a patient with fulminant pseudomembranous colitis. Note the thickened and edematous bowel wall (arrow). Such patients have abdominal pain, fever, leukocytosis, and hypoalbuminemia, but they may not have diarrhea. Many clinicians would suspect ischemic colitis, rather than Clostridium difficile colitis. This form of C. difficile disease has a very high mortality rate and a poor response to vancomycin and metronidazole. In addition to the usual therapy in such patients, physicians might want to consider early use of passive immunotherapy against C. difficile toxins, as described in reference 14 and in several of the treatment references listed at the end of this article. Reprinted from reference 10 with permission from the American Medical Association.
TREATMENT
As mentioned at the beginning of the paper, in the 1970s Dallas physicians gave Lomotil to some patients with this disease. That's a bad choice: Lomotil tends to promote an ileus and to cause retention of toxins in the colon. All antiperistaltic agents should be avoided (16). Treatment of C. difficile diarrhea or colitis should begin with fluid therapy. Precipitating antibiotics should be discontinued or modified if possible.
When an antibiotic against C. difficile is indicated (15, 16), oral metronidazole is the preferred agent except in women who are pregnant or breastfeeding. Metronidazole is absorbed completely and cannot be detected in stools in the absence of diarrhea/colitis. Thus, the drug works in those who have the disease but not in carriers. Carriers do not require treatment, which is fortunate since neither vancomycin nor metronidazole can eliminate the carrier state (20).
In early studies, metronidazole and vancomycin were equally effective. Pépin et al have suggested that vancomycin may be better in very sick patients (35), and vancomycin possesses some theoretical advantages in treatment (16). However, most authorities have favored metronidazole because of its much lower cost and the concern of development of vancomycin-resistant organisms (16). Overall, response rates to these drugs are not as high as they once were, possibly due to increased virulence of C. difficile rather than antibiotic resistance. In some series, 25% of the patients failed to respond to either one of these drugs. Thus, prevention is key (36).
Additional information on treatment is provided in references 15 and 16 and in the treatment references printed after the numbered references.
DIAGNOSTIC TESTS
The antibiotic treatment recommendations given above relate only to C. difficile–associated intestinal disease, not to antibiotic-associated diarrhea in general. This is because most patients with antibiotic-associated diarrhea do not have C. difficile. For example, of a hypothetical 100 patients with antibiotic-associated diarrhea, only 25 would have a bacterial cause. Most of the other 75 patients would have a coincidental association. Of those with a bacterial cause, 20 cases would be from C. difficile, with the disease manifesting as pseudomembranous colitis, colitis without pseudomembranes, or diarrhea without colitis. Four patients would have Klebsiella oxytoca (37), and 1 patient would have Escherichia coli 0157:H7. These latter two organisms usually produce a right-sided colitis that on colonoscopy appears similar to ischemic colitis.
For K. oxytoca–associated diarrhea, which generally follows administration of penicillins, there is no need to administer metronidazole or vancomycin. Once the antibiotic is discontinued, the disease resolves (Högenauer and Krejs, personal communication). For E. coli 0157:H7–associated colitis, administering an antibiotic makes the disease worse. Thus, treatment for these diseases differs significantly from the treatment of C. difficile intestinal disease. Empiric treatment of all patients with antibiotic-associated diarrhea with metronidazole or vancomycin would be a mistake.
To guide treatment, then, a specific test for C. difficile toxins is needed. Several tests are available:
Direct fecal cytotoxicity test. Feces is added to a cell culture system, and the laboratory analyzes whether a demonstrated cytotoxic effect is neutralized by a specific antitoxin. The results are available in about 48 hours. This test is no longer used in most clinical laboratories, including BUMC's.
ELISA test for toxin A or toxins A and B. Results are available in about 2 hours. However, 10% to 30% of results are false-negatives when compared with the direct fecal cytotoxicity assay. Repeat testing is only mildly helpful in identifying additional true positive results (38). This is the only test used at BUMC and at most US hospitals.
“Toxigenic culture”: a stool culture for C. difficile followed by a toxin assay of C. difficile colonies (Figure 6). This method, which takes 48 hours, is highly sensitive. However, in the USA it is not provided by most clinical laboratories.
Polymerase chain reaction and molecular typing. These methods are not available for clinical use, and their accuracy has not been established.
Figure 6.
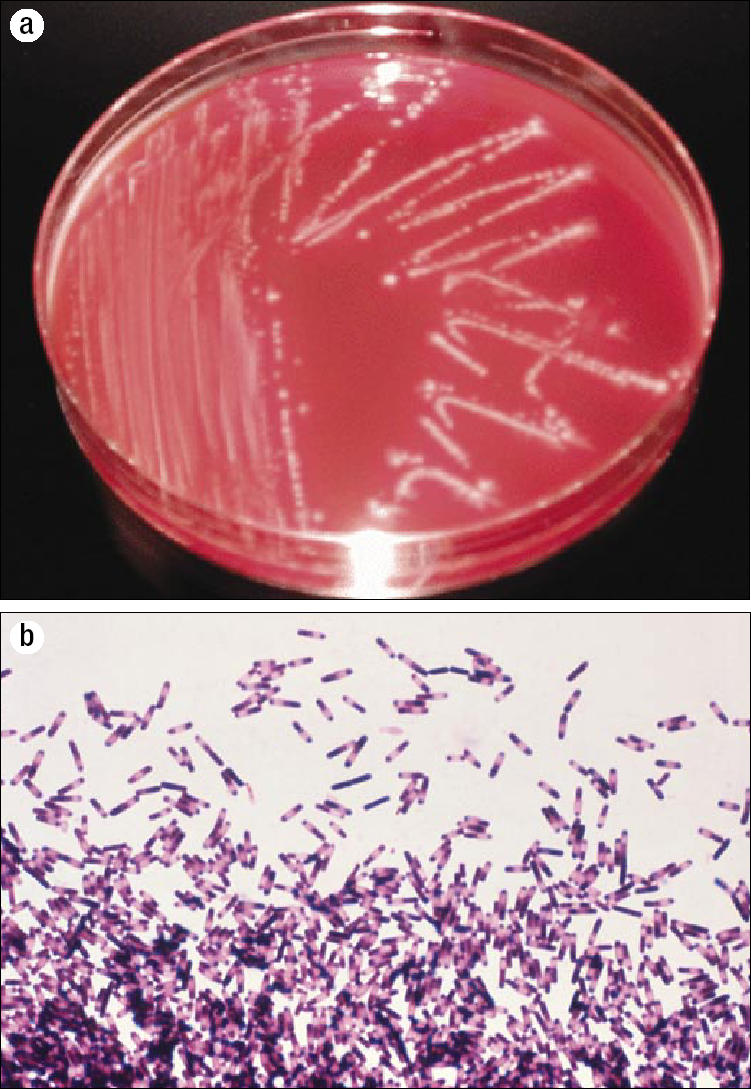
A toxigenic culture for Clostridium difficile. (a) Culture of C. difficile colonies on cycloserine cefoxitin fructose agar under anaerobic conditions. The colonies have a characteristic smell, similar to horse manure. They are nonhemolytic and white to yellowish with a ground glass appearance, and they fluoresce chartreuse under ultraviolet light. Reprinted from reference 11 with permission from The European Society of Clinical Microbiology and Infectious Diseases. (b) The second step, an impression smear from an anaerobic blood agar plate, reveals both spores and vegetative forms of C. difficile. Photo courtesy of CDC/Dr. Gilda Jones/Public Health Image Library. Not shown: In a third step, the culture medium is tested for C. difficile toxins to establish whether or not C. difficile bacteria have the potential for causing colitis.
In 2005, Delmée et al compared the results of the direct fecal cytotoxicity test and toxigenic culture on 10,552 specimens of diarrheal stool (39). About 90% of specimens—9494—were negative with both tests. Another 460 specimens (4.3%) were positive with both tests. However, 355 specimens were negative with the direct fecal cytotoxin assay but were positive with toxigenic culture. Thus, the direct fecal cytotoxin assay missed almost as many cases as it detected. A possible explanation for this lapse is that colonocytes have a receptor for the toxin (and thus are highly sensitive), while fibroblasts used in the cytotoxicity test may not have such receptors.
The authors made a plea for using a toxigenic culture in routine practice (39). Although toxigenic culture requires technical laboratory expertise, adds to the cost of diagnosis, and takes longer, it is beneficial for correct diagnosis and outbreak prevention. Further, culture is the only way to perform surveillance studies, type strains, and test antimicrobial susceptibility.
Some might argue that the cases that are missed by the direct fecal cytotoxicity test represented mild cases that would do well without treatment. However, that is not the case. Johal et al studied a subset of patients with sigmoidoscopy-diagnosed pseudomembranous colitis. In contrast to the several causes of antibiotic-associated diarrhea, C. difficile is considered to be the only cause of antibiotic-associated pseudomembranous colitis, which always reflects severe colitis. In Johal's series of 56 patients with pseudomembranous colitis, the direct fecal cytotoxicity test missed about half of the cases: it identified C. difficile toxin in 27 of the 56, but it missed C. difficile toxin in 29 of the 56. In nine of the latter, toxigenic culture was carried out, and all specimens were positive for toxigenic C. difficile (17).
Thus, physicians face a diagnostic dilemma. The tests available in most hospitals are not sensitive, yet accurate diagnosis seems essential to control this common nosocomial infection. ELISA is the poorest test in terms of accuracy, yet that is the test used at BUMC and most other hospitals. The direct fecal cytotoxin test is also poor; it missed 43% of cases. The toxigenic culture is best, but almost no centers in the USA use it. While flexible sigmoidoscopy would allow the diagnosis of some cases that would be missed by ELISA and other tests, it would add to hospital contamination. Moreover, if the patient had severe disease, a sigmoidoscopy might induce a perforation. The challenge, then, is to curtail the spread of C. difficile within the hospital when the diagnostic test we use detects only about half of infected patients. It is hard for me to understand why we do not use toxigenic culture in patients who develop acute diarrhea in the hospital but who have negative direct fecal cytotoxic tests for C. difficile toxins.
IMPACT OF A C. DIFFICILE EPIDEMIC
When an epidemic of C. difficile disease was in progress in Canada, some physicians wanted to warn the public, but others did not (40). Some of their comments are insightful:
No hospital … wants to be labelled as the C. diff hospital. —Dr. Todd McConnell (40) Patients should be told that taking antibiotics can precipitate
C. difficile disease. —Michael Libman (40) C. difficile would rank as #1 among superbugs like VRE [van-comycin-resistant enterococci] and MRSA [methicillin-resistant Staphylococcus aureus]. It's more virulent than SARS [severe acute respiratory syndrome]. —Dr. Vivian Loo (32)
Even more troublesome is the comment of Dr. Jacques Pépin: “There's less reaction than if it had been an outbreak among younger patients” (41). In other words, epidemics don't create an outcry if only grandmothers are dying (Figure 7).
Figure 7.

Previously healthy 80-year-old Suzanne Cloutier Rocher had emergency abdominal surgery and was admitted to a hospital. She became severely sick and died 5 weeks later of Clostridium difficile intestinal disease (40). Photo: Isabelle Rocher.
CONCLUSION
C. difficile intestinal disease is a serious problem, it is getting worse, and the standard treatments are losing some of their effectiveness. I believe that improvements in prevention and in diagnostic methods would help ensure that BUMC does not become a “C. diff hospital.” In the meantime, the best that individual physicians can do for a grandmother is to keep her out of the hospital whenever possible, avoid giving her unnecessary antibiotics, adhere to the antibiotic management program provided on page 7, avoid combinations of drugs that increase the risk of C. difficile disease, maintain a high index of suspicion for fulminant C. difficile disease that presents without diarrhea, and wash their hands.
Acknowledgments
I am grateful to William Sutker, MD, Louis Sloan, MD, and Christy Vaughan, PharmD, for their careful review of the manuscript and for their many constructive suggestions.
References
- 1.Hoberman LJ, Eigenbrodt EH, Kilman WJ, Hughes LR, Norgaard RP, Fordtran JS. Colitis associated with oral clindamycin therapy. A clinical study of 16 patients. Am J Dig Dis. 1976;21(1):1–17. doi: 10.1007/BF01074133. [DOI] [PubMed] [Google Scholar]
- 2.Larson HE, Parry JV, Price AB, Davies DR, Dolby J, Tyrrell DA. Un- described toxin in pseudomembranous colitis. Br Med J. 1977;1(6071):1246–1248. doi: 10.1136/bmj.1.6071.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rifkin GD, Fekety FR, Silva J., Jr Antibiotic-induced colitis: implication of a toxin neutralised by Clostridium sordellii antitoxin. Lancet. 1977;2(8048):1103–1106. doi: 10.1016/s0140-6736(77)90547-5. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett JG, Onderdonk AB, Cisneros RL, Kasper DL. Clindamycin associated colitis due to a toxin-producing species of Clostridium in hamsters. J Infect Dis. 1977;136(5):701–705. doi: 10.1093/infdis/136.5.701. [DOI] [PubMed] [Google Scholar]
- 5.George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, Burdon DW. Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J. 1978;1(6114):695. doi: 10.1136/bmj.1.6114.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan MY, Hall WH. Staphylococcal enterocolitis—treatment with oral vancomycin. Ann Intern Med. 1966;65(1):1–8. doi: 10.7326/0003-4819-65-1-1. [DOI] [PubMed] [Google Scholar]
- 7.Hall IC, O'Toole E. Intestinal flora in newborn infants with description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child. 1935;49:390. [Google Scholar]
- 8.Allen SD. Pig-bel and other necrotizing disorders of the gut involving Clostridium perfringens. In: Connor DH, Chandler FW, Schwartz DA, Manz HJ, Lack EE, Manz H, editors. Pathology of Infectious Diseases. New York: McGraw-Hill Professional; 1997. [Google Scholar]
- 9.Just I, Gerhard R. Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol. 2004;152(1):23–47. doi: 10.1007/s10254-004-0033-5. [DOI] [PubMed] [Google Scholar]
- 10.Hurley BW, Nguyen CC. The spectrum of pseudomembranous entero colitis and antibiotic-associated diarrhea. Arch Intern Med. 2002;162(19):2177–2184. doi: 10.1001/archinte.162.19.2177. [DOI] [PubMed] [Google Scholar]
- 11.Bouza E, Munoz P, Alonso R. Clinical manifestations, treatment and control of infections caused by Clostridium difficile. Clin Microbiol Infect. 2005;11(Suppl 4):57–64. doi: 10.1111/j.1469-0691.2005.01165.x. [DOI] [PubMed] [Google Scholar]
- 12.Aboudola S, Kotloff KL, Kyne L, Warny M, Kelly EC, Sougioultzis S, Giannasca PJ, Monath TP, Kelly CP. Clostridium difficile vaccine and serum immunoglobulin G antibody response to toxin A. Infect Immun. 2003;71(3):1608–1610. doi: 10.1128/IAI.71.3.1608-1610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sougioultzis S, Kyne L, Drudy D, Keates S, Maroo S, Pothoulakis C, Giannasca PJ, Lee CK, Warny M, Monath TP, Kelly CP. Clostridium difficile toxoid vaccine in recurrent C. difficile–associated diarrhea. Gastroenterology. 2005;128(3):764–770. doi: 10.1053/j.gastro.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Salcedo J, Keates S, Pothoulakis C, Warny M, Castagliuolo I, LaMont JT, Kelly CP. Intravenous immunoglobulin therapy for severe Clostridium difficile colitis. Gut. 1997;41(3):366–370. doi: 10.1136/gut.41.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fekety R, American College of Gastroenterology, Practice Parameters Committee Guidelines for the diagnosis and management of Clostridium difficile–associated diarrhea and colitis. Am J Gastroenterol. 1997;92(5):739–750. [PubMed] [Google Scholar]
- 16.Bartlett JG. Pseudomembranous enterocolitis and antibiotic-associated diarrhea. In: Feldman M, Freidman LS, Sleisenger MH, editors. Sleisenger and Fordtran's Gastrointestinal and Liver Disease Pathophysiology/Diagnosis/Management. 7th ed. Philadelphia: Saunders; 2002. pp. 1914–1931. [Google Scholar]
- 17.Johal SS, Hammond J, Solomon K, James PD, Mahida YR. Clostridium difficile associated diarrhoea in hospitalised patients: onset in the community and hospital and role of flexible sigmoidoscopy. Gut. 2004;53(5):673–677. doi: 10.1136/gut.2003.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McFarland LV, Mulligan ME, Kwok RY, Stamm WE. Nosocomial ac quisition of Clostridium difficile infection. N Engl J Med. 1989;320(4):204–210. doi: 10.1056/NEJM198901263200402. [DOI] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention. Clostridium difficile: information for healthcare providers. Atlanta, GA: CDC, updated July 22, 2005. Available at http://www.cdc.gov/ncidod/hip/gastro/ClostridiumDifficileHCP_print.htm; accessed October 12, 2005.
- 20.National Clostridium difficile Standards Group Report to the Department of Health. J Hosp Infect. 2004;56(Suppl 1):1–38. doi: 10.1016/j.jhin.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Gurian L, Ward TT, Katon RM. Possible foodborne transmission in a case of pseudomembranous colitis due to Clostridium difficile: influence of gastrointestinal secretions on Clostridium difficile infection. Gastroenterology. 1982;83(2):465–469. [PubMed] [Google Scholar]
- 22.Waldum HL, Brenna E, Sandvik AK. Long-term safety of proton pump inhibitors: risks of gastric neoplasia and infections. Expert Opin Drug Saf. 2002;1(1):29–38. doi: 10.1517/14740338.1.1.29. [DOI] [PubMed] [Google Scholar]
- 23.Tryba M, Cook D. Current guidelines on stress ulcer prophylaxis. Drugs. 1997;54(4):581–596. doi: 10.2165/00003495-199754040-00005. [DOI] [PubMed] [Google Scholar]
- 24.Spechler SJ. Peptic ulcer disease and its complications. In: Feldman M, Freidman LS, Sleisenger MH, editors. Sleisenger and Fordtran's Gastrointestinal and Liver Disease Pathophysiology/Diagnosis/Management. 7th ed. Philadelphia: Saunders; 2002. pp. 747–781. [Google Scholar]
- 25.Dial S, Alrasadi K, Manoukian C, Huang A, Menzies D. Risk of Clostridium difficile diarrhea among hospital inpatients prescribed proton pump inhibitors: cohort and case-control studies. CMAJ. 2004;171(1):33–38. doi: 10.1503/cmaj.1040876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah S, Lewis A, Leopold D, Dunstan F, Woodhouse K. Gastric acid suppression does not promote clostridial diarrhoea in the elderly. QJM. 2000;93(3):175–181. doi: 10.1093/qjmed/93.3.175. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham R, Dale B, Undy B, Gaunt N. Proton pump inhibitors as a risk factor for Clostridium difficile diarrhoea. J Hosp Infect. 2003;54(3):243–245. doi: 10.1016/s0195-6701(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 28.Muto CA, Pokrywka M, Shutt K, Mendelsohn AB, Nouri K, Posey K, Roberts T, Croyle K, Krystofiak S, Patel-Brown S, Pasculle AW, Paterson DL, Saul M, Harrison LH. A large outbreak of Clostridium difficile– associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infect Control Hosp Epidemiol. 2005;26(3):273–280. doi: 10.1086/502539. [DOI] [PubMed] [Google Scholar]
- 29.Noble DW. Proton pump inhibitors and stress ulcer prophylaxis: pause for thought? Crit Care Med. 2002;30(5):1175–1176. doi: 10.1097/00003246-200205000-00046. [DOI] [PubMed] [Google Scholar]
- 30.Bliss DZ, Johnson S, Savik K, Clabots CR, Willard K, Gerding DN. Acquisition of Clostridium difficile and Clostridium difficile–associated diarrhea in hospitalized patients receiving tube feeding. Ann Intern Med. 1998;129(12):1012–1019. doi: 10.7326/0003-4819-129-12-199812150-00004. [DOI] [PubMed] [Google Scholar]
- 31.Department of Health and Public Health Laboratory Service Joint Working Party . Clostridium difficile infection: prevention and management. London: Department of Health; 1994. Quoted in reference 11. [Google Scholar]
- 32.Eggertson L. C. difficile: by the numbers. CMAJ. 2004;171(11):1331–1332. doi: 10.1503/cmaj.1041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eggertson L. C. difficile strain 20 times more virulent. CMAJ. 2005;172(10):1279. doi: 10.1503/cmaj.050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warny M, Pépin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- 35.Pépin J, Valiquette L, Alary ME, Villemure P, Pelletier A, Forget K, Pépin K, Chouinard D. Clostridium difficile–associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. CMAJ. 2004;171(5):466–472. doi: 10.1503/cmaj.1041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerding DN. Metronidazole for Clostridium difficile–associated disease: is it okay for mom? Clin Infect Dis. 2005;40(11):1598–1600. doi: 10.1086/430317. [DOI] [PubMed] [Google Scholar]
- 37.Beaugerie L, Metz M, Barbut F, Bellaiche G, Bouhnik Y, Raskine L, Nicolas JC, Chatelet FP, Lehn N, Petit JC, Infectious Colitis Study Group Klebsiella oxytoca as an agent of antibiotic-associated hemorrhagic colitis. Clin Gastroenterol Hepatol. 2003;1(5):370–376. doi: 10.1053/s1542-3565(03)00183-6. [DOI] [PubMed] [Google Scholar]
- 38.Borek AP, Aird DZ, Carroll KC. Frequency of sample submission for optimal utilization of the cell culture cytotoxicity assay for detection of Clostridium difficile toxin. J Clin Microbiol. 2005;43(6):2994–2995. doi: 10.1128/JCM.43.6.2994-2995.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmee M, Van Broeck J, Simon A, Janssens M, Avesani V. Laboratory diagnosis of Clostridium difficile–associated diarrhoea: a plea for culture. J Med Microbiol. 2005;54(2):187–191. doi: 10.1099/jmm.0.45844-0. [DOI] [PubMed] [Google Scholar]
- 40.Eggertson L, Sibbald B. Hospitals battling outbreaks of C. difficile. CMAJ. 2004;171(1):19–21. doi: 10.1503/cmaj.1040979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eggertson L. C. difficile hits Sherbrooke, Que., hospital: 100 deaths. CMAJ. 2004;171(5):436. doi: 10.1503/cmaj.1041250. [DOI] [PMC free article] [PubMed] [Google Scholar]
OTHER ARTICLES THAT WERE CONSULTED Proton pump inhibitors, acid secretion, and acid inhibition
- 42.Allen ME, Kopp BJ, Erstad BL. Stress ulcer prophylaxis in the postoperative period. Am J Health Syst Pharm. 2004;61(6):588–596. doi: 10.1093/ajhp/61.6.588. [DOI] [PubMed] [Google Scholar]
- 43.Cook DJ, Fuller HD, Guyatt GH, Marshall JC, Leasa D, Hall R, Winton TL, Rutledge F, Todd TJ, Roy P, Lacroix J, Griffith L, Willan A, Canadian Critical Care Trials Group Risk factors for gastrointestinal bleeding in critically ill patients. N Engl J Med. 1994;330(6):377–381. doi: 10.1056/NEJM199402103300601. [DOI] [PubMed] [Google Scholar]
- 44.Jung R, MacLaren R. Proton-pump inhibitors for stress ulcer prophylaxis in critically ill patients. Ann Pharmacother. 2002;36(12):1929–1937. doi: 10.1345/aph.1C151. [DOI] [PubMed] [Google Scholar]
- 45.Kantorova I, Svoboda P, Scheer P, Doubek J, Rehorkova D, Bosakova H, Ochmann J. Stress ulcer prophylaxis in critically ill patients: a randomized controlled trial. Hepatogastroenterology. 2004;51(57):757–761. [PubMed] [Google Scholar]
- 46.McCarthy DM. Sucralfate. N Engl J Med. 1991;325(14):1017–1025. doi: 10.1056/NEJM199110033251407. [DOI] [PubMed] [Google Scholar]
- 47.Mutlu GM, Mutlu EA, Factor P. Prevention and treatment of gastrointestinal complications in patients on mechanical ventilation. Am J Respir Med. 2003;2(5):395–411. doi: 10.1007/BF03256667. [DOI] [PubMed] [Google Scholar]
- 48.Nardino RJ, Vender RJ, Herbert PN. Overuse of acid-suppressive therapy in hospitalized patients. Am J Gastroenterol. 2000;95(11):3118–3122. doi: 10.1111/j.1572-0241.2000.03259.x. [DOI] [PubMed] [Google Scholar]
- 49.Parente F, Cucino C, Gallus S, Bargiggia S, Greco S, Pastore L, Bianchi Porro G. Hospital use of acid-suppressive medications and its fall-out on prescribing in general practice: a 1-month survey. Aliment Pharmacol Ther. 2003;17(12):1503–1506. doi: 10.1046/j.1365-2036.2003.01600.x. [DOI] [PubMed] [Google Scholar]
- 50.Rees WD. Mechanisms of gastroduodenal protection by sucralfate. Am J Med. 1991;91(2A):58S–63S. doi: 10.1016/0002-9343(91)90452-4. [DOI] [PubMed] [Google Scholar]
- 51.Stollman N, Metz DC. Pathophysiology and prophylaxis of stress ulcer in intensive care unit patients. J Crit Care. 2005;20(1):35–45. doi: 10.1016/j.jcrc.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Tryba M. Side effects of stress bleeding prophylaxis. Am J Med. 1989;86(6A):85–93. doi: 10.1016/0002-9343(89)90165-4. [DOI] [PubMed] [Google Scholar]
Infection control
- 53.Carling P, Fung T, Killion A, Terrin N, Barza M. Favorable impact of a multidisciplinary antibiotic management program conducted during 7 years. Infect Control Hosp Epidemiol. 2003;24(9):699–706. doi: 10.1086/502278. [DOI] [PubMed] [Google Scholar]
- 54.O'Connor KA, Kingston M, O'Donovan M, Cryan B, Twomey C, O'Mahony D. Antibiotic prescribing policy and Clostridium difficile diarrhoea. QJM. 2004;97(7):423–429. doi: 10.1093/qjmed/hch076. [DOI] [PubMed] [Google Scholar]
- 55.Perez J, Springthorpe VS, Sattar SA. Activity of selected oxidizing microbicides against the spores of Clostridium difficile: relevance to environmental control. Am J Infect Control. 2005;33(6):320–325. doi: 10.1016/j.ajic.2005.04.240. [DOI] [PubMed] [Google Scholar]
- 56.Rutala WA, Gergen MF, Weber DJ. Inactivation of Clostridium difficile spores by disinfectants. Infect Control Hosp Epidemiol. 1993;14(1):36–39. doi: 10.1086/646628. [DOI] [PubMed] [Google Scholar]
- 57.Schaier M, Wendt C, Zeier M, Ritz E. Clostridium difficile diarrhoea in the immunosuppressed patient—update on prevention and management. Nephrol Dial Transplant. 2004;19(10):2432–2436. doi: 10.1093/ndt/gfh428. [DOI] [PubMed] [Google Scholar]
- 58.Thomas C, Riley TV. Restriction of third generation cephalosporin use reduces the incidence of Clostridium difficile–associated diarrhoea in hospitalised patients. Commun Dis Intell. 2003;27(Suppl):S28–S31. doi: 10.33321/cdi.2003.27.17. [DOI] [PubMed] [Google Scholar]
- 59.Wilcox MH. Cleaning up Clostridium difficile infection. Lancet. 1996;348(9030):767–768. doi: 10.1016/S0140-6736(05)65204-X. [DOI] [PubMed] [Google Scholar]
- 60.Wullt M, Odenholt I, Walder M. Activity of three disinfectants and acidified nitrite against Clostridium difficile spores. Infect Control Hosp Epidemiol. 2003;24(10):765–768. doi: 10.1086/502129. [DOI] [PubMed] [Google Scholar]
Clinical manifestations
- 61.Dallal RM, Harbrecht BG, Boujoukas AJ, Sirio CA, Farkas LM, Lee KK, Simmons RL. Fulminant Clostridium difficile: an underappreciated and increasing cause of death and complications. Ann Surg. 2002;235(3):363–372. doi: 10.1097/00000658-200203000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobs A, Barnard K, Fishel R, Gradon JD. Extracolonic manifestations of Clostridium difficile infections. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2001;80(2):88–101. doi: 10.1097/00005792-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Tsutaoka B, Hansen J, Johnson D, Holodniy M. Antibiotic-associated pseudomembranous enteritis due to Clostridium difficile. Clin Infect Dis. 1994;18(6):982–984. doi: 10.1093/clinids/18.6.982. [DOI] [PubMed] [Google Scholar]
Diagnosis and testing
- 64.Livsey S. Clostridium difficile: towards a standard operating procedure. Commun Dis Public Health. 2003;6(3):263–265. [PubMed] [Google Scholar]
- 65.Wilkins TD, Lyerly DM. Clostridium difficile testing: after 20 years, still challenging. J Clin Microbiol. 2003;41(2):531–534. doi: 10.1128/JCM.41.2.531-534.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Treatment
- 66.Aslam S, Hamill RJ, Musher DM. Treatment of Clostridium difficile– associated disease: old therapies and new strategies. Lancet Infect Dis. 2005;5(9):549–557. doi: 10.1016/S1473-3099(05)70215-2. [DOI] [PubMed] [Google Scholar]
- 67.Cavagnaro C, Berezin S, Medow MS. Corticosteroid treatment of severe, nonresponsive Clostridium difficile induced colitis. Arch Dis Child. 2003;88(4):342–344. doi: 10.1136/adc.88.4.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giannasca PJ, Warny M. Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine. 2004;22(7):848–856. doi: 10.1016/j.vaccine.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 69.Johnson S, Homann SR, Bettin KM, Quick JN, Clabots CR, Peterson LR, Gerding DN. Treatment of asymptomatic Clostridium difficile carriers (fecal excretors) with vancomycin or metronidazole. A randomized, placebo-controlled trial. Ann Intern Med. 1992;117(4):297–302. doi: 10.7326/0003-4819-117-4-297. [DOI] [PubMed] [Google Scholar]
- 70.Kyne L, Warny M, Qamar A, Kelly CP. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet. 2001;357(9251):189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 71.Longo WE, Mazuski JE, Virgo KS, Lee P, Bahadursingh AN, Johnson FE. Outcome after colectomy for Clostridium difficile colitis. Dis Colon Rectum. 2004;47(10):1620–1626. doi: 10.1007/s10350-004-0672-2. [DOI] [PubMed] [Google Scholar]
- 72.Musher DM, Aslam S, Logan N, Nallacheru S, Bhaila I, Borchert F, Hamill RJ. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clin Infect Dis. 2005;40(11):1586–1590. doi: 10.1086/430311. [DOI] [PubMed] [Google Scholar]
- 73.Vasa CV, Glatt AE. Effectiveness and appropriateness of empiric metronidazole for Clostridium difficile–associated diarrhea. Am J Gastroenterol. 2003;98(2):354–358. doi: 10.1111/j.1572-0241.2003.07227.x. [DOI] [PubMed] [Google Scholar]
- 74.Wilcox MH. Descriptive study of intravenous immunoglobulin for the treatment of recurrent Clostridium difficile diarrhoea. J Antimicrob Chemother. 2004;53(5):882–884. doi: 10.1093/jac/dkh176. [DOI] [PubMed] [Google Scholar]
Epidemiology
- 75.Al-Jumaili IJ, Shibley M, Lishman AH, Record CO. Incidence and origin of Clostridium difficile in neonates. J Clin Microbiol. 1984;19(1):77–78. doi: 10.1128/jcm.19.1.77-78.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barbut F, Petit JC. Epidemiology of Clostridium difficile–associated infections. Clin Microbiol Infect. 2001;7(8):405–410. doi: 10.1046/j.1198-743x.2001.00289.x. [DOI] [PubMed] [Google Scholar]
- 77.Blot E, Escande MC, Besson D, Barbut F, Granpeix C, Asselain B, Falcou MC, Pouillart P. Outbreak of Clostridium difficile–related diarrhoea in an adult oncology unit: risk factors and microbiological characteristics. J Hosp Infect. 2003;53(3):187–192. doi: 10.1053/jhin.2002.1356. [DOI] [PubMed] [Google Scholar]
- 78.Fawley WN, Parnell P, Verity P, Freeman J, Wilcox MH. Molecular epidemiology of endemic Clostridium difficile infection and the significance of subtypes of the United Kingdom epidemic strain (PCR ribotype 1) J Clin Microbiol. 2005;43(6):2685–2696. doi: 10.1128/JCM.43.6.2685-2696.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kyne L, Warny M, Qamar A, Kelly CP. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 2000;342(6):390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 80.Samore MH, Venkataraman L, DeGirolami PC, Arbeit RD, Karchmer AW. Clinical and molecular epidemiology of sporadic and clustered cases of nosocomial Clostridium difficile diarrhea. Am J Med. 1996;100(1):32–40. doi: 10.1016/s0002-9343(96)90008-x. [DOI] [PubMed] [Google Scholar]
Pathophysiology
- 81.Brito GA, Carneiro-Filho B, Oria RB, Destura RV, Lima AA, Guerrant RL. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: role of Gln and Ala-Gln in toxin A effects. Dig Dis Sci. 2005;50(7):1271–1278. doi: 10.1007/s10620-005-2771-x. [DOI] [PubMed] [Google Scholar]
- 82.Gerding DN. Clindamycin, cephalosporins, fluoroquinolones, and Clostridium difficile–associated diarrhea: this is an antimicrobial resistance problem. Clin Infect Dis. 2004;38(5):646–648. doi: 10.1086/382084. [DOI] [PubMed] [Google Scholar]
- 83.Justus PG, Martin JL, Goldberg DA, Taylor NS, Bartlett JG, Alexander RW, Mathias JR. Myoelectric effects of Clostridium difficile: motility-altering factors distinct from its cytotoxin and enterotoxin in rabbits. Gastroenterology. 1982;83(4):836–843. [PubMed] [Google Scholar]
- 84.McVey DC, Vigna SR. The role of leukotriene B4 in Clostridium difficile toxin A–induced ileitis in rats. Gastroenterology. 2005;128(5):1306–1316. doi: 10.1053/j.gastro.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 85.Schirmer J, Aktories K. Large clostridial cytotoxins: cellular biology of Rho/ Ras-glycosylating toxins. Biochim Biophys Acta. 2004;1673(1–2):66–74. doi: 10.1016/j.bbagen.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 86.Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding DN. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet. 1998;351(9103):633–636. doi: 10.1016/S0140-6736(97)08062-8. [DOI] [PubMed] [Google Scholar]
- 87.Tang-Feldman YJ, Henderson JP, Ackermann G, Feldman SS, Bedley M, Silva J, Jr, Cohen SH. Prevalence of the ermB gene in Clostridium difficile strains isolated at a university teaching hospital from 1987 through 1998. Clin Infect Dis. 2005;40(10):1537–1540. doi: 10.1086/428835. [DOI] [PubMed] [Google Scholar]
- 88.Wilcox MH, Fawley WN, Settle CD, Davidson A. Recurrence of symptoms in Clostridium difficile infection—relapse or reinfection? J Hosp Infect. 1998;38(2):93–100. doi: 10.1016/s0195-6701(98)90062-7. [DOI] [PubMed] [Google Scholar]
Reviews
- 89.Allen SD, Emery CL, Lyerly DM. Clostridium. In: Murray PR, Baron EJ, Jorgensen JH, Pfaller MA, Yolken RH, editors. Manual of Clinical Microbiology. 8th ed. Washington: ASM Press; 2003. pp. 835–856. [Google Scholar]
- 90.Gerding DN, Johnson S, Peterson LR, Mulligan ME, Silva J., Jr Clostridium difficile–associated diarrhea and colitis. Infect Control Hosp Epidemiol. 1995;16(8):459–477. doi: 10.1086/648363. [DOI] [PubMed] [Google Scholar]
- 91.Poutanen SM, Simor AE. Clostridium difficile–associated diarrhea in adults. CMAJ. 2004;171(1):51–58. doi: 10.1503/cmaj.1031189. [DOI] [PMC free article] [PubMed] [Google Scholar]