

Abstract
Epidemiological evidence suggests that nonsteroidal anti-inflammatory drugs (NSAIDs) decrease the risk for Alzheimer's disease (AD). Certain NSAIDs can activate the peroxisome proliferator-activated receptor-γ (PPARγ), which is a nuclear transcriptional regulator. Here we show that PPARγ depletion potentiates β-secretase [β-site amyloid precursor protein cleaving enzyme (BACE1)] mRNA levels by increasing BACE1 gene promoter activity. Conversely, overexpression of PPARγ, as well as NSAIDs and PPARγ activators, reduced BACE1 gene promoter activity. These results suggested that PPARγ could be a repressor of BACE1. We then identified a PPARγ responsive element (PPRE) in the BACE1 gene promoter. Mutagenesis of the PPRE abolished the binding of PPARγ to the PPRE and increased BACE1 gene promoter activity. Furthermore, proinflammatory cytokines decreased PPARγ gene transcription, and this effect was supressed by NSAIDs. We also demonstrate that in vivo treatment with PPARγ agonists increased PPARγ and reduced BACE1 mRNA and intracellular β-amyloid levels. Interestingly, brain extracts from AD patients showed decreased PPARγ expression and binding to PPRE in the BACE1 gene promoter. Our data strongly support a major role of PPARγ in the modulation of amyloid-β generation by inflammation and suggest that the protective mechanism of NSAIDs in AD involves activation of PPARγ and decreased BACE1 gene transcription.
Keywords: amyloid, inflammation, Alzheimer's disease, transgenic mice
Formation of amyloid peptides [amyloid-β (Aβ)] by neurons is generally thought to be a prime trigger of the pathogenesis of Alzheimer's disease (AD). The generation of Aβ is initiated by a protease that cleaves a larger precursor protein, the β-amyloid precursor protein (βAPP), at the N terminus side of the Aβ peptide. β-secretase or β-site APP-cleaving enzyme (BACE1) was cloned and identified as a transmembrane aspartyl protease (1). BACE1 deficiency precludes Aβ formation in transgenic mice (2) and does not cause or promote any neurological or phenotypic abnormalities. Moreover, BACE1 inactivation rescues memory deficits in transgenic mice (3), strongly supporting the importance of BACE1 as a therapeutic target in AD.
Several studies have demonstrated that BACE1 expression can be modulated by various factors. For instance, oxidative stress increases the expression and activity of BACE1 in NT2 neurons (4), whereas it is up-regulated in chronic models of gliosis (5) and experimental traumatic brain injury (6). In addition, BACE1 protein levels and the β-secretase product (β-C-terminal fragment) are increased in brain of sporadic AD patients (7). Although BACE1 expression increases with age in mice (8), there is no consensus regarding BACE1 mRNA levels in AD patients (7, 9). Evidence has been presented for regulation of BACE1 expression at the transcriptional as well as the translational levels. The untranslated 5′-BACE1 transcript leader contains upstream ORFs that can reduce the translation of the main ORF (10). The BACE1 gene promoter region was recently cloned, and several putative transcription factor-binding sites were identified (11-13).
We recently demonstrated that BACE1 mRNA and protein levels are increased by proinflammatory mediators and down-regulated by nonsteroidal anti-inflammatory drugs (NSAIDs) (14). Because certain NSAIDs are agonists for peroxisome proliferator-activated receptor-γ (PPARγ) (15), we hypothesized that the protective mechanisms by which NSAIDs mediate reduction of Aβ via BACE1 could involve activation of PPARγ (14). PPARγ represent ligand-activated transcription factors that belong to a nuclear receptor superfamily, and two isoforms, i.e., PPARγ1 (16) and PPARγ2 (17), are formed from the same gene by alternative mRNA splicing. PPARγ forms heterodimers with retinoid X receptors (RXR) and upon ligand activation, the PPAR/RXR heterodimer recruits coactivators and binds to sequence-specific PPRE present in the promoter region of its target genes (18). Alternatively, PPARγ can inhibit specific gene expression without direct binding to the gene promoter, because transrepression of several genes, i.e., iNOS and COX-2, is achieved in part by antagonizing the activities of transcription factors STAT1, NF-κB, and AP-1 (19).
In the present work, we analyzed the promoter region of the BACE1 gene and identified a functional PPRE. Further, we examined whether PPARγ is altered in AD by determination of PPARγ levels in brain of AD patients and AD transgenic mice. We demonstrate that NSAIDs modulate BACE1 transcription by repressing its promoter activity specifically through PPARγ activation.
Materials and Methods
Materials and Antibodies. Immunostimulants and inhibitors tested were IFN-γ (Sigma), TNF-α (Roche, Mannheim, Germany), ibuprofen (Sigma), pioglitazone (Takeda, Osaka), indomethacin, naproxen, sulindac sulfide, BAY11-7082 (Alexis Biochemicals, Grünberg, Germany), and the PPARγ antagonist GW0072 (provided by Timothy Willson, Glaxo). The monoclonal antibody 6E10 was obtained from Signet Laboratories (Dedham, MA), polyclonal pan-Aβ antibody was obtained from Calbiochem, polyclonal antibody for PPARγ from Alexis Biochemicals, as well as polyclonal H-100 from Santa Cruz Biotechnology, which is highly specific for PPARγ and does not crossreact with PPARα or PPARβ. The polyclonal antibody 369 for APP was a generous gift from Sam Gandy (Thomas Jefferson University, Philadelphia) and polyclonal antibody against CT-BACE1 7520 from C. Haass (Ludwig-Maximilians-Universität, Munich). Tissue culture reagents were obtained from Invitrogen. All other chemicals were purchased from Sigma.
Human Postmortem Brain Samples. Human brains were obtained from routine autopsies at the Huddinge Brain Bank in accordance with the laws and the permission of the ethical committee. The control group included brains from subjects who died either of nonneurological diseases or traffic accidents and had no history of long-term illness or dementia (seven male, three female; mean age was 77 ± 4 years, range 62-91years). The AD group included the brains from patients with clinically and pathologically confirmed AD (four male, six female, mean age 83 ± 2 years, range 74-93 years).
Animals. The transgenic mice used in this study were of the FVB/N genetic background and expressed APPV717I under the control of the mouse thy1 gene promoter. Mice 10 months of age were used, because APPV717I mice begin to deposit amyloid peptides at this age. Mice were fed Purina 5002 rodent chow ad libitum supplemented with either ibuprofen or pioglitazone for 7 days. There were six animals per group. The final dosage of drug was computed to be 62.5 mg/kg per day of ibuprofen and 40 mg/kg per day of pioglitazone.
Cell Lines, Cell Culture, and Transient Transfection. Mouse embryonic fibroblast (MEF) knockout from PPARγ were obtained as described (20). Mouse neuroblastoma N2a cells stably transfected with APP695 containing the Swedish mutation (K595N/M596L), so-called APPsw, were obtained from G. Thinakaran (University of Chicago, Chicago). Human embryonic kidney (HEK)293 cells stably transfected with APPsw were kindly supplied by C. Haass (University of Munich, Munich). Transfections were carried out by using Lipofectamine 2000 (Invitrogen) or FuGene (Roche). PPARγ1 reporter plasmid pGL3-γP3000 was obtained from Johan Auwerx, Strasbourg, France). Cells were incubated for 20 h with OPTIMEM alone or with cytokines IFN-γ (1 ng/ml) + TNF-α (30 ng/ml).
PPARγ Small Interfering (si)RNA. siRNA duplex to down-regulate PPARγ expression was directed to the target sequence 5′-AAGACCACTCGCATTCCTTTG-3′ of the mouse PPARγ cDNA and to the sequence 5′-GACCACTCCCACTCCTTTG-3′ of the human PPARγ cDNA. As a control, the following target sequence was used: 5′-AAGAGGTGGCCATCCGAATTT-3′, which did not modify PPARγ expression.
Site-Directed Mutagenesis. Site-directed mutagenesis in the 1.5-kb BACE promoter fragment cloned in pGL3 vector was carried out by using the QuickChange Site-Directed Mutagenesis Kit (Stratagene).
Western Blotting and Determination of Secreted Aβ and CTF-β. Determination of Aβ and CTFs and Western blotting were done as described (14).
Luciferase Assay. The luciferase reporter assay was performed according to the instructions of the manufacturer (Promega).
EMSAs. Nuclear proteins (10 μg) were incubated for 20 min on ice with double-stranded 32P-labeled oligonucleotides in a final volume of 15 μl. Oligonucleotides with the consensus PPRE site present in BACE1 promoter were: 5′-ATCAGGTGGGTCATGAGG-3′ 3′-TAGTCCACCCAGTACTCC-5′ and the mutant 5′-ATCAGCAGGCACATGAGG-3′ 3′-TAGTCGTCCGTGTACTCC-5′.
RNA Preparation and RT-PCR. Total RNA was extracted by using TRIzol reagent according to the manufacturer's instructions (Sigma). RT-PCR was performed as described (21). The primers used for mouse BACE were: 5′-CCGGCGGGAGTGGTATTATGAAGT-3′ and 5′-GATGGTGATGCGGAAGGACTGATT-3′; for murine GAPDH: 5′-ACGACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′; and for murine PPARγ: 5′-ATGCTGGCCTCCCTGATGAATAAA-3′ and 3′-ACAAGCGGTTCCACGAGGTC-5′.
Immunohistochemistry. Mouse brain sections were stained with antibody pan-Aβ. For quantitative image analysis of subiculum immunostaining, serial sagittal sections were examined. All images were acquired by using a standard light and immunofluorescence microscope (Nikon, Eclipse E-800) connected to a digital camera (DXC-9100P, Sony, Cologne, Germany) and to a personal computer system with lucia imaging software (lucia 32G, Version 4.11; Laboratory Imaging, Düsseldorf, Germany).
Statistical Evaluation. Data were statistically analyzed by systat (Systat, Evanston, IL) by using ANOVA followed by the Tukey post hoc test.
Results
Effect of PPARγ Supression on Aβ Secretion and BACE1 Expression. We sought to determine the specificity of the effect of inflammatory cytokines and ibuprofen (14, 22) in MEFs either wild type or deficient in PPARγ (PPARγ-/- MEF). The latter cells lack PPARγ protein and mRNA as assessed by Western blotting and semiquantitative RT-PCR, respectively (Fig. 1A). Western blotting analysis with two different antibodies showed specificity for PPARγ, because there was no signal detected in PPARγ null cells.
Fig. 1.

APP metabolism in cells that do not express PPARγ. (A) PPARγ expression and mRNA representation of MEF wild-type cells, heterozygous and knockout for PPARγ. PPARγ expression in N2a-sw cells transfected with siRNA for PPARγ.(B) Quantification of Aβ levels (n = 5) in MEF cells nonstimulated, stimulated with IFN-γ (1 ng/ml) + TNF-α (30 ng/ml) overnight, and then incubated with ibuprofen (IBU) (10 μM) for 4 h. (C) Quantification of BACE1 protein expression in MEF knockout for PPARγ with the same conditions as above. (D) Quantification of Aβ levels (n = 5) in N2a-sw cells transfected with siRNA stimulated as above. (E) Quantification of BACE1 expression in N2a-sw cells with the same conditions as above. Columns represent mean ± SEM. (F) Analysis of Aβ1-40 and Aβ1-42 secretion in HEK293 APPsw cells transfected with siRNA for PPARγ by ELISA (n = 3). Asterisks, significant differences between control and treatment. #, significant differences between treatment with cytokines alone and with NSAIDs. *, P ≤ 0.05; #, P ≤ 0.05, ANOVA followed by a Tukey post hoc test.
MEF were transiently transfected with APPsw (APP695 containing the Swedish mutation) and incubated with a combination of cytokines (TNF-α + IFN-γ). PPARγ-/- MEF did not show any modulation of Aβ secretion and BACE1 protein expression by immunostimulation and/or incubation with ibuprofen (10 μM) (Fig. 1 B and C), and this effect was reversed by PPARγ transfection. Interestingly, the levels of BACE1 protein in PPARγ knockout cells were increased compared with wild-type cells and cells retransfected with PPARγ cDNA, indicating that PPARγ affected BACE1 cytokine-induced expression (Fig. 1C).
Additionally, we determined whether the suppression of PPARγ by siRNA affected the levels of BACE1 protein and the secretion of Aβ. In N2a cells with stable expression of APPsw, transfection of siRNA against mouse PPARγ effectively decreased PPARγ expression (Fig. 1A). Unlike control cells, siRNA-treated cells showed no modulation of Aβ secretion or of BACE1 protein levels by incubation with proinflammatory cytokines or ibuprofen (Fig. 1 D and E). We compared our results using another sequence of siRNA, which did not decrease PPARγ levels, and observed no change in BACE1 expression (data not shown). Specific ELISA for Aβ1-40 and Aβ1-42 in media from HEK293 cells stably transfected with APPsw and transiently transfected with siRNA against human PPARγ showed the same effects (Fig. 1F).
Because PPARγ transcriptional activation depends on ligand binding, we determined whether transfection of N2a-APPsw cells with a mutant inactive PPARγ affected the production of Aβ. The N2a-APPsw cells transfected with PPARγ2-E499Q did not result in any change in Aβ secretion following stimulation by proinflammatory cytokines or by incubation with ibuprofen, relative to N2a-APPsw cells transfected with wild-type PPARγ2 (see Fig. 5 and Supporting Text which are published as supporting information on the PNAS web site).
Effect of Inflammatory Cytokines and Ibuprofen on BACE1 Transcription in PPARγ Knockout Cells. To define whether transcriptional regulation of BACE1 was modified in cells lacking PPARγ, we performed semiquantitative RT-PCR on wild-type and PPARγ-/- MEF cells treated with TNF-α + IFN-γ, with or without ibuprofen (10 μM). Significantly, in MEF lacking PPARγ, the endogenous levels of BACE1 mRNA present 4-fold higher levels than wild-type cells (Fig. 2 A and B). Similar to the results for BACE1 protein expression, the level of BACE1 mRNA increased under inflammatory conditions in wild-type MEF but remained constant in PPARγ-/- MEF (Fig. 2A). The same results were obtained by using Northern blot analysis (data not shown). Conversely, the steady-state PPARγ mRNA levels decreased when cells were incubated with TNF-α + IFN-γ, and this effect was partially abolished by ibuprofen, in agreement with data in adipocytes (23) (Fig. 2 A and C).
Fig. 2.

Transcriptional regulation of PPARγ and BACE1 by inflammatory cytokines and NSAIDs. (A) Modulation of BACE1 and PPARγ steady-state mRNA levels by IFN-γ (1 ng/ml) + TNF-α (30 ng/ml) is reversed with ibuprofen (IBU) (10 μM) in wild-type but not in knockout cells, shown by semiquantitative RT-PCR analysis of BACE mRNA. (B) Quantification of BACE1 steady-state mRNA levels in four experiments performed in PPARγ wild-type, heterozygous, and knockout cells. (C) Quantification of PPARγ steady-state mRNA levels in four experiments performed with the same samples. Columns represent mean ± SEM. (D) N2a-Sw cells transfected with PPARγ1 luciferase reporter construct were stimulated with IFN-γ (1 ng/ml) + TNF-α (30 ng/ml) with or without IBU (10 μM) or INDO(10 μM) (n = 3). *, P ≤ 0.05 ANOVA followed by a Tukey post hoc test.
To determine whether incubation with cytokines modulated PPARγ transcription, we transfected N2a cells with a plasmid containing a 3-kb fragment of the human PPARγ1 gene promoter upstream of a luciferase reporter gene. Incubation of transiently transfected N2a cells with TNF-α + IFN-γ considerably reduced PPARγ1 promoter activity (Fig. 2D). This suppression of PPARγ1 expression by inflammatory cytokines was partially reversed by treatment with indomethacin or ibuprofen (Fig. 2D). Moreover, to define the capability of PPARγ to modulate transcriptional transactivation, we examined the activity of a PPRE reporter construct, transiently transfected in N2a cells, and observed similar effects (see Fig. 5).
Effect of Inflammatory Cytokines and NSAIDs on BACE1 Promoter Activity. We determined whether BACE1 promoter activity was modulated by PPARγ-dependent transactivation. MEF were transfected with a construct containing a 1.5-kb fragment of the rat BACE1 gene promoter in conjunction with luciferase to compare BACE1 gene promoter activity in the presence or absence of PPARγ. In wild-type MEF, the BACE1 promoter activity was not affected by the proinflammatory cytokines, whereas it was decreased by ibuprofen (Fig. 3A). In PPARγ-/- MEF, the activity of the BACE1 gene promoter was increased ≈3.5-fold (Fig. 3A), paralleling the increase in BACE1 mRNA and protein in PPARγ-/- cells, strongly suggesting that PPARγ is a powerful repressor of the BACE1 gene (Fig. 3A). Reconstitution of PPARγ-/- MEF by transfection with PPARγ cDNA reversed the effects and demonstrated the specificity of the action of PPARγ on BACE1 promoter activity (Fig. 3A).
Fig. 3.

PPARγ modulates BACE1 promoter activity. (A) Luciferase activities of a 1.5-kb BACE1 promoter/luciferase reporter construct transfected in PPARγ wild-type, knockout, and knockout transfected with PPARγ cDNA MEF. Cells were treated with by IFN-γ (1 ng/ml) + TNF-α (30 ng/ml) with or without ibuprofen (IBU) (10 μM), n = 5. (B) NSAIDs and PPARγ agonists inhibit transcription of BACE1 promoter. N2a-sw cells transfected with BACE1 promoter and incubated with IBU, Pio, Indo, Napro, GW0072X (1 μM), BAY11-7082, and sulindac sulfide n = 4, all at 10 μM concentration. (C) PPARγ transfection inhibits BACE1 promoter activity. N2a-sw cells were transfected with BACE1 promoter construct and PPARγ1, PPARγ2, and PPARγ2 E499Q cDNA and incubated with or without IBU (10 μM); n = 4. (D) MEF transfected with BACE1 promoter control and mutated at the PPRE site were incubated with IBU (10 μM). (E) N2a cells transfected with BACE1 promoter control and mutated at the PPRE site were incubated with IBU (10 μM). Columns represent mean ± SEM, n = 4. Asterisks, significant differences between wild-type cells and treated cells. #, differences between transfected cells treated or untreated. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; #, P ≤ 0.05; ##, P ≤ 0.01, ANOVA followed by a Tukey post hoc test. (F) Gel-shift analysis with the BACE1-PPRE probe using nuclear extract from MEF cells. The major PPARγ-containing complex is indicated by the arrow. Lane 1, MEF wild-type cells; lane 2, MEF PPARγ knockout cells; lane 3, MEF PPARγ knockout cells transfected with PPARγ1 cDNA; lane 4, MEF wild-type cells incubated with a excess of unlabeled BACE1-PPRE probe. (G) Gel shift using HEK293 cells transfected with human PPARγ2 cDNA. Lane 1, control; lane 2, supershift analysis; lane 3, molar excess of unlabeled BACE1-PPRE probe; lane 4, labeled mutant BACE1-PPRE probe (BACE1-PPREM).
To define the elements in the BACE1 gene promoter that are critical for the PPARγ repression, we analyzed truncated constructs by removing sequence segments that appeared potentially important for the regulation of the BACE1 gene (see Fig. 6, which is published as supporting information on the PNAS web site). The promoter activity of BPR.Del (containing residues -1 to -753) was significantly higher than that of the original 1.5- kb promoter construct, suggesting suppressor elements to be located between -754 and -1,541 bp (11). The activity of this construct was high when tested in cells with or without PPARγ, indicating that the deleted region could contain consensus-binding sites for PPARγ or other transcription factors modulated by PPARγ.
We then tested a number of NSAIDs and PPARγ agonists on BACE1 promoter activity in N2a neuroblastoma cells. Ibuprofen, indomethacin, naproxen, or the PPARγ agonist pioglitazone all repressed BACE1 promoter activity, whereas coincubation with the PPARγ antagonist GW0072 reversed the suppressive effect of ibuprofen (Fig. 3B). We also measured the effect of the NF-κB inhibitor BAY11-7082 and sulindac sulfide, which is an NSAID that does not activate PPARγ, and observed no change in BACE1 promoter activity (Fig. 3B). On the other hand, overexpression of PPARγ1 and of PPARγ2 in N2a cells robustly repressed BACE1 gene promoter activity in a ligand-dependent manner (Fig. 3C). Ibuprofen had no effect on BACE1 gene promoter activity in cells transfected with the mutant PPARγ2-E499Q.
The Rat BACE1 Gene Promoter Contains a PPARγ Responsive Element PPRE. The increase in promoter activity of the BPR-Del(-1 to -753) mutant suggested the presence of suppressors of BACE1 transcription between -754 and -1,541 bp. Inspection of the sequence of this region revealed a possible consensus-binding site for PPARγ between -1356 and -1338, 5′-GGTGGGTCATGAGGTTCA-3′, which was experimentally analyzed by introducing point mutations in the BACE1 gene promoter reporter. Mutation of four nucleotides of the putative PPARγ site resulted in up-regulation of BACE1 gene promoter activity in wild-type MEF cells as opposed to unchanged activity in PPARγ-/- MEF (Fig. 3D). Furthermore, ibuprofen failed to decrease the promoter activity of the putative PPRE mutant BACE1 gene promoter construct (Fig. 3D). The same results were observed in N2a cells transfected with wild-type BACE1 gene promoter and mutated in the PPRE (Fig. 3E).
To define further the specificity of the putative PPRE, we performed gel-shift analysis with a double-stranded oligonucleotide probe corresponding to the BACE1 promoter region containing the PPRE. The radiolabeled double-stranded probe (PPREwt) was incubated with nuclear extracts from MEF and HEK293 cells transfected with human PPARγ2, resulting in the formation of several high-molecular complexes (Fig. 3 F and G). The formation of one of the complexes was efficiently reduced by an excess of unlabeled probe, indicating the specificity of the PPARγ bound to this consensus motif (Fig. 3 F, lane 4, and G, lane 3). Incubation with an antiserum to PPARγ resulted in the disappearance of the same complex (Fig. 3G, lane 2). The substitution of four bases in the synthetic oligonucleotide (PPREM) abolished the binding (Fig. 3G, lane 4). Nuclear extracts from PPARγ-/- MEF cells and PPARγ-/- MEF transfected with PPARγ1 cDNA were also used as controls (Fig. 3F, lanes 2 and 3).
Treatment with PPARγ Agonists Affects the Levels of PPARγ, BACE1, and Aβ Generation in APPV717I Mice. To prove the effects of PPARγ activation in vivo, we performed experiments using the APPV717I mouse model of AD. In wild-type animals, the levels of PPARγ mRNA and protein decreased with age and were higher compared with APPV717I transgenic mice, in agreement with the existence of an inflammatory component in AD mice (Fig. 4A). We have recently observed that ibuprofen and pioglitazone treatment in APP transgenic mice results in a reduction of glial inflammation and Aβ levels in plaques as measured by ELISA (24). Therefore, we sought the underlying mechanism behind these effects and detected increased PPARγ transcript and reduced BACE1 mRNA levels (Fig. 4B) as well as reduced CTF-β levels (Fig. 4E). To validate that PPARγ activation decreases total Aβ generation in vivo, we determined the intracellular Aβ levels by staining brain sections with antibody pan-Aβ. Treatment with ibuprofen and pioglitazone dramatically reduced the levels of intracellular Aβ (Fig. 4 C and D), without affecting the APP levels (data not shown), suggesting that PPARγ activation could regulate Aβ generation.
Fig. 4.

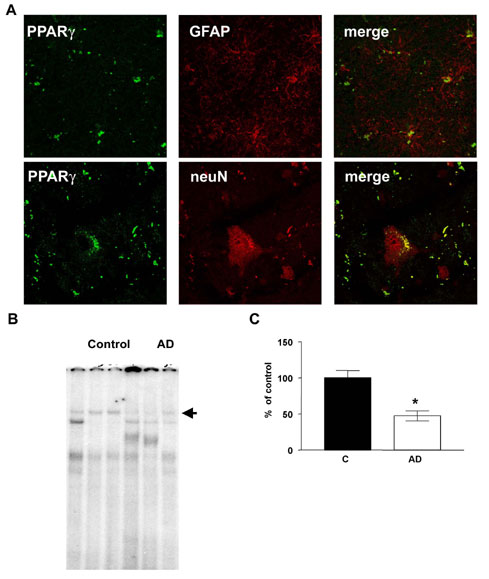
PPARγ in the brain of APPV717I transgenic mice and in AD patients. (A) Quantification of PPARγ expression in cortex from mouse brain control and APPV717I of 3 and 16 months of age. (B) Quantification of the mRNA levels of PPARγ and BACE1 APPV717I mice treated with ibuprofen (IBU) and pioglitazone (Pio). (C) Immunostaining of intracellular Aβ in subiculum from APPV717I mice treated with IBU and Pio. (D) Quantification of percentage of intracellular Aβ in subiculum from APPV717I mice treated with IBU and Pio. Bar, 100 μM. Columns represent mean ± SEM, n = 4. (E) Representative Western blot for CTF-β fragments in cortex from APPV717I mice treated with IBU and Pio. (F) Representative Western blot for PPARγ expression in brain lysates from two control and two AD patients. As a negative control, β-actin expression was analyzed. (G) Quantification of the levels of β-actin, PPARγ, and BACE1 in the frontal cortex of 10 controls and 10 AD patients. Asterisks, significant differences between control and AD. *, P ≤ 0.05; **, P ≤ 0.01. ANOVA followed by a Tukey post hoc test or Student's t test for human brains.
PPARγ and BACE1 Expression in AD Brain. To determine whether PPARγ is important for BACE1 regulation in humans, we examined the levels of expression of PPARγ in postmortem brain sections from AD patients (see Fig. 7, which is published as supporting information on the PNAS web site). We quantified the protein levels of PPARγ and BACE1 in the frontal cortex of 10 AD patients compared with 10 controls. In AD brain, PPARγ protein levels were reduced by 40% compared with controls (Fig. 4 F and G), whereas BACE1 expression was increased (Fig. 4F). To investigate whether the binding of PPARγ to the BACE1 gene promoter was affected, we performed gel-shift analysis with the PPREwt probe after incubation with nuclear extracts from brain of five AD patients and five controls. The retarded band corresponding to the PPRE complex was reduced by 50% (see Fig. 7), indicating that PPARγ protein levels and its binding are decreased in AD brain. Combined with all of the other data, these findings strongly point to a direct role of PPARγ in the increased BACE1 transcription and activity in AD, facilitating the generation of Aβ.
Discussion
Epidemiological and experimental evidence suggested a significant inflammatory component in AD and documented the beneficial effect of NSAIDs in this pathology (25, 26), stating an anti-inflammatory effect. The underlying molecular mechanism was related to decreased secretion of Aβ peptides in cultured cells and in amyloid mouse models (14, 22, 24, 27-29). Some NSAIDs, but not all, have been shown to be able to activate PPARγ (15). Ibuprofen, indomethacin, and naproxen are among the five most-prescribed NSAIDs, which potentially decrease the risk for AD (30). In this paper, we suggest that the NSAIDs that are PPARγ activators reduce Aβ levels by repressing BACE1 promoter activity.
Because it is widely recognized that a local inflammatory response within the brain contributes to the pathophysiology of AD, we performed our assays under inflammatory stimuli. For this reason, our data related to NSAIDs on APP processing are not comparable to those previously reported by using non-inflammatory conditions (28, 29, 31, 32). We recently observed that proinflammatory cytokines activate BACE1 expression, transcription, and activity in cultured neuroblastoma cells (14). The increase was reversed after treatment with NSAIDs or PPARγ agonists. Therefore, we sought to define whether this effect was specific for PPARγ and to determine the molecular mechanisms by which PPARγ decreases BACE1 transcription and thereby Aβ generation. Here we show that the effect of ibuprofen and other NSAIDs on BACE1 expression and Aβ secretion is specifically mediated by PPARγ. Importantly, BACE1 transcription was found to be up-regulated in MEF PPARγ knockout cells compared with wild-type cells. These results were confirmed by reporter gene assays, which demonstrated that lack of endogenous PPARγ facilitates BACE1 promoter activity, suggesting that PPARγ could be a suppressor for BACE1 promoter. Moreover, incubation of N2a cells with NSAIDs that are PPARγ activators showed a decrease in BACE1 gene promoter activity, and this effect was not reproduced by NSAIDs that are not PPARγ agonists, such as sulindac sulfide and aspirin. Equally robust effects were detected by overexpression of PPARγ1 and PPARγ2, which significantly reduced BACE1 promoter activity.
The upstream sequences of the BACE1 gene promoter are highly conserved among rat, mouse, and human, indicating that this region contains regulatory elements that modulate BACE1 gene transcription (11-13). We identified a putative PPARγ consensus sequence in the region located between nucleotides -1356 and -1338. The activity of the BACE1 gene promoter mutated in the putative consensus sequence for PPARγ was increased in MEF cells relative to the wild-type promoter and was not affected by ibuprofen, strengthening the hypothesis of the negative modulation of BACE1 transcription by PPARγ. That PPARγ binds to the putative PPRE in the BACE1 gene promoter was demonstrated directly by mobility-shift assays. The BACE1 gene promoter contains other putative transcription factor-binding sites, i.e., for NF-κB, SP-1, AP1, and AP2. Because PPARγ can modulate the activity of different transcription factors, we cannot exclude completely that PPARγ agonists also affect BACE1 gene promoter activity by antagonizing the activity of other transcriptional regulators.
It has been suggested that inflammatory cytokines and oxidative stress decrease PPARγ mRNA in adipocytes (23), and thiazolidinediones reverse this effect (33). Similarly, we demonstrate here that certain combinations of inflammatory cytokines are able to decrease PPARγ mRNA levels and PPRE activity in neuronal cells, and this effect is suppressed by incubation with NSAIDs. In addition, we show that PPARγ gene transcription is strongly reduced by inflammatory cytokines, and this can be prevented by incubation with ibuprofen or indomethacin. In this line, it has been recently shown in adipocytes that TNF-α suppresses PPARγ2 transcription by inhibiting the binding of C/EBPδ to the PPARγ2 promoter (34). Interestingly, we observed strongly decreased PPARγ protein levels in the frontal cortex of AD brain and in transgenic mice. An increase of the release of inflammatory cytokines by glial cells could be the determinant for the decrease in PPARγ levels in the brain and consequently could alter BACE1 transcription. This would support the hypothesis of the existence of a vicious cycle that accelerates the progression of AD (Fig. 8, which is published as supporting information on the PNAS web site). We believe that PPARγ is herein a major factor, because in the absence of PPARγ, there is no modulation of Aβ secretion by inflammation. The decrease of PPARγ levels detected in the brain of AD patients is not likely to be solely due to neuronal loss or secondary effects, i.e., loss of synapses. We have observed a similar decrease of PPARγ levels in lymphocytes from multiple sclerosis patients compared with healthy controls (35). It is therefore tempting to speculate that patients, even with minor signs of cerebral inflammation, could be predisposed to develop AD earlier due to reduced PPARγ levels that would consequently increase BACE1 expression and Aβ generation.
Our in vivo results support the in vitro observations, indicating that the PPARγ mRNA levels are up-regulated in transgenic mice after treatment with PPARγ agonists and as a consequence, decreased BACE1 expression leads to reduced total intracellular Aβ. This indicates that PPARγ activation is indeed involved in the regulation of Aβ generation, in contrast with another hypothesis that suggests PPARγ activation could cause an increase of extracellular Aβ degradation (31). Our data are in agreement with previous publications describing that treatment with certain NSAIDs and PPARγ agonists in AD mouse models decreased Aβ1-40 and Aβ1-42 levels (24, 27, 29, 36, 37). We do not exclude that some NSAIDs could show a selective reduction in Aβ1-42, but that effect could be independent of PPARγ activation. In addition, the concentrations of NSAIDs required to detect an effect in Aβ1-40/Aβ1-42 ratio are considerably higher (28), whereas the concentrations of NSAIDs to start the activation of PPARγ and decrease BACE1 transcription are in the same range as active concentrations of NSAIDs found in human plasma and cerebrospinal fluid (38-40).
Conclusion
We have shown that PPARγ represses BACE1 gene promoter activity in response to ligand binding to a PPRE located in the BACE1 gene promoter. This could be a potential mechanism by which NSAIDs have a protective effect against the development of AD. Moreover, we demonstrate that inflammatory cytokines modulate PPARγ transcription and PPRE activity, suppressing the negative regulation of PPARγ on BACE1 gene expression in vivo and in vitro. These effects may explain the overexpression of BACE1 in the brain under inflammatory conditions and emphasize the hypothesis that neuroinflammatory mechanisms significantly contribute to the pathogenesis of AD.
Supplementary Material
Acknowledgments
We thank Dr. Sam Gandy (Thomas Jefferson University, Philadelphia), Dr. Rotraut Mossner (University of Göttingen, Göttingen, Germany), and Dr. Christian Haass (University of Munich, Munich) for generous gifts of antibodies and cells; Dr. Gopal Thinakaran (University of Chicago, Chicago) for providing cells; Dr. Timothy M. Willson (Glaxo Wellcome) for PPARγ antagonist GW0072; Dr. Efrat Levy (Nathan Kline Institute for Psychiatric Research, Orangeburg, NY) for APPsw cDNA; Dr. Ron Evans (The Salk Institute for Biological Studies, San Diego) for PPAR-γ1 cDNA; Dr. Bruce Spiegelman (Harvard Medical School, Boston) for mouse PPARγ2- E499Q cDNA; Dr. Walter Wahli (University of Lausanne, Lausanne, Switzerland) for human PPARγ2 cDNA; and Dr. Andreas von Knethen (University of Kaiserslautern, Kaiserslautern, Germany) for the PPARγ responsive element (PPRE) reporter plasmid. This investigation was supported by the Sonderforschungsbereich 400 [SFB 400, Teilprojekt A8 (to M.T.H. and T.K.) and DFG WA1477/3 (to J.W.)], by Fonds voor Wetenschappelijk Onderzoek Vlaanderen, by the Katholieke Universiteit Leuven Research Fund, and by Katholieke Universiteit Leuven Research and Development. I.D. is a postdoctoral fellow at Fonds voor Wetenschappelijk Onderzoek Vlaanderen.
Author contributions: M.S., S.R., B.O.E., F.v.L., M.T.H., and T.K. designed research; M.S., I.D., P.B., and L.D.-O. performed research; M.S., I.D., and P.B. analyzed data; S.R., B.O.E., G.L., J.W., N.B., E.R., D.R.T., P.B., and F.v.L. contributed new reagents/analytic tools; M.S. wrote the paper; and T.K. and M.T.H. provided laboratory and personal financial support.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Aβ, amyloid-β; AD, Alzheimer's disease; βAPP, β-amyloid precursor protein; APPsw, APP695 containing the Swedish mutation; BACE, β-site APP cleaving enzyme; MEF, mouse embryonic fibroblasts; NSAIDs, nonsteroidal antiinflammatory drugs; PPARγ, peroxisome proliferator-activated receptor-γ; PPRE, PPAR responsive element; siRNA, small interfering RNA; HEK, human embryonic kidney.
References
- 1.Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S, Amarante, P., Loeloff, R., et al. (1999) Science 286, 735-741. [DOI] [PubMed] [Google Scholar]
- 2.Luo, Y., Bolon, B., Damore, M. A., Fitzpatrick, D., Liu, H., Zhang, J., Yan, Q., Vassar, R. & Citron, M. (2003) Neurobiol. Dis. 14, 81-88. [DOI] [PubMed] [Google Scholar]
- 3.Ohno, M., Sametsky, E. A., Younkin, L. H., Oakley, H., Younkin, S. G., Citron, M., Vassar, R. & Disterhoft, J. F. (2004) Neuron 41, 27-33. [DOI] [PubMed] [Google Scholar]
- 4.Tamagno, E., Bardini, P., Obbili, A., Vitali, A., Borghi, R., Zaccheo, D., Pronzato, M. A., Danni, O., Smith, M. A., Perry, G. & Tabato, M. (2002) Neurobiol. Dis. 10, 279-288. [DOI] [PubMed] [Google Scholar]
- 5.Hartlage-Rubsamen, M., Zeitschel, U., Apelt, J., Gartner, U., Franke, H., Stahl, T., Gunther, A., Schliebs, R., Penkowa, M., Bigl, V. & Rossner, S. (2003) Glia 4, 169-179. [DOI] [PubMed] [Google Scholar]
- 6.Blasko, I., Beer, R., Bigl, M., Apelt, J., Franz, G., Rudzki, D., Ransmayr, G., Kampfl, A. & Schliebs, R. (2004) J. Neural Transm. 111, 523-536. [DOI] [PubMed] [Google Scholar]
- 7.Holsinger, R. M., McLean, C. A., Beyreuther, K., Masters, C. L. & Evin, G. (2002) Ann. Neurol. 51, 783-786. [DOI] [PubMed] [Google Scholar]
- 8.Fukumoto, H., Rosene, D. L., Moss, M. B., Raju, S., Hyman, B. T. & Irizarry, M. C. (2004) Am. J. Pathol. 164, 719-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Ca, H., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 3632-3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogers, G. W., Jr., Edelman, G. M. & Mauro, V. P. (2003) Proc. Natl. Acad. Sci. USA 101, 2794-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lange-Dohna, C., Zeitschel, U., Gaunitz, F., Perez-Polo, J. R., Bigl, V. & Rossner, S. (2003) J. Neurosci. Res. 73, 73-80. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, M. A., Zhou, W., Qing, H., Lehman, A., Philipsen, S. & Song, W. (2004) Mol. Cell. Biol. 24, 865-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sambamurti, K., Kinsey, R., Maloney, B., Ge, Y. W. & Lahiri, D. K. (2004) FASEB J. 18, 1034-1036. [DOI] [PubMed] [Google Scholar]
- 14.Sastre, M., Dewachter, I., Landreth, G. E., Willson, T. M., Klockgether, T., van Leuven, F. & Heneka, M. T. (2003) J. Neurosci. 23, 9796-9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann, J. M., Lenhard, J. M., Oliver, B. B., Ringold, G. M. & Kliewer, S. A. (1997) J. Biol. Chem. 272, 3406-3410. [DOI] [PubMed] [Google Scholar]
- 16.Kliewer, S. A., Forman, B. M., Blumberg, B., Ong, E. S., Borgmeyer, U., Mangelsdorf, D. J., Umesono, K. & Evans, R.M. (1994) Proc. Natl. Acad. Sci. USA 91, 7355-7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tontonoz, P., Hu, E. & Spiegelman, B. M. (1994) Cell 79, 1147-1156. [DOI] [PubMed] [Google Scholar]
- 18.Tugwood, J. D., Issemann, I., Anderson, R. G., Bundell, K. R., McPheat, W. L. & Green, S. (1992) EMBO J. 11, 433-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, A. C., Brown, K. K., Silvestre, M. J., Willson, T. M., Palinski, W. & Glass, C. K. (2000) J. Clin. Invest. 106, 523-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen, E. D., Hsu, C. H., Wang, X., Sakai, S., Freeman, M. W., Gonzalez, F. J. & Spiegelman, B. M. (2002) Genes Dev. 16, 22-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heneka, M. T., Klockgether, T. & Feinstein, D. L. (2000) J. Neurosci. 20, 6862-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasko, I. Apochal, A., Boeck, G., Hartmann, T., Grubeck-Loebenstein, B. & Ransmayr, G. (2001) Neurobiol. Dis. 8, 1094-1101. [DOI] [PubMed] [Google Scholar]
- 23.Xing, H. Northrop, J. P., Grove, J. R., Kilpatric, K. E., Su, J.-L. & Ringold, G. M. (1997) Endocrinology 138, 2776-2783. [DOI] [PubMed] [Google Scholar]
- 24.Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O'Banion, K., Klockgether, T., Van Leuven, F. & Landreth, G. E. (2005). Brain 128, 1442-1453. [DOI] [PubMed] [Google Scholar]
- 25.Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., et al. (2000) Neurobiol. Aging 21, 383-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szekely, C. A., Thorne, J. E., Zandi, P. P., Ek, M., Messias, E., Breitner, J. C. & Goodman, S. N. (2004) Neuroepidemiology 23, 159-169. [DOI] [PubMed] [Google Scholar]
- 27.Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K. H., Frautschy, S. A. & Cole, G. M. (2000) J. Neurosci. 20, 5709-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., et al. (2001) Nature 414, 212-216. [DOI] [PubMed] [Google Scholar]
- 29.Eriksen, J. L., Sagi, S. A., Smith, T. E., Weggen, S., Das, P., McLendon, D. C., Ozols, V. V., Jessing, K. W., Zavitz, K. H., Koo, E. H., et al. (2003) J. Clin. Invest. 112, 440-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.In't Veld, B. A., Ruitenberg, A., Hofman, A., Launer, L. J., van Duijn, C. M., Stijnen, T., Breteler, M. M. B. & Stricker, B. H. C. (2001) N. Engl. J. Med. 345, 1515-1521. [DOI] [PubMed] [Google Scholar]
- 31.Camacho, I. E., Serneels, L., Spittaels, K., Merchiers, P., Dominguez, D. & De Strooper, B. (2004) J. Neurosci. 24, 10908-10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lleo, A., Berezovska, O., Herl, L., Raju, S., Deng, A., Bacskai, B. J., Frosch, M. P., Irizarry, M. & Hyman, B. T. (2004) Nat. Med. 10, 1065-1066. [DOI] [PubMed] [Google Scholar]
- 33.Gimble, J. M., Robinson, C. E., Wu, X., Kelly, K. A., Rodriguez, B. R., Kliewer, S. A., Lehmann, J. M. & Morris, D. C. (1996) Mol. Pharmacol. 50, 1087-1094. [PubMed] [Google Scholar]
- 34.Kudo, M., Sugawara, A., Uruno, A., Takeuchi, K. & Ito, S. (2004) Endocrinology 145, 4948-4956. [DOI] [PubMed] [Google Scholar]
- 35.Klotz, L., Schmidt, M., Sastre, M., Giese, T., Knolle, P., Klockgether, T. & Heneka, M. T. (2005) J. Immunol. 35, 4948-4955. [DOI] [PubMed] [Google Scholar]
- 36.Yan, Q., Zhang, J., Liu, H., Babu-Khan, S., Vassar, R., Biere, A. L., Citron, M. & Landreth, G. (2003) J. Neurosci. 23, 7504-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sung, S., Yang, H., Uryu, K., Lee, E. B., Zhao, L., Shineman, D., Trojanowski, J. Q., Lee, V. M. & Pratico, D. (2004) Am. J. Pathol. 165, 2197-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaradat, M. S., Wongsud, B., Phornchirasilp, S., Rangwala, S. M., Shams, G., Sutton, M., Romstedt, K. J., Noonan, D. J. & Feller, D. R. (2001) Biochem. Pharmacol. 62, 1587-1595. [DOI] [PubMed] [Google Scholar]
- 39.Bannwarth, B., Netter, P., Lapicque, F., Pere, P., Thomas, P. & Gaucher, A. (1990) Eur. J. Clin. Pharmacol. 38, 343-346. [DOI] [PubMed] [Google Scholar]
- 40.Bannwarth, B., Lapicque, F., Pehourcq, F., Gillet, P., Schaeverbeke, T., Laborde, C., Dehais, J., Gaucher, A. & Netter, P. (1995) Br. J. Clin. Pharmacol. 40, 266-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}