

Abstract
Ero1p is a key enzyme in the disulfide bond formation pathway in eukaryotic cells in both aerobic and anaerobic environments. It was previously demonstrated that Ero1p can transfer electrons from thiol substrates to molecular oxygen. However, the fate of electrons under anaerobic conditions and the final fate of electrons under aerobic conditions remained obscure. To address these fundamental issues in the Ero1p mechanism, we studied the transfer of electrons from recombinant yeast Ero1p to various electron acceptors. Under aerobic conditions, reduction of molecular oxygen by Ero1p yielded stoichiometric hydrogen peroxide. Remarkably, we found that reduced Ero1p can transfer electrons to a variety of small and macromolecular electron acceptors in addition to molecular oxygen. In particular, Ero1p can catalyze reduction of exogenous FAD in solution. Free FAD is not required for the catalysis of dithiol oxidation by Ero1p, but it is sufficient to drive disulfide bond formation under anaerobic conditions. These findings provide insight into mechanisms for regenerating oxidized Ero1p and maintaining disulfide bond formation under anaerobic conditions in the endoplasmic reticulum.
Keywords: electron transfer, Ero1, flavoenzyme, hydrogen peroxide
The thiol oxidase enzyme Ero1p (1, 2), conserved across eukaryotes, generates disulfide bonds de novo in the endoplasmic reticulum (ER) by catalyzing the transfer of electrons from dithiols to molecular oxygen (3). The Ero1p active site consists of a Cys-Xaa-Xaa-Cys amino acid sequence motif (in which Xaa is a non-Cys amino acid) juxtaposed with the isoalloxazine ring system of a bound FAD cofactor (4) (Fig. 1). An additional redox center, a Cys-Xaa4-Cys disulfide, has been proposed to accept electrons from substrate proteins and transfer them to the Cys-Xaa-Xaa-Cys disulfide (4, 5).
Fig. 1.

Structure of the Ero1p active site. A ribbon diagram of the active-site region of yeast Ero1p (4) is shown with the bound FAD in orange and disulfides in a ball-and-stick representation. The Cys sulfurs are numbered according to the position of the residue in the yeast Ero1p sequence (RefSeq accession no. NP_013576). The disulfide between Cys-352 and Cys-355 abuts the flavin. The loop containing Cys-100 and Cys-105 is flexible (only one of the two conformations observed in the crystal structures is shown) and may be involved in shuttling electrons from substrate to the Cys-352-Cys-355 disulfide. The structural or functional role of the Cys-90-Cys-349 disulfide has yet to be determined.
The arrangement of bound flavin, fixed active-site disulfide, and flexible shuttle disulfide is also found in Erv2p (6), a second yeast ER thiol oxidase with no sequence similarity to Ero1p (7, 8). Erv2p is a member of the QSOX/ALR enzyme family (9). The overall reaction of the QSOX/ALR enzymes can be divided into two half-reactions. In the reductive half-reaction, the enzyme accepts electrons from reducing substrates, resulting in a reduction of the bound flavin cofactor. In the oxidative half-reaction, the enzyme deposits the electrons on an acceptor, such as molecular oxygen, to restore the bound cofactor to its initial state, as shown in Eqs. 1 and 2.
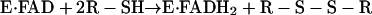 |
[1] |
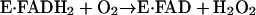 |
[2] |
Although this scheme describes the simplest scenario, four-, six-, and even eight-electron-reduced states of these f lavin-dependent sulfhydryl oxidase enzymes may also be possible, depending on the number of redox-active Cys pairs and the rate of internal equilibration between them (10, 11).
It is likely that the basic reaction scheme described above for the QSOX/ALR family applies to Ero1p as well. First, the similar arrangements of the functional groups in the active sites of Ero1p and Erv2p (4) suggest that the two thiol oxidase families may share features of their catalytic mechanisms in common. Second, depletion of oxygen in the solution, which should disable the oxidative half-reaction in the above scheme, traps the Ero1p-bound FAD in its reduced state (4). Third, as shown herein, stoichiometric transfer of electrons from thiol substrates to Ero1p occurs under such conditions.
In this study, we focus primarily on the oxidative half-reaction (Eq. 2) to address two questions regarding the mechanism of Ero1p. The first concerns the product of the reaction when molecular oxygen is the direct oxidative substrate of Ero1p. A two-electron reduction of molecular oxygen is expected to yield hydrogen peroxide, and the potential for Ero1p-mediated thiol oxidation to contribute to the pool of reactive oxygen species in the cell has been appreciated (12). However, only low levels of hydrogen peroxide were detected in vitro as a result of Ero1p activity (3). Because any peroxide produced can potentially go on to react with electron donors present in the solution, we sought assay conditions to maximize peroxide recovery or detection.
The second question we addressed is whether electron acceptors other than oxygen can reoxidize Ero1p. It was reported that the addition of free FAD, in excess of the FAD bound by the purified enzyme, was essential for RNaseA refolding in vitro catalyzed by Ero1p and protein disulfide isomerase (3). One hypothesis explaining this observation was that Ero1p has a “sensor” site for exogenous FAD, which may function as an allosteric effector to modulate disulfide bond formation according to the cellular metabolic status (3). To investigate the possibility of another role for FAD in the Ero1p-mediated reaction, we tested whether free FAD could be reduced directly by FADH2 of the Ero1p holoenzyme as an alternative to the reduction of molecular oxygen at the enzyme active site. We then went on to determine whether nonflavin compounds could also serve as electron acceptors under anaerobic conditions.
In live yeast cells, the ero1-1 mutation causes temperature sensitivity under either aerobic or anaerobic conditions (8, 13). This observation suggests that Ero1p is required for disulfide bond formation in both the presence and the absence of oxygen and that the enzyme has the ability to transfer electrons to acceptors other than oxygen in vivo. In this article, we demonstrate that various routes for regeneration of an oxidized Ero1p active site are biochemically feasible, laying the groundwork for the search for physiological electron acceptors other than oxygen in the ER. Furthermore, our observation that aerobic Ero1p activity does indeed produce H2O2, a reactive oxygen species potentially damaging to the cell, supports proposals that futile cycling of Ero1p-mediated thiol oxidation and glutathione-mediated reduction in the ER leads to oxidative stress and cell death (14, 15). The question remains how normal cells with heavy loads of protein thiols in their secretory pathways accommodate or eliminate the byproducts of disulfide bond formation.
Materials and Methods
Protein Production and Purification. Two versions of the catalytic domain of Saccharomyces cerevisiae Ero1p were produced in Escherichia coli for this study. One, spanning amino acid residues 56-424, was produced as described in ref. 4, except that induction of protein expression was done at 15°C for 48 h. The fusion protein consists of GST, a thrombin cleavage site, a His6-tag, a second thrombin cleavage site, and the crystallized domain of Ero1p (4). The second thrombin cleavage site was not recognized by the protease, so the amino terminus of the cleaved product was GSHHHHHHSSGLVPRGS, followed by the Ero1p sequence, and finally the sequence LERPHRD derived from the vector. The molecular mass measured by mass spectrometry was 45,354 Da, whereas the calculated molecular mass for this fragment is 45,294 Da, taking into account the loss of two protons for every disulfide bond formed. Unless otherwise noted, the data shown here were obtained by using this version of Ero1p. A second construct, spanning S. cerevisiae Ero1p residues 10-424, was also produced and tested. The relevant segment of the Ero1p coding sequence was inserted into the pGEX-4T1 vector (Amersham Pharmacia Biosciences). After cleavage of the protein product with thrombin, an additional Gly and Ser remained at the amino terminus, and the carboxy terminus included the LERPHRD sequence derived from the vector. In all cases tested, the longer construct behaved qualitatively similar to the shorter Ero1p construct but with a slightly faster rate. The longer construct was expressed under the same conditions as the shorter construct. Cells were then lysed in PBS, and the insoluble material was removed by centrifugation. Triton X-100 was added to the supernatant to a final concentration of 1%, and after 20 min, the clarified cell extract was loaded onto a glutathione-Sepharose column. The column was washed with PBS, and the protein was eluted in PBS containing 10 mM reduced glutathione. The buffer was exchanged to 100 mM Tris (pH 8.0)/2.5 mM CaCl2/25 mM NaCl, and thrombin was added (≈10 units per liter of original cell culture volume). After 1.5 h, the solution was passed through a glutathione-Sepharose column, and the unbound material was collected. The buffer was exchanged to 10 mM potassium phosphate (pH 6.8)/50 mM NaCl and loaded onto a hydroxyapatite column. Protein was eluted with increasing phosphate. The molecular mass measured for the longer construct was 48,659 Da compared with a calculated molecular mass of 48,662 Da.
After purification, Ero1p was applied to a PD-10 desalting column (Amersham Pharmacia Biosciences) equilibrated with 50 mM KPO4 (pH 7.5)/65 mM NaCl/0.5 mM EDTA (buffer A). The extinction coefficient of FAD bound to the enzyme was determined by releasing the FAD with the detergent cetyltrimethylammonium bromide (CTAB). The absorbance spectrum of 990 μl of an Ero1p solution was recorded, 10 μl of a 100 mM solution of CTAB was added to the cuvette, and the absorbance was recorded again. The original absorbance was corrected for the subsequent 1% dilution. This procedure was repeated with a solution of a similar and known concentration (16 μM) of FAD as a control for the effects of CTAB on the spectrum of FAD, which were found to be negligible. The addition of CTAB to the enzyme resulted in a loss of features and the appearance of a smooth spectrum resembling that of free FAD, an indication that the cofactor was effectively removed from the enzyme active site. The maximum absorbance in the visible region shifted from 454 to ≈446-448 nm. From this procedure, the extinction coefficient of the FAD bound to the enzyme was determined to be 12.5 mM-1·cm-1 at 454 nm, assuming an extinction coefficient of 11.3 mM-1·cm-1 at 446 nm for free FAD (16). The cofactor/enzyme ratio was determined by measuring absorbance in 6 M guanidine/20 mM NaPO4 (pH 6.5) at 446 nm (ε = 11.3 mM-1·cm-1) and 280 nm [calculated ε = 79.1 mM-1·cm-1 (17), which includes the contribution of the FAD moiety at this wavelength: ε = 21.3 mM-1·cm-1]. The concentration of protein was thus measured to be 14.4 μM, and the concentration of FAD was measured to be 13.9 μM, a ratio of 1.04.
An expression vector for E. coli thioredoxin I (Trx1) was constructed by amplifying the Trx1 coding sequence from bacterial lysate and inserting it between the NdeI and BamHI sites in the pET-28 vector (Novagen). Protein was expressed in BL21(DE3)pLysS (Novagen) bacterial cells grown at 37°C to 0.7 OD units and induced with 0.5 mM isopropyl β-d-thiogalactoside. After 3 h, cells were harvested, lysed by sonication in 50 mM NaPO4 (pH 8.0)/300 mM NaCl/10 mM imidazole, and centrifuged to remove insoluble material. Triton X-100 was added to a concentration of 1% to the supernatant, which was then applied to a Ni-NTA column. The column was washed first with 50 mM NaPO4 (pH 8.0)/300 mM NaCl/20 mM imidazole/20% glycerol to remove the Triton X-100, and washed again with the same solution lacking glycerol. Protein was eluted with a step gradient of increasing imidazole. DTT was added to the eluted Trx1 to a concentration of 100 mM, and the protein was incubated for 1 h at room temperature. The reduced Trx1 (0.5-0.7 ml) was applied to a PD-10 column equilibrated with buffer A. Fractions were collected and tested qualitatively for thiol content by using Ellman's assay (18). A clear separation was seen between the peak corresponding to reduced Trx1 (Trxred) and that of DTT. Trx1red peak fractions were pooled, and the reported Trxred concentrations were determined quantitatively by Ellman's assay. All oxygen consumption and spectroscopic assays were performed within 6 h of measuring the thiol concentration of Trx1red.
Bovine liver catalase and horse heart cytochrome c were purchased from Sigma. The concentration of cytochrome c was determined by subtracting the absorbance at 550 nm of oxidized protein from that of dithionite-reduced protein (Δε = 21.1 mM-1·cm-1). S. cerevisiae cytochrome b5 was amplified by PCR from yeast genomic DNA, inserted between the NdeI and BamHI sites in the pET-15 vector (Novagen), and expressed and purified by using the procedure for Trx1. Pseudomonas aeruginosa azurin was a gift of Israel Pecht (The Weizmann Institute of Science). Its concentration was determined at 630 nm (ε = 5.6 mM-1·cm-1).
Anaerobic Titration. Ero1p (7.4 μM) in buffer A was deoxygenated in the main space of an anaerobic cuvette by repeated cycles of vacuum and flushing with nitrogen (19). A solution of Trxred (0.65 mM in buffer A) was deoxygenated by cycles of degassing and flushing with nitrogen for >1 h and then used to fill a 50-μl gas-tight syringe (19). The syringe was loaded onto a ground-glass port of the cuvette, and an initial spectrum of the anaerobic enzyme was recorded. The spectral changes were completed within 5 min for steps in the titration up to 2 eq of Trxred. Toward the end of the experiment, the interval increased to 40 min per addition.
Oxygen Consumption Assays. Oxygen consumption was measured by using an Oxygraph Clark-type oxygen electrode (Hansatech Instruments, Pentney King's Lynn, U.K.). FAD, FMN, and riboflavin concentrations were determined by absorbance at 446 nm in 6 M guanidine/20 mM NaPO4 (pH 6.5) by using extinction coefficients of 11.3, 12.5, and 12.2 mM-1·cm-1, respectively. The oxygen electrode chamber was covered with aluminum foil during reactions to avoid photoreduction of the flavins. DTT solutions were prepared by weight and then corrected by using Ellman's assay. All experiments were performed in buffer A unless otherwise noted. All components of each reaction, except Ero1p, were mixed fresh in a total volume of 1.0 ml. Oxygen levels were monitored until a linear baseline was established, and the reaction was initiated by injection of Ero1p, typically in a volume of 20 μl, to yield a final enzyme concentration of 1 μM. For determination of the apparent Km for oxygen by using Trxred as the reducing substrate, the Trxred solution was stirred in the oxygen electrode chamber under a gentle stream of nitrogen until the oxygen concentration dropped to 30 μM (≈15 min) before Ero1p was injected.
Hydrogen Peroxide Assays. Hydrogen peroxide production was detected by monitoring oxygen evolution upon addition of catalase. After the thiol oxidation reaction was complete, 10 μl of a 2-mg/ml stock of catalase was injected into the reaction vessel of the oxygen electrode. In parallel, hydrogen peroxide production was followed kinetically by using the PeroXOquant quantitative peroxide assay kit (Pierce). H2O2 standards were made by using an ≈30% H2O2 stock solution (10.8 M). The stock solution concentration was standardized by absorbance at 240 nm of a 1:1,000 dilution by using an extinction coefficient of 43.6 M-1·cm-1. All PeroXOquant measurements, including the standard curve, were made in buffer A lacking EDTA. The standard curve was derived from the following H2O2 concentrations: 0, 1, 5, and 10-100 μM in intervals of 10 μM. The plot of absorbance vs. H2O2 concentration was linear in this range. For this assay, Trxred was prepared as described above except that after reduction, the protein was exchanged into buffer A lacking EDTA and used within 2 h. Ero1p was dialyzed against buffer A lacking EDTA. The enzymatic reaction was performed in a total volume of 750 μl while being stirred in a glass vial. The concentration of Trxred was 100 μM, and the enzyme concentration was 1 μM. Samples of the reaction mixture (20 μl) were taken every 5 s for the first minute and then every 10 s up to 5 min total and added to 200 μl of working reagent in a 96-well plate. Working reagent was prepared according to the PeroXOquant manual (1 vol of reagent A in 100 vol of reagent B). After a 30-min incubation, absorbance was measured in a Tecan SPECTRAFluor Plus ELISA reader (Durham, NC) at 595 nm. Standards were added to a similar volume of working reagent immediately after the end of the reaction.
Results
Enzyme and Substrates. For enzymatic assay of Ero1p, we used forms of the S. cerevisiae enzyme that could be efficiently expressed as recombinant proteins, comprising Ero1p residues 56-424 (shorter construct) or 10-424 (longer construct). The full-length yeast enzyme is 563 aa in length, but the carboxy terminus of our fragments corresponds to that of other Ero1p orthologs (i.e., human), which lack the ≈130-aa extension found in yeast Ero1p (2). Enzyme was produced in the Origami B(DE3)pLysS E. coli bacterial strain as detailed in an earlier article describing the high-resolution structure of Ero1p (4) and in Materials and Methods. Previous biochemical experiments on full-length S. cerevisiae Ero1p purified from yeast were conducted by using enzyme preparations that bound the FAD cofactor substoichiometrically (13). In contrast, our preparations of Ero1p contained equimolar amounts of protein and FAD. The extinction coefficient for FAD bound to Ero1p is 12.5 mM-1·cm-1 at 454 nm (Fig. 2).
Fig. 2.

Absorbance spectrum of Ero1p-bound FAD. The spectrum of Ero1p was measured (black), CTAB was then added to release the bound FAD, and the spectrum was measured again (red). The original spectrum was corrected for the 1% dilution when adding the CTAB before the two spectra were superposed. The absorbance peak of the bound flavin is red-shifted from ≈448 nm to 454 nm, and a shoulder appears at ≈485 nm. Inset is an expansion of the absorbance scale in the visible region.
Enzyme activity was directly monitored by oxygen consumption or reduction of the substrate of the second half-reaction (Eq. 2). Ero1p can oxidize a variety of small and macromolecular substrates (data not shown). In all cases, a lag phase is observed in the progress curves. Because the lag can be eliminated by preincubation of the enzyme with substrate but not by incubation with reaction products (data not shown), we attribute the lag to reductive activation of the enzyme. We found that Ero1p has a higher turnover number for E. coli Trxred than for DTT at equivalent substrate concentrations. For assays in which the reducing substrate would be stoichiometrically limiting, we therefore used Trxred. Although Trxred is a nonphysiological substrate, its reaction rate was suitable, and the protein could be produced easily in large quantities.
Reductive Titration of Ero1p with Trxred. Fig. 3 shows a titration of Ero1p with Trxred under anaerobic conditions. Substantially complete reduction of the enzyme-bound flavin required approximately four electrons, indicating that at least two molecules of Trxred can be oxidized by Ero1p in this static experiment. Similar results were obtained previously for egg white sulfhydryl oxidase, which required ≈2 eq of DTT (four electrons) to completely reduce the flavin (20). In both cases, the additional two electrons are taken up by disulfides. The Ero1p enzyme used in this study (shorter construct) contains five disulfide bonds in total, so it is possible that additional electrons may be transferred from Trxred to Ero1p after the bound flavin is reduced, although these later events would be largely silent spectroscopically. There is a noticeable lag in Fig. 3 Inset, consistent with the involvement of additional redox centers beside the flavin in the early stages of the experiment. During the titration, a rise in long-wavelength absorbance, exhibiting distinct shoulders at 580 and 630 nm, is observed. These features indicate the generation of the blue neutral flavin radical. Much higher levels of this semiquinone species are observed in another flavin-linked sulfhydryl oxidase, the human augmenter of liver regeneration, upon the addition of DTT (21).
Fig. 3.

Ero1p (7.4 μM in buffer A) was titrated anaerobically with Trxred added from a gas-tight syringe. Inset is a plot of absorbance at 454 nm recorded after each addition. Selected spectra from the titration (curves 1-7, corresponding to 0.0, 0.5, 1.0, 1.5, 2.1, 2.5, and 2.9 eq of Trxred per mol of flavin) are plotted in the main panel.
Hydrogen Peroxide Is a Product of Ero1p-Catalyzed Thiol Oxidation. Previous studies reported only low levels of hydrogen peroxide formation during catalysis by Ero1p (3), although typical FAD-binding oxidases produce hydrogen peroxide (22). Because the generation of reactive oxygen species during oxidative folding in the ER is of considerable potential significance (12, 14, 15), we reinvestigated this issue by using two independent methods. Trxred was chosen as the thiol substrate because it is rapidly and completely oxidized at low concentrations. Fig. 4A illustrates oxygen consumption during Ero1p-mediated oxidation of 50 μM Trxred. After a drop of 49 μM oxygen, the addition of catalase restored 23 μM oxygen to the solution, indicating that 46 μM hydrogen peroxide [stoichiometry 2H2O2 ↔ 2H2O + O2 (reviewed in ref. 23)] had been present. Thus, the generation of one disulfide yielded 0.94 molecules of hydrogen peroxide.
Fig. 4.

Oxygen is reduced by Ero1p activity to form hydrogen peroxide. (A) The reaction was initiated at time 0 by injecting the shorter construct of Ero1p to a final concentration of 1 μM into a solution containing 50 μM Trxred, and oxygen consumption was monitored. Approximately half of the oxygen originally consumed during Trxred oxidation was restored with the addition of catalase (arrow), indicating that oxygen has been reduced to H2O2 in the initial reaction. Similar results were obtained for the longer construct. (B) The reaction was initiated at time 0 by injecting the shorter form of Ero1p to a final concentration of 1 μM into a rapidly stirred solution containing 100 μM Trxred. Samples were withdrawn and mixed with the PeroXOquant detection reagent. Absorbance values were converted to H2O2 concentration according to a standard H2O2 calibration curve.
As an independent measure of peroxide formation, aliquots from a Trxred oxidation reaction were mixed with a colorimetric reagent for the quantitation of hydrogen peroxide (Fig. 4B). Because EDTA interferes with the development of the Fe(III)·xylenol orange chromagen, it was eliminated from the reaction. The absence of EDTA leads to a decreased peroxide yield (≈0.4 molecules per disulfide formed). A correspondingly lower recovery of hydrogen peroxide was obtained when EDTA was omitted from the experiment in Fig. 4A (data not shown). Although the peroxide yield is reduced, the lag phase in Fig. 4B mirrors that seen in the oxygen consumption curve obtained in Fig. 4A. This lag reflects the reductive activation of Ero1p; it is not seen when the Erv2p enzyme is assayed by using DTT as the thiol substrate, in which case hydrogen peroxide is produced with single-exponential kinetics (data not shown).
Km Value for Ero1p Reduction of Oxygen. Once we determined that hydrogen peroxide is the product of oxygen reduction during Ero1p-mediated thiol oxidation, we sought to determine the Km value for oxygen. The use of 12.5 mM DTT such that the thiol substrate concentration remained effectively unchanged upon oxygen depletion yielded an approximate Km value of 4 μM for oxygen (Fig. 5). Similar values were obtained for the long and short Ero1p constructs. Because the rate of the reaction is initially determined by the nature of the reducing substrate, the apparent Km value for oxygen will be different for different reductants. Using the longer Ero1p construct, we performed a similar experiment with 300 μM Trxred as the substrate of the reductive half-reaction and an initial oxygen concentration of around 30 μM such that the Trxred would be depleted by 10% during the course of the reaction. Under these conditions, the Km value of Ero1p for oxygen was found to be ≈10 μM (data not shown). These low Km values suggest that Ero1p can function efficiently at low oxygen tension.
Fig. 5.

Determination of the Km value for oxygen. Ero1p (shorter construct) was injected into a solution containing 12.5 mM DTT in the oxygen electrode chamber. The graph shows the end of the reaction, when the oxygen has been depleted to <10% of its initial value. Inset is the first derivative of the smoothed data, which was used to identify the point at which the rate was half maximal.
Effect of FAD on the Rates of Reactions Catalyzed by Ero1p. Because of previous reports that the addition of FAD to the enzyme solution increased activity in an RNase refolding assay (3), we tested the effect of FAD on the rate of oxygen consumption. We found that 100 μM FAD did not affect the oxygen consumption rate during Ero1p-catalyzed oxidation of either Trxred (Fig. 6A) or DTT (Fig. 6B). Similar results were obtained for 100 μM FMN and 100 μM riboflavin (Fig. 6). Because riboflavin and FMN are readily photo-reduced (24), all reactions were performed in the dark. Results were qualitatively similar for both Ero1p constructs and are shown for the longer version.
Fig. 6.

Effect of exogenous flavin compounds on the rate of oxygen consumption during Ero1p catalysis of disulfide formation. The experiments shown were performed by using the longer Ero1p construct, but the shorter version gave similar results. At time 0, Ero1p was injected to a final concentration of 1 μM into a solution containing 98 μM Trxred (A)or10mMDTT(B) to obtain the curves indicated by filled symbols. Traces measured in the absence of enzyme are indicated by open symbols. The black curves show the kinetics of oxygen consumption without added flavin compounds. The red, orange, and gold curves show the kinetics of oxygen consumption under similar conditions, except that the indicated flavin compounds were present in the reaction (100 μM FAD, 100 μM FMN, and 100 μM riboflavin, respectively).
In the presence of Trxred, Ero1p catalyzed the transfer of reducing equivalents to both free FAD and FMN (Fig. 7). Experiments were performed anaerobically and monitored by a decrease in absorbance at 446 nm. The concentration of free flavin in these experiments was at least 20-fold greater than the concentration of enzyme, indicating that reduction of the flavin occurred catalytically. In the absence of Ero1p, direct reduction of free flavin by Trxred was negligible.
Fig. 7.

Reduction of flavins in the absence of oxygen. The reductant was 50μM Trxred, and the electron acceptors were 21 μM FAD or 30 μM FMN. When present, the shorter construct of Ero1p was at a concentration of 1 μM. The longer version of Ero1p also reduces exogenous flavins (data not shown).
Ero1p Transfer Electrons to Nonflavin Exogenous Acceptors. In addition to reducing free flavin in solution, Ero1p could also catalyze transfer of electrons to nonflavin acceptors. Although many potential small-molecule electron acceptors had to be excluded from our studies because of high rates of nonenzymatic reduction by the thiol substrates, certain metalloproteins [i.e., horse cytochrome c, yeast cytochrome b5 (data not shown), and bacterial azurin] exhibited sufficiently slow noncatalytic reduction that they could be tested in our system. These proteins were reduced catalytically in the presence of Ero1p with Trxred as the reducing substrate (Fig. 8). Regardless of the electron acceptor, its reduction was detected only after a lag, similar to the kinetics observed for consumption of oxygen, when present (Fig. 6). The abrupt decrease in reaction rate as the concentrations of azurin or cytochrome c approached zero indicates qualitatively that the Km values for these proteins are very low.
Fig. 8.

Ero1p-catalyzed reduction of protein metal sites in the absence of oxygen. The reductant was 50 μM Trxred, and the electron acceptors were cytochrome c (open circles) or azurin (filled circles) at 25 μM. In both cases, the uncatalyzed transfer of electrons from donor to acceptor was negligible.
Discussion
In this study, we demonstrate that Ero1p generates hydrogen peroxide when acting on thiol substrates in the presence of oxygen, as expected of a typical flavin-containing oxidase (22). This observation contrasts with a reported stoichiometry of ≈0.05 hydrogen peroxide molecules per disulfide bond introduced into reduced RNase (3). Although differences in the Ero1p constructs could conceivably influence the reaction stoichiometry, we note that hydrogen peroxide production is frequently underestimated because it can decompose in the presence of trace metals or can react with thiols (25). In addition, thiols interfere with quantitation of hydrogen peroxide by peroxidase-mediated chromagen formation (26). The low Km value of Ero1p for oxygen suggests that the reduction of oxygen to hydrogen peroxide is a relevant route for electron transfer during Ero1p-catalyzed disulfide formation, except under severe hypoxic conditions. That reactive and potentially damaging redox species are the outcome of catalysis of disulfide bond formation may explain why induction of Ero1p by the ER stress response pathway leads to the production of excess peroxides in Caenorhabditis elegans (14).
In the absence of oxygen, however, we find that flavin compounds can also serve as electron acceptors for Ero1p. This reduction of free flavin by Ero1p cannot be the result of an exchange between bound and free flavin because, under the conditions of the assay, no loss of the stoichiometrically bound FAD cofactor could be detected in dialysis measurements (data not shown). Furthermore, Erv2p, an enzyme with no sequence homology to Ero1p but containing a similar core structure and arrangement of functional groups, also catalyzes reduction of free flavins (data not shown). Precedents for reduction of exogenous flavins by flavoproteins exist, including the reductase modules of two-component monooxygenases (reviewed in ref. 27) and an Amphibacillus xylanus NADH oxidase (28). Although the capacity for electron transfer between flavoproteins and exogenous flavins in vitro appears to be a general phenomenon (24), its physiological implications in the present case are less clear. The ability of flavins to function as electron acceptors for Ero1p may be shared by other chromophores and does not necessarily indicate a specific role for free flavins in thiol oxidation by this enzyme. To resolve this issue, two questions need to be addressed in future work. The first question is whether free flavin is indeed present at low micromolar concentrations in the ER (3) and, if so, whether it is a kinetically competent oxidant of Ero1p under such conditions. The second question is how reduced free flavin might be reoxidized during oxidative folding under anaerobic conditions.
Experiments showing that Ero1p activity in vivo is tied to the amount of free intracellular FAD have been interpreted as evidence that Ero1p activity can be allosterically controlled by FAD (3). However, the importance of intracellular FAD levels is also consistent with a role for FAD as a cofactor and/or possible electron acceptor for Ero1p. An explanation for the synthetic lethality of the ero1-1 and fad1-1 mutants (3) may be that the folding and, hence, active levels of the temperature-sensitive Ero1p variant are particularly sensitive to the amount of FAD available during its biosynthesis. The Gly-to-Ser mutation at position 229 encoded by ero1-1 is adjacent to the FAD-binding site and is likely to affect the coupling of folding and cofactor binding.
Our experiments also raise the possibility that nonflavin molecules may be on the pathway for Ero1p catalysis of disulfide bond formation under anaerobic conditions. Two heme-binding proteins and a protein with a copper center all served as efficient electron acceptors from Ero1p under anaerobic conditions in vitro. It was recently shown that ALR, a mammalian homolog of Erv2p found in the mitochondrial intermembrane space, can use cytochrome c as an electron acceptor (21). In this article, we demonstrate that an ER thiol oxidase can also transfer electrons to cytochrome c and to other proteins that function in electron transport chains. One significant difference between Ero1p and ALR, however, is that ALR uses molecular oxygen as an electron acceptor relatively poorly, ≈100 times less efficiently than it uses cytochrome c (21), whereas Ero1p can use both oxygen and other electron acceptors efficiently. Although the metalloproteins tested in this study are not known to function in the lumen of the ER, the capacity of Ero1p to use diverse electron acceptors leads us now to consider a variety of redox active molecules in the search for the physiological electron acceptors for Ero1p.
Acknowledgments
We are grateful to Amnon Horovitz and Israel Pecht for helpful discussions and to Milko Van Der Boom for providing access to an anaerobic chamber. This work was supported by a U.S.-Israel Binational Science Foundation grant (to D.F. and C.A.K.) and National Institutes of Health Grant GM26643 (to C.T.). D.F. is an incumbent of the Lilian and George Lyttle Career Development Chair.
Author contributions: E.G., C.S.S., C.A.K., C.T., and D.F. designed research; E.G., N.H., E.V., M.B., C.T., and D.F. performed research; C.A.K., C.T., and D.F. analyzed data; and C.A.K., C.T., and D.F. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ER, endoplasmic reticulum; CTAB, cetyltrimethylammonium bromide; Trxred, reduced Trx1.
References
- 1.Frand, A. R. & Kaiser, C. A. (1998) Mol. Cell 1, 161-170. [DOI] [PubMed] [Google Scholar]
- 2.Pollard, G. M., Travers, J. K. & Weissman, J. S. (1998) Mol. Cell 1, 171-182. [DOI] [PubMed] [Google Scholar]
- 3.Tu, B. P. & Weissman, J. S. (2002) Mol. Cell 10, 983-994. [DOI] [PubMed] [Google Scholar]
- 4.Gross, E., Kastner, D. B., Kaiser, C. A. & Fass, D. (2004) Cell 117, 601-610. [DOI] [PubMed] [Google Scholar]
- 5.Frand, A. R. & Kaiser, C. A. (2000) Mol. Biol. Cell 11, 2833-2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross, E., Sevier, C. S., Vala, A., Kaiser, C. A. & Fass, D. (2002) Nat. Struct. Biol. 9, 61-67. [DOI] [PubMed] [Google Scholar]
- 7.Gerber, J., Mühlenhoff, U., Hofhaus, G., Lill, R. & Lisowsky, T. (2001) J. Biol. Chem. 276, 23486-23491. [DOI] [PubMed] [Google Scholar]
- 8.Sevier, C. S., Cuozzo, J. W., Vala, A., Åslund, F. & Kaiser, C. A. (2001) Nat. Cell Biol. 3, 874-882. [DOI] [PubMed] [Google Scholar]
- 9.Thorpe, C., Hoober, K. L., Raje, S., Glynn, N. M., Burnside, J., Turi, G. K. & Coppock, D. L. (2002) Arch. Biochem. Biophys. 405, 1-12. [DOI] [PubMed] [Google Scholar]
- 10.Hoober, K. L. & Thorpe, C. (1999) Biochemistry 38, 3211-3217. [DOI] [PubMed] [Google Scholar]
- 11.Raje, S. & Thorpe, C. (2003) Biochemistry 42, 4560-4568. [DOI] [PubMed] [Google Scholar]
- 12.Tu, B. P. & Weissman, J. S. (2004) J. Cell Biol. 164, 341-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu, B. P., Ho-Schleyer, S. C., Travers, K. J. & Weissman, J. S. (2000) Science 290, 1571-1574. [DOI] [PubMed] [Google Scholar]
- 14.Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R., et al. (2003) Mol. Cell 11, 619-633. [DOI] [PubMed] [Google Scholar]
- 15.Haynes, C. M., Titus, E. A. & Cooper, A. A. (2004) Mol. Cell 15, 767-776. [DOI] [PubMed] [Google Scholar]
- 16.Beinert, H. (1960) The Enzymes (Academic, New York), 2nd Ed., Vol. 2, pp. 339-416. [Google Scholar]
- 17.Gill, S. C. & von Hippel, P. H. (1989) Anal. Biochem. 182, 319-326. [DOI] [PubMed] [Google Scholar]
- 18.Ellman, L. G. (1959) Arch. Biochem. Biophys. 82, 70-77. [DOI] [PubMed] [Google Scholar]
- 19.Williams, C. H., Arscott, L. D., Matthews, R. G., Thorpe, C. & Wilkinson, K. D. (1979) Methods Enzymol. 62, 185-198. [DOI] [PubMed] [Google Scholar]
- 20.Hoober, K. L., Joneja, B., White, H. B., III, & Thorpe, C. (1996) J. Biol. Chem. 271, 30510-30516. [DOI] [PubMed] [Google Scholar]
- 21.Farrell, S. R. & Thorpe, C. (2005) Biochemistry 44, 1532-1541. [DOI] [PubMed] [Google Scholar]
- 22.Massey, V. (1994) J. Biol. Chem. 269, 22459-22462. [PubMed] [Google Scholar]
- 23.Chelikani, P., Fita, I. & Loewen, P. C. (2004) Cell. Mol. Life Sci. 61, 192-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massey, V., Stankovich, M. & Hemmerich, P. (1978) Biochemistry 17, 1-8. [DOI] [PubMed] [Google Scholar]
- 25.Winterbourn, C. C. & Metodiewa, D. (1999) Free Radical Biol. Med. 27, 322-328. [DOI] [PubMed] [Google Scholar]
- 26.Raje, S., Glynn, N. & Thorpe, C. (2002) Anal. Biochem. 307, 266-272. [DOI] [PubMed] [Google Scholar]
- 27.Tu, S. C. (2001) Antioxid. Redox Signaling 3, 881-897. [DOI] [PubMed] [Google Scholar]
- 28.Niimura, Y., Nishiyama, Y., Saito, D., Tsuji, H., Hidaka, M., Miyaji, T., Watanabe, T. & Massey, V. (2000) J. Bacteriol. 182, 5046-5051. [DOI] [PMC free article] [PubMed] [Google Scholar]