

Abstract
Allopolyploid formation requires the adaptation of two nuclear genomes within a single cytoplasm, which may involve programmed genetic and epigenetic changes during the initial generations following genome fusion. To study the dynamics of genome change, we synthesized 49 isogenic Brassica napus allopolyploids and surveyed them with 76 restriction fragment length polymorphism (RFLP) probes and 30 simple sequence repeat (SSR) primer pairs. Here, we report on the types and distribution of genetic and epigenetic changes within the S1 genotypes. We found that insertion/deletion (indel) events were rare, but not random. Of the 57,710 (54,383 RFLP and 3,327 SSR) parental fragments expected among the amphidiploids, we observed 56,676 or 99.9%. Three loci derived from Brassica rapa had indels, and one indel occurred repeatedly across 29% (14/49) of the lines. Loss of one parental fragment was due to the 400-bp reduction of a guanine-adenine dinucleotide repeat-rich sequence. In contrast to the 4% (3/76) RFLP probes that detected indels, 48% (35/73) detected changes in the CpG methylation status between parental genomes and the S1 lines. Some loci were far more likely than others to undergo epigenetic change, but the number of methylation changes within each synthetic polyploid was remarkably similar to others. Clear de novo methylation occurred at a much higher frequency than de novo demethylation within allopolyploid sequences derived from B. rapa. Our results suggest that there is little genetic change in the S0 generation of resynthesized B. napus polyploids. In contrast, DNA methylation was altered extensively in a pattern that indicates tight regulation of epigenetic changes.
A very large number of species are of allopolyploid origin and allopolyploidization is thus one of the most widespread modes of speciation in higher plants. In a successful allopolyploid, two parental genomes must function within a common cytoplasm and be transmitted through meiotic divisions. Developmentally, this process requires both heritable stabilization of the two disparate genomes and the establishment of proper gene transcription levels and timing. One means of this stabilization may be through epigenetic changes that alter chromatin structure and affect gene expression. For example, hypermethylated genes are generally transcribed at a lower level than hypomethylated genes, and the methylation status and gene expression profiles of many allopolyploids differ from their parental genomes at both repetitive and low-copy-number sequences (e.g. Song et al., 1995; Chen and Pikaard, 1997a; Kashkush et al., 2002; Madlung et al., 2002, 2005). Genetic changes, including deletions of sequences that are deleterious within the allopolyploid, may also contribute to genome stability (e.g. Feldman et al., 1997). In wheat (Triticum aestivum), DNA sequences that are genome and chromosome specific within established allopolyploids are deleted at high frequency from parental genomes within synthetic allopolyploids (Feldman et al., 1997; Liu et al., 1998b). An analysis of Aegilops sharonensis × Aegilops umbellata F1 plants showed that as much as 14% of the DNA sequences from one parent were eliminated in the synthetic (Shaked et al., 2001). Similar results have been seen in synthesized allopolyploid triticale (Triticum-Secale hexaploids and octaploids) that show that 97% of the Triticum coding sequences, but only 61.6% of the Secale sequences, are maintained after 15 to 35 generations (Ma et al., 2002, 2004).
There has been some debate as to whether observed epigenetic and genetic changes are stochastic or nonrandom. Many of the same sequences that are absent from polyploid bread wheat are eliminated rapidly among a small number of synthetic polyploids generated from different parents (Liu et al., 1998b; Ozkan et al., 2001; Shaked et al., 2001). These nonrandom changes suggest that characteristics of these sequences target them for elimination. However, other differences between parental and polyploid genomes are more variable. Changes may only occur within a cross between two certain species or they may appear within one synthetic polyploid derived from two species, but not a second synthetic derived from the same species. Liu et al. (1998a) proposed that these variable changes correspond to random changes within the genome and that they can introduce novel, heritable variation. We also have found that variation among Brassica polyploid lines derived from a single synthetic polyploid can lead to heritable, phenotypic differences for traits such as flowering time (Schranz and Osborn, 2000, 2004; Schranz et al., 2002; Pires et al., 2004), as well as molecular variation (Song et al., 1995). However, none of these studies on Brassica or wheat analyzed a large number of isogenic allopolyploids derived from a single pair of diploid parents. Such an investigation would permit a rigorous statistical analysis to determine whether changes are directed or random.
In this work, we investigate the frequency of genetic and epigenetic changes among 49 newly synthesized Brassica napus allopolyploid lines across a large number of loci. These lines were derived by hybridizing double haploids of Brassica oleracea and Brassica rapa. Thus, they represent independent polyploidization events that are expected to be genetically identical. We investigated DNA sequence and methylation changes among the bulked progeny of S0 polyploids to assay changes in the first generation of polyploidy. Our results show that genetic changes in S0 polyploid genomes are rare. However, epigenetic changes in CpG methylation occur at a much higher frequency, and the frequency and genomic location of methylation changes are tightly regulated.
RESULTS
Loss of Parental Loci within Allopolyploid Genomes
We assayed for genetic changes within 49 unique synthetic polyploids by comparing the EcoRI, HindIII, and DraI restriction fragments of the pooled progeny of B. napus S0 plants with the restriction fragments of their diploid progenitors, B. oleracea (TO1000C) and B. rapa (IMB218A). The 49 S0 lines were generated by interspecific hybridization followed by either colchicine treatment or spontaneous doubling (Fig. 1). Seventy-six probes were hybridized to each genomic DNA digest. We detected a total of 1,115 fragments within both the B. oleracea and B. rapa genomes. Only 255 (23%) restriction fragments were shared between the two species. Eight-hundred-sixty fragments (77%) were unique to a single species. B. oleracea had a larger number of fragments (718) relative to B. rapa (652). In addition, EcoRI restriction sites were more highly conserved between species than HindIII and DraI restriction sites. Thus, across both species, EcoRI fragments (255) made up a significantly lower number of the unique fragments than HindIII (307) and DraI (298) fragments (P < 0.02). We observed an average of 5.09 fragments per probe-enzyme combination in the B. napus genome. This level of redundancy is consistent with the hypothesis that the two parental genomes are ancient polyploids (Lagercrantz, 1998; Lukens et al., 2003, 2004).
Figure 1.

Pedigree of lines used in this study. Plants of two double-haploid genotypes (Brassica oleracea, TO1000C and Brassica rapa, IMB218A) were crossed to generate a population of 49 hybrid (CA) plantlets. Eight of these plantlets spontaneously generated putative allopolyploid tissues; 39 plantlets were treated with colchicine. Two hybrid plantlets (EL570 and EL780) were chimeric and used to generate colchicine-doubled and spontaneously doubled allopolyploids. Thus, the number of putative S1 allopolyploids is 2 greater than the number of hybrid plants (see text). Allopolyploidy was assessed using molecular markers, flow cytometry, pollen fertility, and morphological characteristics. Allopolyploid S0 plants were self pollinated to generate S1 progeny, and eight to 12 plants were grown from each family for a bulk analysis.
Because both parental lines were derived from double-haploid plants, we expected that all fragments detected within the genomes of TO1000C and IMB218A would also be detected within the progeny representing the 49 S0 allopolyploids. Except for a few cases, this expectation was fulfilled. Out of the 54,383 expected parental fragments (54,635 expected minus 252 unreadable fragments), we observed 54,349 (99.94%), within the polyploids (Table I). Figure 2A shows a typical additive pattern of fragments within the diploid and polyploid lines. Figure 2, B to D, illustrates the rare situations (0.06%) where parental genome-specific restriction fragments were not inherited as expected. Among the 34 cases in which a parental fragment was lost (Table I), all 34 were lost from the B. rapa genome (0.18% of all B. rapa-specific fragments).
Table I.
Summary of B. oleracea and B. rapa fragment changes among the amphidiploid lines as compared with diploid progenitors
Total expected count is the number of polyploid fragments that are expected if fragments are conserved between the diploid and polyploid genomes. Note that because fragments are counted across all three enzymes, a single deletion and or insertion event may produce several fragment changes (see “Results”). The number of parental fragments lost may be greater than the number of nonparental fragments gained (given in parenthesis) because one genotype lost two parental fragments and simultaneously gained only one novel fragment for a particular probe.
| Fragments Present in Both Parents
|
Fragments Specific to B. oleracea
|
Fragments Specific to B. rapa
|
Novel Fragments
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Expected | Total Observed | Loss (No Gain) | Loss (Gain) | Total Expected | Total Observed | Loss (No Gain) | Loss (Gain)b | Total Expected | Total Observed | Loss (No Gain) | Loss (Gain) | Gain | |
| HindIII, EcoRI, and DraI | 12,436 | 12,436 | 0 | 0 (0) | 22,584 | 22,584 | 0 | 0 (0) | 19,363 | 19,329 | 4 | 30 (30) | 16 |
| HpaII | 3,143 | 3,140 | 3 | 0 (0) | 7,266 | 6,949 | 302 | 15 (14) | 6,258 | 6,031 | 209 | 18 (17) | 38 |
| MspI | 5,194 | 5,194 | 0 | 0 (0) | 8,132 | 6,947 | 0 | 2 (2) | 5,928 | 5,928 | 0 | 0 (0) | 2 |
Figure 2.

Comparisons of EcoRI, HindIII, and DraI fragments among allopolyploids and their diploid progenitors. Southern-blot analysis of genomic DNA restricted with EcoRI, HindIII, and DraI revealed four types of relationships between S1 B. napus genomes and their diploid progenitor genomes, B. rapa and B. oleracea. Each EL lane contains a bulked sample of DNA extracted from eight to 12 individual S1 plants. A, Hybridization of EcoRI-restricted DNA with pW212 shows that the progeny of the S0 synthetic allopolyploids have the same fragments as the diploid progenitors. Two of three polymorphic fragments are shown. B, Hybridization of HindIII-restricted DNA with pW220 shows that one line, EL600B, has lost a parental fragment. No novel fragment appears. All five fragments observed on the Southern blot are shown. C, Hybridization of pW241 to DNAs restricted with HindIII shows a parental fragment is absent from line EL9100A and a novel HindIII fragment appears. Two of three parental polymorphic fragments observed on the blot are shown. D, Hybridization of pW241 to DNAs restricted with HindIII shows that all donor genome fragments are present and there is a novel fragment in two lines, EL2700A and EL3400A. Two of three parental polymorphic fragments are shown.
All but three of the 34 parental fragment changes across the population of nascent polyploids are due to insertions or deletions (indels), and these were not evenly distributed across loci. Two or more restriction digests that detected missing parental fragments indicated an indel. Sometimes, digestion with the third restriction enzyme revealed all parental fragments. In this case, we observed the quantitative loss of the hybridization intensity of one fragment and the increase in the hybridization intensity of another parental fragment (i.e. pX124, HindIII fragments; data not shown). The same change occurred independently 14 times at a locus detected by pX124 (Table II, multilocus genotypes [MGs] 2 and 4). Other changes were rare. A large deletion occurred one time within line EL600B (MG 6) within a sequence similar to pW220 (Table II; Fig. 2B). A small deletion occurred one time in a sequence similar to pW241 in EL9100 (MG 5). Three fragment patterns could not be explained by indels. Probe pW241 detected a novel HindIII fragment in EL3400A and EL2700A (Table II, MG 3 and MG 4; Fig. 2D), but hybridization of DNA restricted by all other enzymes showed parental types. In addition, EL9100A had a pX124 DraI fragment loss that was not accompanied by the loss of any other parental fragments (Table II, MG 5). These fragments may have been generated by star activity or base pair substitution within the recognition sequence. Because of genome redundancy and a lack of knowledge of the restriction sites overlapped by a single probe at a single locus, we cannot tally the proportion of loci that changed among the polyploids. A very conservative estimate is that each probe detected a single locus in each parental genome. Thus, the maximal frequency of indels per locus is 0.2% (16 changes/[49 lines × 2 loci × 76 probes]). Two-thirds of all probes were significantly similar to predicted genes within the Arabidopsis (Arabidopsis thaliana) genome, including the probes pW241, pW220, and pX124.
Table II.
Characterization of six restriction fragment length polymorphism multilocus genotypes observed in the S1 progeny of S0 plants for EcoRI (E), HindIII (H), and DraI (D) enzymes
+, Expected fragment detected in DNAs surveyed by a specific probe/enzyme combination. In the case of a nonparental fragment (n), a fragment is absent within genotypes with the plus symbol and present within genotypes with the minus symbol; for all other rows, a minus sign indicates that the bulk sample of S1 progeny does not contain this fragment. Expected, Expected fragments for these probe/enzyme combinations. DNAs surveyed with all other probe/enzyme combinations revealed parental fragments (data not shown). No. PL, Number of polyploid lines.
| Polymorphic Probes
|
Enzyme-Fragment
|
Expected
|
Multilocus Genotype
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | Totals | |||
| No. PL: 32 | 13 | 1 | 1 | 1 | 1 | 49 | |||
| pW241 | H-1 | + | + | + | + | + | − | + | |
| pW241 | H-2n | + | + | + | − | − | + | + | |
| pW241 | H-3n | + | + | + | + | + | − | + | |
| pW241 | D-1 | + | + | + | + | + | − | + | |
| pW241 | D-2n | + | + | + | + | + | − | + | |
| pW220 | E-1 | + | + | + | + | + | + | − | |
| pW220 | H-1 | + | + | + | + | + | + | − | |
| pW220 | D-1 | + | + | + | + | + | + | − | |
| pX124 | E-1 | + | + | − | + | − | + | + | |
| pX124 | E-2n | + | + | − | + | − | + | + | |
| pX124 | H-1n | + | + | − | + | − | + | + | |
| pX124 | D-1 | + | + | − | + | − | − | + | |
| pX124 | D-2n | + | + | − | + | − | + | + | |
| Changes: | |||||||||
| Gain | 0 | 1 | 1 | 2 | 0 | 0 | 16 | ||
| Loss | 0 | 0 | 0 | 0 | 1 | 3 | 4 | ||
| Loss/Gain | 0 | 2 | 0 | 2 | 2 | 0 | 30 | ||
| Events | 0 | 1 | 1 | 2 | 2 | 1 | 19 | ||
To confirm and characterize one unique deletion, we cloned and sequenced a 3.6-kb fragment within line EL9100A (Table II, MG 5) that had a 600-bp deletion relative to its 4.2-kb B. rapa parent as identified by pW241 (Figs. 2C and 3A). Genomic DNA restricted with HindIII of approximately 3,600 bp and 4,200 bp was isolated from both EL9100A and a pooled sample of wild-type polyploid DNA, respectively. We ligated adapters onto the fragments and amplified the allele using primers specific to pW241 and to the adapters. Fragments generated from the primers at the ends of pW241 and the external adapters were the same length in both samples (Fig. 3, B and C), indicating that the deletion occurred within the sequence homologous to pW241. As expected, the internal pW241 fragment derived from EL9100A was 600 bp shorter than the parental control fragment (Fig. 3D). Nucleotide sequences of both fragments were identical at the 5′ end except for a guanine at position 491 present within the control and absent from EL9100. From the 5′ end starting at position 873, the sequence contained solely guanine-adenine dinucleotides (GpA; Fig. 3E). From the 3′ end, both large and small fragments were 99.5% identical for 381 bp. From 1,819 bp toward the 5′ end, both sequences were composed of the thymine-cytosine dinucleotide. This result demonstrates that the deletion in EL9100A occurred within a GpA/thymine-cytosine dinucleotide repeat-rich region.
Figure 3.

Characterization of deletion within allotetraploid genomic fragment. A, Graphic model of a deletion event within the allopolyploid EL9100A relative to the parental type. The dark horizontal line represents genomic DNA from the two types. H represents the HindIII recognition sites. Primers are numbered 1 to 8 and designated by a short arrow; primers external to the HindIII sites match an external adapter. Gray regions refer to areas of homology between the genomic DNA fragment and the 5′ and 3′ ends of pW241. The two slash marks in EL9100A represent the 600-bp deletion within this line. The control is a bulk sample of polyploid DNAs that did not have a deletion. Primers 2 and 4 are one set of genome-walking, gene-specific primers. Primers 1 and 3 are the second set of genome-walking, adapter-specific primers from BD Bioscience. Primers 5, 6, 7, and 8 amplify the interior of the fragment similar to pW241. B, Fragments amplified from primers 1 and 2 (flanking region 1, F1) are the same size between EL9100A (lane 1) and the control (lane 2). C, Fragments amplified from primers 3 and 4 (flanking region 2, F2), are also the same size between EL9100A (lane 1) and the control (lane 2). D, Amplification of EL9100A genomic DNA and control DNA with primers 5 and 6 (lanes 1 and 3) and primers 7 and 8 (lanes 2 and 4). Lanes 1 and 2 contain amplification products from EL9100A. Lanes 3 and 4 contain products from the control. The arrows depict the different sizes of these fragments. There is a 600-bp difference in amplification product size between EL9100A and the control for both primer pairs (compare lanes 1 and 3 and lanes 2 and 4, respectively). E, Graphic model of sequence comparison from the internal region (I) derived from EL9100A and the control. Base pairs 1 to 873 were identical between EL9100A and the control except for a single guanine indel event at position 491. The final 381 bp at the 3′ end were identical between EL9100A and the control. At position 873, numerous copies of the dinucleotide GA were present within both sequences. We sequenced a total of 70 dinucleotides at the 5′ end and 133 dinucleotides at the 3′ end within this region where the 600-bp deletion occurred. The 2,200- and 1,600-bp fragments were amplified with primers 5 and 6 as shown in plates A and D (lanes 1 and 3).
Overall, restriction fragment length polymorphism (RFLP) analyses detected very few genetic differences between parental genomes and synthetic genomes. Over 99% of expected parental fragments were inherited in an additive manner across all 49 allopolyploids (Table I), and 32 of 49 allopolyploids exhibited the expected, parental genotypes (Table II). Sixteen of the remaining 17 allopolyploid progeny each revealed a clear, single indel event. To confirm that these results were representative even if a different screening measure was applied, we also assayed the 49 synthetic genotypes and the parental lines with 30 simple sequence repeat (SSR) primer pairs using DNA from both the S0 plants and S1 pools. Among the S0 plants, the SSR primers amplified 73 different fragments (mean 2.4 and range 1–12 fragments per primer pair that were detected both within a parental genome and within a mix of parental DNA). Of the 3,577 fragments expected among all polyploids (73 codominant fragments × 49 polyploids), 250 were unreadable, but all others were the parental type. Three fragments detected by SSR primer pair snra94 in B. rapa were absent from the control mix of parental genomic DNA due to dominance of the B. oleracea allele. Analyses of the bulked progeny of each S0 plant also revealed no genetic changes. These results confirm both that the genetic structure of B. napus is highly conserved within the S0 and S1 generations and that microsatellite length variation is not a general response to hybridization or polyploidization.
Methylation Status of Loci within Synthetic Polyploids
We assayed epigenetic changes within the S0 polyploids by analyzing changes in restriction fragments generated by two isoschizomers, HpaII and MspI. B. oleracea and B. rapa were highly polymorphic for HpaII and MspI recognition sites and differed for 596 fragments. One-hundred-fourteen MspI fragments were present in both genomes, and 103 HpaII fragments were shared as well. Likely because HpaII is sensitive to internal cytosine methylation, we detected a smaller number of polymorphic HpaII fragments than MspI fragments within both B. oleracea (157 versus 172) and B. rapa (129 versus 138). A total of 73 probes were used in this analysis. The three probes that revealed indels (pW241, pW220, and pX124) were not included in the MspI/HpaII survey.
Allopolyploid lines were expected to have the same HpaII and MspI fragment pattern as both diploid parents, indicating no change in methylation status. To test this hypothesis, we made 14,060 pairwise comparisons between polymorphic parental MspI fragments and allopolyploid fragments and 13,524 pairwise comparisons between polymorphic parental HpaII fragments and fragments within the allopolyploids (Table I). Two lines, EL7900A and EL8400C, were missing parent-specific MspI fragments (Table I). Both MspI changes involved the loss of a single MspI fragment derived from B. oleracea and the gain of a novel fragment within a pX144-similar genomic segment. This sequence also changed in our previous analysis of synthetic Brassicas (EC3E12 [Song et al., 1995]). Two novel MspI fragments also appeared within two lines.
Hybridization of DNAs restricted with HpaII showed that cytosine CpG methylation changes between polyploid and diploid genomes were much more frequent than CpCpG changes, and fragments often did not have expected hybridization patterns. In total, 544 of 13,524 parent-specific HpaII fragments present within either B. oleracea or B. rapa were not detected within the polyploids (Table I). A loss of a fragment due to methylation or demethylation may be expected to co-occur with the appearance of a novel fragment. However, within 94% (511/544) of the cases, a loss of a species-specific parental fragment within the allopolyploids was not accompanied by the gain of a novel fragment (Table I). Novel fragments were rare, likely because of genome redundancy (Lukens et al., 2004), and an increased copy number of an existing fragment was apparent within several blots. For example, in Figure 4A, methylation of a B. rapa restriction site has caused a greater hybridization signal of an existing 2-kb fragment among the lines with missing fragments than among those lines without a missing fragment. The number of lost HpaII fragments did not differ greatly between the parental genomes. Out of 7,266 and 6,258 pairwise comparisons, B. oleracea fragments were absent in 302 (4.2%) cases and B. rapa fragments were absent in 209 (3.3%) cases, respectively (Table I). In 38 cases, a novel HpaII restriction fragment appeared within the polyploid sample and was not accompanied by loss of a parental fragment (Table I; Fig. 4B). This pattern again suggests that genome redundancy may have masked the loss or gain of a restriction site (Fig. 4B).
Figure 4.

Changes in the restriction fragment pattern of HpaII digests between parental and polyploid genomes. Observed fragment patterns on Southern blots of HpaII restriction digests from B. oleracea, B. rapa, and pooled DNAs from the progeny of individual S0 polyploids (right). All S0 progeny DNAs had parental MspI restriction fragments. Models of the putative methylation changes that generated the observed hybridization pattern are depicted to the left. The two horizontal rectangles associated with a single genome represent homoeologous loci. Each vertical line represents a HpaII recognition site, the sequence CCGG. A solid vertical line represents a site that does not have cytosine C5 methylation within the CpG dinculeotide sequence. A vertical line with a methyl group represents a CpG methylated site. A, Loss of a B. rapa-specific fragment (1 kb) is not accompanied by the gain of a novel fragment, or the novel fragment comigrates with preexisting fragments and cannot be unambiguously scored. Here, hybridization of HpaII-restricted DNA with pX144 shows a high-Mr fragment (2 kb) has a greater hybridization intensity among lines with a missing 1-kb fragment (EL4400B, EL6800A, EL7300A, and EL7400A) than among lines without the missing fragment. This pattern suggests that de novo methylation occurred within the polyploid A genome. All parental fragments are shown. B, Gain of a novel fragment within allopolyploids as seen by hybridization of HpaII-restricted DNA with pW157. This pattern may suggest that demethylation occurred at one of two or more homoeologous loci. All parental fragments are shown. C, Loss of a parental fragment and gain of a larger, novel fragment within the allopolyploid as seen by hybridization of HpaII-restricted DNA with pW125. De novo cytosine methylation within the S0 parent at a site unmethylated within the diploid B. rapa progenitor explains this pattern. All parental fragments are shown.
Six percent (33/544) of the specific parental HpaII fragment losses were accompanied by the gain of a novel fragment (Table I; Fig. 4C). With this class, we can unambiguously infer whether genes tend to be hyper- or hypomethylated within the polyploid as compared to the diploid. A higher level of CpG methylation within the polyploid genome at a site that was unmethylated within the diploid progenitors would yield novel fragments larger than the missing parental fragments. Sixteen of 17 novel fragments within the B. rapa genome of the allopolyploids were larger than the fragments that were lost, a pattern that differs significantly from the expectation of equality (P < 0.0001). In contrast, within the 14 novel fragments identified in B. oleracea comparisons, eight fragments were larger than the fragments lost, while six novel fragments were smaller. This finding is not significantly different from the expectation of equality (P < 0.42). Thus, de novo CpG methylation primarily occurs within B. rapa homoeologs; while both de novo methylation and demethylation occur within B. oleracea homoeologs.
The distribution of CpG methylation changes across loci shows that specific loci repeatedly underwent epigenetic modification in the S0. Although each of the 73 RFLP probes detected a mean of 7.8 differences in HpaII restriction fragments between the polyploid and diploid genomes, 38 probes (52%) did not reveal any changes across the 49 polyploid lines, and 61 probes (84%) detected fewer changes than the mean (Fig. 5A). In contrast, pX146 detected 89 changes within the polyploid lines. Eight additional markers, EZ3, pW216, pW219, pW134, pW149, pW170, pX126, and pX128, detected more than 35 changes each. Unexpectedly, noncoding sequences were not over-represented among the nine markers that each accounted for more than 35 changes. The regions of the Arabidopsis genome similar to Brassica probe sequences are given in Supplemental Table I. Brassica probe sequences had only weak similarity to DNA repeats (Supplemental Table II). The variability of genomic changes across probes was much greater than expected (Fig. 5A; χ2 = 18.91, P < 0.0001). This result clearly shows that DNA sequence identity has a strong influence on the number of changes observed.
Figure 5.

Distribution of HpaII methylation changes between S1 progeny and parental genomes across lines and across probes. A, Frequency distribution of the number of changes per probe. The distribution is overdispersed relative to the expected Poisson distribution (dotted line). B, Frequency distribution of the number of changes per line. The distribution is underdispersed relative to the expected Poisson distribution (dotted line).
The frequency of epigenetic changes was remarkably uniform across lines (Fig. 5B), consistent with the finding that a small number of loci are preferentially, but not invariably, targeted for epigenetic modification. Methylation changes affected each of the 49 polyploid lines, and each line had eight to 17 fragment changes relative to the diploid progenitors, corresponding to 1.4% to 2.9% of total changes. The mean number of changes per line was 11.61, with a variance of 4.56. The variability of genomic changes across polyploids was much lower than expected (χ2 = 18.91, P < 0.0001).
Polyploidization and Colchicine Effects on Genome Structure
Based on the analysis of lines derived from S0 plants, we suggest that interspecific hybridization and allopolyploidization are associated with rare, targeted indels and more frequent, targeted epigenetic changes. However, indels and epigenetic changes could occur at a similar rate within a population of diploids, could be caused by colchicine treatment, or could be caused by ovule culture. In addition, a B. napus population that had not undergone recent resynthesis may have similar levels of change. To address these questions, we included three controls. First, to assay the frequency of genetic and epigenetic changes between diploids and within the ovule culture, two separate diploid parents were surveyed at all loci. One plant underwent ovule culture; the other was generated from seed. Two TO1000C diploids were identical at all loci. Two IMB218A diploids differed for three novel fragments (at loci detected by pW188 [Hpa II], pW221 [Hpa II], and pW237 [Msp I]). Second, to estimate colchicine effects, we compared two, resynthesized allopolyploid genotypes, EL5700P and EL7800P, that occurred spontaneously (i.e. without colchicine treatment) with resynthesized genotypes EL5700 and EL7800, that were generated by colchicine treatment. Between EL5700 and EL5700P, 1,924 fragments were identical and no fragments differed. Between EL7800 and EL7800P, 1,911 fragments were identical and six fragments differed. Both the diploid pairs and colchicine-treated plants and noncolchicine-treated plants from the same genotype were significantly more similar than were different allopolyploids to each other (α = 0.05). Only 48 of 1,176 (0.04) possible pairwise comparisons between polyploid lines had fewer than six fragment differences and only 1 of 1,176 (<0.001) possible pairwise comparisons had no differences. Finally, to test the possibility that cytosine methylation may vary naturally within the polyploid B. napus, we analyzed DNA of 50 individuals from two mapping populations, one derived from natural × natural and the other derived from natural × resynthesized B. napus parents (Udall et al., 2005). Four probes that detected many novel HpaII restriction fragments within the resynthesized allopolyploids (pW134, pX126, pX128, and pX146) were hybridized to HpaII- and MspI-digested DNAs of the mapping population. These probes detected two and three novel fragments among the 50 individuals of each of the two mapping populations. Thus, the recently derived allopolyploids had over 8 times the number of novel fragments than did the allopolyploids from the mapping populations. In summary, epigenetic change occurred significantly less frequently within control diploids, colchicine-treated plants, and control polyploids than among the de novo polyploid lines.
DISCUSSION
Allopolyploidy is ubiquitous among plants (Levin, 1983) and, in many cases, involves large-scale DNA deletions and alterations in cytosine methylation patterns that may cause gene silencing as well as developmental changes (for review, see Wendel, 2000; Liu and Wendel, 2002, 2003; Osborn et al., 2003; Adams and Wendel, 2005). These changes may be highly targeted responses to hybridization and polyploidization or changes may be the result of a less precisely regulated or repeatable process. Studies to date of a small number of individuals have found evidence for both directed and variable changes. In this study, we analyzed a large number of de novo polyploids to investigate the frequency and distribution of the genomic effects that accompany the fusion and doubling of two genomes.
Genetic Changes in Brassica S0 Allopolyploids
Genetic changes were rare and targeted in Brassica S0 allopolyploids. Of the 76 RFLP probes, 73 detected parental fragments, and the frequency of indels was at most 0.2% per locus. Sixteen indels explained all but three of the 80 fragment differences between the S1 progeny of 49 synthetic S0 polyploids and parental genomes (Table I). One probe (pX124) revealed 14 indels across independently derived individuals, indicating that sequences homologous to this probe are targeted for change (Table II). The estimated frequency of genetic change in S0 Brassica allopolyploids is higher than some species but lower than the frequency of DNA indels from allopolyploids generated within and between the genera Aegilops and Triticum (Ozkan et al., 2001; Shaked et al., 2001). Polyploid genomes such as Gossypium spp. have very few, if any, genetic changes at low-copy sequences compared to donor genomes (Liu et al., 2001), and Spartina polyploids show infrequent changes within both low- and high-copy sequences (Baumel et al., 2001; Ainouche et al., 2004). In contrast, within wheat allotetraploid genomes, Shaked and colleagues (2001) found that between 6.7% and 11.8% of parental fragments had disappeared by the S0 generation. DNA deletions within wheat polyploids are mostly in noncoding sequences but include some coding sequences as well (Liu et al., 1998a, 1998b; Kashkush et al., 2002). Although one may expect indels to be more common among noncoding than coding sequences in Brassica, we found that probes similar to predicted genes did not reveal a smaller number of changes than probes without similarity to predicted genes. All genetic changes occurred within the B. rapa genome and may be the result of maternal cytoplasmic effects (e.g. Dahleen, 1996).
Several mechanisms could cause the elimination of parental sequences within polyploid genomes, including gene conversion-like events (Wendel et al., 1995), transposon excision or insertion (Baumel et al., 2002; Zhao et al., 1998), chromosomal breakage and repair, and mitotic or meiotic crossing over between direct repeats that flank a sequence. Probe pW220 revealed a large deletion, while pX124 and pW241 revealed small deletions or insertions. One small deletion that we investigated further (pW241 within line EL9100A) revealed the loss of a GpA/CpT microsatellite-like motif. Microsatellite-like sequences may decrease in length by recombination or polymerase slippage (Richard and Paques, 2000; Eckert et al., 2002). Nonetheless, not one of 3,327 microsatellite amplification fragments from polyploids in our study differed in size from the parental fragment. Thus, deletions of microsatellite-like repeats are not a hallmark of interspecific hybridization of polyploidization per se in Brassica.
Frequency and Genome Specificity of Epigenetic Alterations between S0 Brassica Allopolyploids and Their Parents
The survey of the population of 49 resynthesized B. napus plants for CpG and CpCpG methylation changes using the 73 RFLP probes that did not show indels revealed 622 total fragment differences between parental and polyploid genomes (Table I). CpCpG methylation changes were very rare, while CpG methylation changes were relatively frequent. In only two cases out of 14,060 pairwise comparisons (0.01%) was a parent-specific MspI fragment lost within a polyploid genome, and only four novel fragments appeared within the polyploids. The maintenance of CpNpG methylation in Arabidopsis involves chromomethylase3 (CMT3; Bartee et al., 2001; Lindroth et al., 2001) and methyltransferases Drm1 and Drm2 (Cao and Jacobsen, 2002). Thus, our results suggest that the targeting of CpNpG methyltransferase remains largely the same in hybrid and polyploid genomes as in parental genomes. In contrast, 544 parent-specific HpaII fragments differed between diploids and polyploids at loci that were unchanged in MspI digests (Table I). CpG methylation is maintained by a subfamily of methyltransferases similar to MET1 (Finnegan et al., 1996; Ronemus et al., 1996), indicating that the targeting of CpG methyltransferase changes within the polyploids. We can only estimate the proportion of loci that were affected by genomic CpG methylation changes because HpaII restriction sites are unknown. Nonetheless, we may assume that all HpaII fragment differences between a specific parent and polyploid detected by a single probe are due to a single methylation change. If we assume that each probe detects a single locus in each parent, then for HpaII, 518/(2 × 49 × 73), or 7% of the loci are affected. Because probes typically detect several loci within the diploid progenitors (e.g. Lagercrantz, 1998; Lukens et al., 2003), this percentage is high. A low-bound estimate of the number of loci modified within the S0 would be 2.4% (518/21,462), assuming each probe detected six loci within the allopolyploid instead of two (e.g. Lagercrantz, 1998).
Models that take into account the redundancy of the Brassica genome can explain many of the HpaII fragment changes observed within the polyploids. In most cases, the loss of parental fragments did not accompany the gain of a nonparental novel fragment. Out of 544 parent-specific HpaII fragments absent from S0 progeny, 511 (94%) did not co-occur with the gain of a novel fragment. Instead, lost fragments frequently generated an additional fragment that was the same size as an existing fragment. In 38 cases, a nonparental fragment appeared and did not co-occur with the loss of a parental fragment (Table I). Here, it is likely that the polyploid genome had homoeologous loci that generated fragments of the same length. At least one locus was maintained in the parental methylation state after modification of another locus, creating a novel fragment without the loss of a parental fragment. This interpretation is consistent with the highly replicated genome of the Brassicas (e.g. Lukens et al., 2004). Similarly, Liu et al. (1998a) investigated the hybridization pattern of 43 coding sequences across DNAs from nine newly synthesized wheat amphiploids of different ploidy levels. In their study, the most frequent change was the loss of parental fragments within the allopolyploid, likely due to changes in DNA methylation. The appearance of a novel fragment or the concurrent loss of a parental fragment and gain of a novel fragment occurred rarely. Nonetheless, we cannot eliminate other mechanisms. For example, differences in methylation uniformity between diploid parents and polyploid offspring would explain the loss of a parental fragment and the failure to gain a novel fragment within the polyploid (e.g. Lund et al., 1995; Xiong et al., 1999; Yamada et al., 2004).
In 6% (33/544) of the cases in which a parent-specific fragment was absent from the polyploid, the disappearance of parental fragments coincided with the appearance of a novel fragment (Fig. 4C). This pattern arises because a CpG site that was fully methylated within the diploid progenitor became demethylated or because a site that was demethylated within the diploid progenitor became fully methylated. Within this class, B. rapa loci were preferentially methylated in the polyploid, whereas B. oleracea loci were both methylated and demethylated. De novo cytosine methylation requires both de novo DNA methyltransferases and RNA-silencing genes (Chan et al., 2004). Thus, perhaps B. rapa-specific repeats are transcribed within the nascent hybrid or polyploid, thereby causing chromatin level silencing of B. rapa genomic sequences.
Methylation Changes Occur at Specific Loci and Are Largely Uniform among Lines
In our large population of resynthesized polyploids, we had the sample size to detect rare epigenetic changes and the distribution of common changes. One finding from this analysis is that CpG methylation changes occurred at a high frequency among some loci (nine probes detected >35 changes) and did not occur among others (38 probes detected no changes). Using smaller populations, several authors also noted the repeatability of epigenetic changes among allopolyploids. For example, Chen and Pikaard (1997b) showed that natural and newly resynthesized Brassica polyploids had the same pattern of nucleolar dominance. Shaked et al. (2001) also found that the same sequence changes occurred in a methylation-sensitive amplified fragment length polymorphism analysis of three different amphiploids generated from the same parents. In our population, despite the strong bias for specific loci, it is nonetheless important to note that not one locus showed the same change among all the polyploids. These differences in epigenetic changes may explain both phenotypic variation and variation in gene expression within allopolyploids (Comai et al., 2000; Wang et al., 2004).
Our finding that CpG methylation changes are relatively rare (between 2% and 7%) is consistent with previous analyses of polyploid gene expression and the known targets of methylation. Within maize (Zea mays) polyploids, the expression of most genes maintains a similar per genome level within the polyploid as in the diploid (Guo et al., 1996), suggesting there is no widespread epigenetic silencing. Likewise, expression differences of Arabidopsis genes and wheat genes in polyploid genomes are relatively infrequent (Comai et al., 2000; Lee and Chen, 2001; Kashkush et al., 2003). These studies suggest that methylation changes are more likely due to local or cryptic heterochromatin changes caused by flanking sequence repeats rather than to large-scale heterochromatin changes (Martiennsen and Colot, 2001; Lippman et al., 2004). Other aspects of our findings are consistent with this conclusion. The methylation changes reported here do not extend over large, chromosomal intervals. The 35 probes that detected changes within the population were not clustered together within the genome, as measured by comparing the position of Arabidopsis sequences homologous to Brassica probe sequences (data not shown) whose linkage relationships are often highly conserved between the species (Lukens et al., 2003). In addition, a very similar number of methylation changes occurred within each lineage of Brassica allopolyploids (Fig. 5B). This finding is due in part to tightly controlled, locus-specific methylation occurring repeatedly across lines.
Timing of Genetic and Epigenetic Changes following Polyploidization
Several molecular genetic analyses of polyploids have investigated the genomic changes that occur in the initial and subsequent generations following the formation of a small number of allopolyploids. A large number of differences between the parental and polyploid genomes in wheat are detectable within the interspecific F1 hybrid (Shaked et al., 2001), and differences accumulate between the F1 and S3 generations (Ozkan et al., 2001). A similar pattern of genomic change has been found in triticale, with a great degree of the sequence modification/elimination occurring in the F1 hybrid and additional sequence modification/elimination over subsequent generations after polyploidization (Ma et al., 2002, 2004). These types of rapid changes could affect the processes of both chromosome diploidization and genetic diploidization by which duplicate genes are silenced or differentially regulated (Feldman et al., 1997).
Both the genetic and epigenetic changes reported here very likely occurred within the S0 generation, perhaps prior to polyploidization. In this analysis, we examined changes within the pooled S1 progeny of self-pollinated, individual S0 plants. Because RFLP and SSR fragments are codominant, the fact that a parental fragment was absent from the bulk of S1 plant DNA indicates that the modification had occurred either within the S0 genome or is a programmed response that occurs many times within or after meiosis of the S0 plant. The most parsimonious explanation is that each modification occurred within an S0 plant prior to flowering. One finding further suggests that most epigenetic changes observed within the S0 progeny had occurred in the S0 hybrid state prior to genome doubling. In our analysis, progeny of two S0 hybrid plants that had one sector treated with colchicine and one sector that spontaneously doubled had either a few or no differences. Prior analyses have shown that hybridization may influence genome epigenetic status (Xiong et al., 1999; Shaked et al., 2001). The pooled sample of S1 plants may differ because of large deletions or homoeologous recombination events that occurred within S0 meiosis (e.g. Parkin et al., 1995). These events would not be detected here but may be visible in subsequent generations.
By investigating a large number of polyploids, we show that the fusion of distinct genomes in a common nucleus is characterized by targeting specific loci for both genetic and epigenetic change and by maintaining the methylation status of other loci. Interestingly, this targeting is not exact. Many loci are altered infrequently and those loci that are commonly altered are not invariably so. We have previously reported both shared and variable molecular and phenotypic characters in smaller populations of synthesized polyploid Brassicas in later generations (S1–S4 [Song et al., 1995]; S3–S6 [Pires et al., 2004]). The molecular changes we report here (in the S1) contribute to both the stabilization and diversity of polyploid genomes and help explain the remarkable success of polyploids within agriculture and the environment. Analysis of these 49 allopolyploid lines at later generations will allow more detailed comparisons to other studies and provide a more complete picture of the dynamics of genome change in new allopolyploids.
MATERIALS AND METHODS
Plant Material
Resynthesized Brassica napus allopolyploid plants (CCAA) were developed by hybridizing plants of Brassica oleracea (CC) genotype TO1000 DH 3, herein TO1000C, and plants of Brassica rapa (AA), genotype IMB 218 DH 1, herein IMB218A. The two parental genotypes (TO1000C and IMB218A) were double-haploid lines of the inbred, self-compatible, and rapid flowering lines of B. oleracea (TO1000) and B. rapa (IMB218). Double-haploid plants were developed by the selection of single plants from microspore culture and the doubling of their chromosomes. Both double-haploid plants were self pollinated, and six male S1 B. rapa plants of IMB218A and 10 female S1 B. oleracea plants of TO1000C were used as parents for interspecific crosses. Siliques were harvested 6 to 7 d after pollination and sterilized siliques were incubated in darkness on White's basal media supplemented with casein at 21°C for 14 d. Embryos were then removed from the siliques, transferred to Murashige and Skoog media, and incubated at 21°C in 16-h-light/8-h-dark cycles. After approximately 21 d, plantlets were transplanted to sterile soil. Forty-nine unique hybrid (CA) plants were generated in this fashion. Embryos from two parental diploid controls were cultured and handled in the same fashion. Separate diploid controls were generated from seed.
A total of 51 allopolyploid plants were generated from the 49 hybrid plants using colchicine (Fig. 1). Two of the 49 plants appeared to be mosaics of allopolyploid (CCAA) and hybrid (CA) tissues prior to colchicine treatment. Cuttings were taken from the two regions of both plants and rooted. The hybrid cuttings were subsequently treated with colchicine, generating the allopolyploid plants EL5700 and EL7800. The allopolyploid cuttings generated the plants EL5700P and EL7800P. The two spontaneously doubled lines (EL 5700P and EL 7800P) were excluded from the overall analysis, but used in a later comparison. Of the remaining 47 hybrid plants, eight spontaneously doubled and 39 were treated with colchicine to produce allopolyploid plants. Plants were treated by soaking roots in 0.3% colchicine for 2 h.
To compare rates of cytosine methylation between a control group of polyploids and de novo polyploids, we determined cytosine methylation differences within two segregating populations of double-haploid lines. The Udall population was a cross between a natural B. napus (P1804) and a resynthesized B. napus (JU504). The Quijada population was a cross between two natural B. napus lines (P1804 and RV128). The development of these populations for genetic mapping was described previously (Udall et al., 2005).
Southern Blotting, Genome Walking, and Microsatellite Fragment Amplification
Genomic DNA was extracted from leaf tissue of young plants using the cetyl-trimethyl-ammonium bromide method (Kidwell and Osborn, 1992). For one SSR survey, DNA was extracted from the 51 S0 plants. For a RFLP and a second SSR survey, DNA was extracted from a bulk of eight to 12 S1 plants derived from self pollinating each S0 plant. Thus, the samples represented the parental S0 plant. DNA was digested with restriction endonucleases HpaII and MspI (Promega) for 8 h at 37°C to survey methylation changes. HpaII is sensitive to methylation of either cytosine at its recognition site 5′-CCGG, whereas the isoschizomer MspI is sensitive to methylation of only the external cytosine. DraI, EcoRI, and HindIII were used to assay for indels. EcoRI cleavage is inhibited by cytosine methylation within its recognition sequence GAATTC. According to the manufacturer, HindIII is insensitive to CpG and CpNpG cytosine methylation, but there have been reports of sensitivity (Huang et al., 1982). Southern blots were probed with 76 Brassica cDNA (denoted pX_) and genomic DNA (denoted pW_) fragments used in previous studies (Ferreira et al., 1994; Butruille et al., 1999; Udall et al., 2005). Southern blotting, probe radiolabeling, and membrane hybridization are as described by Ferreira et al. (1994). The probe nomenclature of Parkin et al. (1995), Sharpe et al. (1995), and Udall et al. (2005) was used.
Genome walking was utilized to isolate both upstream and downstream genomic DNA fragments homologous to probe sequences (Universal GenomeWalker kit; BD Biosciences). Amplification products were sequenced using fluorescence-based cycle sequencing and electrophoresis separation (Big Dye Sequencing kit and ABI Prism 377; ABI).
Thirty A or C genome- and chromosome-specific SSR primer pairs (developed by D. Lydiate and A. Sharpe, personal communication) were used to assay genome structure within the synthetic allopolyploids. Fragments were amplified in 10-μL reactions with 1× buffer, 0.2 mm deoxyribonucleotide triphosphate, 0.3 μm of both primers, 2.0 mm MgCl2, 0.5 units Taq polymerase, 40 ng genomic DNA, and 0.1 μL of [α-33P]dCTP. The amplification of fragments by these primers confirmed the chromosomal complement of the 51 putative allopolyploids. Each marker survey was performed with a negative control (a reaction without DNA), two replicates of reactions with parental DNA, and a sample with both parental DNAs within the same reaction. All reactions that did not generate a fragment of the expected size or that generated an unclear fragment were repeated for confirmation. The PCR conditions were as follows: 95°C for 10 min; eight cycles of 94°C for 30 s, 50°C for 30 s, 72°C for 50 s; 27 cycles of 89°C for 30 s, 50°C for 30 s, and 72°C for 50 s; and ending at 72°C for 10 min. Labeled SSR fragments were separated on a 4% acrylamide sequencing gel for 3 h at 90 V and 11 mA. Fragments were visualized by film (Kodak Biomax MR-1 film) after exposure for 1 to 3 d. The expected DNA content of one polyploid and both diploid parents was also confirmed with flow cytometry.
Fragment Comparison and Statistical Analyses
Differences in restriction fragments and SSR amplification products between parental, diploid genomes, and the DNA of S1 allopolyploid plants were tallied to estimate the number of genetic and epigenetic changes between the parental and S0 genomes. The absence of a parental fragment in the allopolyploid was counted as a fragment loss. The presence of a fragment within allopolyploids that was absent from both diploid parents was counted as a gain (as in Table I). Hybridizations of HindIII, DraI, and EcoRI restriction digests were used to identify indels (see “Results”). To estimate the number of epigenetic changes, we compared parental and allopolyploid HpaII and MspI fragments among loci with no evidence of genetic changes. Differences between the S1 allopolyploids and parental fragments for MspI fragments are indicative of changes in CpCpG cytosine methylation. Differences between HpaII fragments at loci that do have parental MspI fragment types are indicative of changes in CpG cytosine methylation.
All analyses were done with the statistical software R (R Development Core Team, 2004). We used the prop.test function of R to determine the probability that the difference between two counts was significantly greater than zero. For example, we tested whether probes with high similarity to coding sequences within the Arabidopsis (Arabidopsis thaliana) genome detect significantly more differences between allopolyploid and parental genomes than probes with little similarity to coding sequences. A dispersion test was used to evaluate whether the number of epigenetic changes per marker or the number of epigenetic changes per line follow a Poisson distribution. Both of these measures are expected to be Poisson distributed in the first case if markers are homogeneous and cytosine methylation changes are independent and, in the second case, if lines are homogeneous and cytosine methylation changes are independent. Under the null hypothesis, the ratio 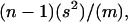 where n is the number of counts, s2 is the variance estimate, and m is the estimate of the mean, will follow a chi-squared distribution with n − 1 degrees of freedom.
where n is the number of counts, s2 is the variance estimate, and m is the estimate of the mean, will follow a chi-squared distribution with n − 1 degrees of freedom.
Sequences from RFLP probes have been deposited in GenBank (accession nos. DT469117–DT467171 for the pX probes and CZ 906364–CZ906485 for the pW probes). Sequences were compared with the RefSeq GenBank records NC_003070.4 (ch 1), NC_003071.3 (ch2), NC_003074.4 (ch3), NC_003075.3 (ch4), and NC_003076.4 (ch5) using BLASTn with default parameters. Sequences were also compared with Arabidopsis and Brassica transposable elements downloaded from The Institute for Genomic Research Brassicaceae Repeat Database. All bioinformatics analyses were done with Bioperl.
Supplementary Material
Acknowledgments
We thank Dr. Nicole Riddle and two anonymous reviewers for their comments on this manuscript.
This work was supported by the National Science Foundation Plant Genome Program (grant no. 0077774 to T.C.O.), and by the Natural Sciences and Engineering Research Council of Canada and the Ontario Ministry of Agriculture and Food (grants to L.L.).
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) are: Lewis N. Lukens (llukens@uoguelph.ca) and J. Chris Pires (piresjc@missouri.edu).
The online version of this article contains Web-only data.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.105.066308.
References
- Adams KL, Wendel J (2005) Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8: 135–141 [DOI] [PubMed] [Google Scholar]
- Ainouche ML, Baumel A, Salmon A (2004) Spartina anglica: a natural model system for studying early evolutionary changes that affect allopolyploid genomes. Biol J Linn Soc 82: 475–484 [Google Scholar]
- Bartee L, Malagnac F, Bender J (2001) Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev 15: 1753–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumel A, Ainouche M, Kalendar R, Schulman A (2002) Retrotransposons and genomic stability of the young allopolploid species Spartina anglica C.E. Hubbard (Poaceae). Mol Biol Evol 19: 1218–1227 [DOI] [PubMed] [Google Scholar]
- Baumel A, Ainouche M, Levasseur JE (2001) Molecular investigations in populations of Spartina anglica C.E. Hubbard (Poaceae) invading coastal Brittany (France). Mol Ecol 10: 1689–1701 [DOI] [PubMed] [Google Scholar]
- Butruille DV, Guries RP, Osborn TC (1999) Linkage analysis of molecular markers and quantitative trait loci in populations of inbred backcross lines of Brassica napus L. Genetics 153: 949–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Jacobsen S (2002) Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci USA 99: 16491–16498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SWL, Zilberman D, Xie Z, Johansen LK, Carrington JC, Jacobsen SE (2004) RNA silencing genes control de novo DNA methylation. Science 303: 1336 [DOI] [PubMed]
- Chen ZJ, Pikaard C (1997. a) Epigenetic silencing of RNA polymerase I transcription: a role for DNA methylation and histone modification in nucleolar dominance. Genes Dev 11: 2124–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Pikaard C (1997. b) Transcriptional analysis of nucleolar dominance in polyploid plants: biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc Natl Acad Sci USA 94: 3442–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B (2000) Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12: 1551–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahleen LS (1996) Molecular marker analysis of hypoploid regenerates from cultures of barley × Canada wild rye. Genome 39: 367–372 [DOI] [PubMed] [Google Scholar]
- Eckert KA, Mowery A, Hile SE (2002) Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite frame-shift error rates using a forward mutation assay. Biochemistry 41: 10490–10498 [DOI] [PubMed] [Google Scholar]
- Feldman M, Liu B, Segal G, Abbo S, Levy A, Vega J (1997) Rapid elimination of low-copy sequences in polyploid wheat: a possible mechanism for differentiation of homoeologous chromosomes. Genetics 147: 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ME, Williams PH, Osborn TC (1994) RFLP mapping of Brassica napus using F1-derived doubled haploid lines. Theor Appl Genet 89: 615–621 [DOI] [PubMed] [Google Scholar]
- Finnegan EJ, Peacock WJ, Dennis E (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci USA 93: 8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Davis D, Birchler J (1996) Dosage effects on gene expression in a maize ploidy series. Genetics 142: 1349–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L-H, Farnet CM, Ehrlich KC, Ehrlich M (1982) Digestion of highly modified bacteriophage DNA by restriction endonucleases. Nucleic Acids Res 10: 1579–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy A (2002) Gene loss, silencing, and activation in newly synthesized wheat allopolyploid. Genetics 160: 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy A (2003) Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet 33: 102–106 [DOI] [PubMed] [Google Scholar]
- Kidwell KK, Osborn TC (1992) Simple plant DNA isolation procedures. In JS Beckmann, TC Osborn, eds, Plant Genomes: Methods for Genetic and Physical Mapping. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 1–13
- Lagercrantz U (1998) Comparative mapping between Arabidopsis thaliana and Brassica nigra indicates that Brassica genomes have evolved through extensive genome replication accompanied by chromosome fusions and frequent rearrangements. Genetics 150: 1217–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-S, Chen J (2001) Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc Natl Acad Sci USA 98: 6753–6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin DA (1983) Polyploidy and novelty in flowering plants. Am Nat 122: 1–25 [Google Scholar]
- Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE (2001) Requirement of chromomethylase3 for maintenance of CpXpG methylation. Science 292: 2077–2080 [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn M, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau K, et al (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476 [DOI] [PubMed] [Google Scholar]
- Liu B, Brubaker CL, Mergeai G, Cronn RC, Wendel JF (2001) Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome 44: 321–330 [PubMed] [Google Scholar]
- Liu B, Vega JM, Feldman M (1998. a) Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome 41: 535–542 [DOI] [PubMed] [Google Scholar]
- Liu B, Vega JM, Segal S, Abbo S, Rodova M, Feldman M (1998. b) Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. I. Changes in low-copy noncoding DNA sequences. Genome 41: 272–277 [DOI] [PubMed] [Google Scholar]
- Liu B, Wendel JF (2002) Non-Mendelian phenomena in allopolyploid genome evolution. Curr Genomics 3: 489–505 [Google Scholar]
- Liu B, Wendel JF (2003) Epigenetic phenomena and the evolution of plant allopolyploids. Mol Phylogenet Evol 29: 365–379 [DOI] [PubMed] [Google Scholar]
- Lukens L, Quijada PA, Udall J, Pires JC, Schranz ME, Osborn T (2004) Genome redundancy and plasticity within ancient and recent Brassica crop species. Biol J Linn Soc 82: 675–688 [Google Scholar]
- Lukens L, Zou F, Parkin I, Lydiate D, Osborn T (2003) Comparison of the Brassica oleracea genetic map with the Arabidopsis thaliana physical map. Genetics 164: 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund G, Messing J, Viotti A (1995) Endosperm-specific demethylation and activation of specific alleles of alpha-tubulin genes of Zea mays L. Mol Gen Genet 246: 716–722 [DOI] [PubMed] [Google Scholar]
- Ma X-F, Fang P, Gustafson JP (2004) Polyploidization-induced genome variation in triticale. Genome 47: 839–848 [DOI] [PubMed] [Google Scholar]
- Ma X-F, Rodrigues Milla MA, Gustafson JP (2002) AFLP-based genome studies of triticale following polyploidization. In A Aniol, E Arseniuk, K Cooper, N Darvey, P Gustafson, R Jessop, A Lukaszewski, B Myer, M Mergoum, G Oettler, D. Salmon, eds, Proceedings of the Fifth International Triticale Symposium, Vol 1. Plant Breeding and Acclimatization Institute, Radzikow, Poland, pp 89–94
- Madlung A, Masuelli R, Watson B, Reynolds S, Davison J, Comai L (2002) Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol 129: 733–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlung A, Tyagi AP, Watson B, Jiang H, Kagochi T, Doerge RW, Martienssen R, Comai L (2005) Genomic changes in synthetic Arabidopsis polyploids. Plant J 41: 221–230 [DOI] [PubMed] [Google Scholar]
- Martienssen RA, Colot V (2001) DNA methylation and epigenetic inheritance in plants and filamentous fungi. Plant Physiol 129: 733–746 [DOI] [PubMed] [Google Scholar]
- Osborn TC, Pires JC, Birchler JA, Auger D, Chen ZJ, Lee H, Comai L, Madlung A, Doerge R, Colot V, et al (2003) Understanding mechanisms of novel gene expression in polyploids. Trends Genet 19: 141–147 [DOI] [PubMed] [Google Scholar]
- Ozkan H, Levy A, Feldman M (2001) Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops-Triticum) group. Plant Cell 13: 1735–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin IAP, Sharpe AG, Keith DJ, Lydiate DJ (1995) Identification of the A and C genomes of amphidiploid Brassica napus (oilseed rape). Genome 38: 1122–1131 [DOI] [PubMed] [Google Scholar]
- Pires JC, Zhao J, Schranz ME, Leon E, Quijada P, Lukens L, Osborn TC (2004) Flowering time divergence and genomic rearrangements in resynthesized Brassica polyploids (Brassicaceae). Biol J Linn Soc 82: 675–688 [Google Scholar]
- R Development Core Team (2004) R: A language and environment for statistical computing. R Foundation for Statistical Computing. http://www.R-project.org, Version 1.8.1
- Richard GF, Paques F (2000) Mini- and microsatellite expansions: the recombination connection. EMBO Rep 1: 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M, Galbiati M, Ticknor C, Chen J, Dellaporta S (1996) Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273: 654–657 [DOI] [PubMed] [Google Scholar]
- Schranz ME, Osborn TC (2000) Novel flowering time variation in the resynthesized polyploid Brassica napus. J Hered 91: 242–246 [DOI] [PubMed] [Google Scholar]
- Schranz ME, Osborn TC (2004) De novo variation in life-history traits and responses to growth condition of resynthesized polyploid Brassica napus (Brassicaceae). Am J Bot 92: 174–183 [DOI] [PubMed] [Google Scholar]
- Schranz ME, Quijada P, Sung SB, Lukens L, Amasino R, Osborn TC (2002) Characterization and effects of the replicated flowering time gene FLC in Brassica rapa. Genetics 162: 1457–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked H, Kashkush K, Ozkan H, Feldman M, Levy A (2001) Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13: 1749–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AG, Parkin IAP, Keith DJ, Lydiate DJ (1995) Frequent nonreciprocal translocations in the amphidiploid genome of oilseed rape (Brassica napus). Genome 38: 1112–1121 [DOI] [PubMed] [Google Scholar]
- Song K, Lu P, Tang K, Osborn T (1995) Rapid genomic change in synthetic polyploids of Brassica and its implication for polyploid evolution. Proc Natl Acad Sci USA 92: 7719–7723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udall JA, Quijada PA, Osborn TC (2005) Detection of chromosomal rearrangements derived from homoeologous recombination in four mapping populations of Brassica napus L. Genetics 169: 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tian L, Madlung A, Lee H, Chen M, Lee J, Watson B, Kagochi T, Comai L, Chen ZJ (2004) Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167: 1961–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF (2000) Genome evolution in polyploids. Plant Mol Biol 42: 225–249 [PubMed] [Google Scholar]
- Wendel JF, Schnabel A, Seelanan T (1995) Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci USA 92: 280–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong LZ, Xu CG, Saghai Maroof MA, Zhang Q (1999) Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261: 439–446 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Watanabe H, Miura F, Soejima H, Uchiyama M, Iwasaka T, Mukai T, Sakaki Y, Ito T (2004) A comprehensive analysis of allelic methylation status of CpG islands on human chromosome 21q. Genome Res 14: 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XP, Si Y, Hanson RE, Crane CF, Price HJ, Stelly DM, Wendel JF, Paterson AH (1998) Dispersed repetitive DNA has spread to new genomes since polyploid formation in cotton. Genome Res 8: 479–492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.