

Abstract
The binding of small molecules to distinctive three-dimensional structures in mRNA provides a new dimension in RNA control, previously limited to the targeting of secondary structures with antisense and RNA interference; such targeting can modulate mRNA function and rates of protein biosynthesis. Small molecules that selectively bind the iron-responsive element (IRE), a specific three-dimensional structure in the noncoding region of the ferritin mRNA model that is recognized by the iron-regulatory protein repressor, were identified by using chemical footprinting. The assay used involved an oxoruthenium(IV) complex that oxidizes guanine bases in RNA sequences. Small molecules that blocked oxidation of guanines in the internal loop region were expected to selectively increase the rate of ferritin synthesis, because the internal loop region of the ferritin IRE is distinctive from those of other IREs. The natural product yohimbine was found (based on gel mobility shifts) to block cleavage of the internal loop RNA site by >50% and seemed to inhibit protein binding. In the presence of yohimbine, the rate of biosynthesis of ferritin in a cell-free expression system (rabbit reticulocyte lysate) increased by 40%. Assignment of the IRE–yohimbine interaction as the origin of this effect was supported by a similar increase in synthesis of luciferase protein in a chimera of the IRE and luciferase gene. The identification of a small, drug-like molecule that recognizes a naturally occurring three-dimensional mRNA structure and regulates protein biosynthesis rates raises the possibility that small molecules can regulate protein biosynthesis by selectively binding to mRNA.
Keywords: footprinting, protein synthesis, RNA structure, RNA-binding drugs, gene expression
The targeting of small molecules to three-dimensional structures in RNA has the potential to change the synthesis rate of the encoded proteins (1–4); an example is alteration of the control of reverse-transcription rate rates of HIV-1 RNA by trans-activation responsive region RNA, a target of many drug-like molecules (5–9). A principal challenge in targeting mRNA is identifying three-dimensional RNA structures that are functionally relevant and exhibit structures amenable to selective binding in the presence of large quantities of other nucleic acids (5–7). Recent findings on the activation of bacterial riboswitches by metabolites provide incentive to pursue mRNA binding as a new avenue for pharmaceutical research (10), particularly given the intriguing possibility that such elements exist in eukaryotes (11, 12). The advantages of using drug-like molecules to target three-dimensional RNA targets include the ability to escape biological systems that respond to targeting helical RNA structures (13), the ability to identify potentially useful RNA targets in genome sequences (14, 15), and the possibility of synthesizing reasonably large quantities of RNA for screening by chemical methods (2, 16). An additional appeal is that chemical-footprinting techniques, which are currently more effective for nucleic acids than proteins, allow for determining the precise site of binding of a small molecule on an RNA target by using routine sequencing methods on small quantities of material (17–19). We demonstrate here that such a chemical-footprinting technique can be used to identify a drug-like, RNA-binding molecule that is selective for a physiologically relevant site in mRNA and induces a change in the synthesis rate of the encoded protein.
There have been a limited number of reported ligand–RNA interactions that affect protein synthesis (20–22). Werstuck and Green (20) described such a system in which RNA aptamers selected for aminoglycoside antibiotics were inserted within the 5′ untranslated region (UTR) of a gene; subsequent translation in the presence of the selected antibiotic showed a dose-dependent inhibitory effect on translation of protein both in vitro and in living cells. In addition, it has been reported that forcing a nonphysiologically relevant RNA–protein interaction in the 5′ UTR of a gene by using a secondary ligand–RNA interaction can diminish protein synthesis dramatically (22). Another study identified an intercalator that targeted the hepatitis C virus internal ribosome-entry site to repress protein synthesis (21). To date, we are not aware of a system wherein an RNA–ligand interaction was used to increase the synthesis rate of an encoded protein.
A family of related, folded, stem-loop structures in mRNAs called iron-responsive elements (IREs; Fig. 1) coordinate the synthesis of proteins required for iron homeostasis (23, 24). Protein repressors called iron-regulatory proteins (IRPs) selectively recognize the IRE RNA structures and prevent protein synthesis by inhibiting ribosome binding (25–27). The ferritin IRE RNA folded in vivo has the same structure as in vitro, based on results with chemical nuclease probes for three-dimensional RNA structure protein in cultured mammalian cells (28). At normal cell iron concentrations, the ferritin mRNA is repressed by IRP binding but is activated when iron concentrations increase through changes in the IRE–IRP complex (17). Physiologically, such a situation occurs when genetic diseases such as sickle cell disease are treated with regular blood transfusions, leading to iron overload and saturation of available ferritin with iron (17); excess iron then has to be removed by cumbersome chelation therapies. Because only ≈50% of the ferritin mRNA is activated (29, 30), a strategy for increasing ferritin synthesis is finding a small molecule that binds to the ferritin IRE, blocks or alters binding of the IRP, and allows translation of the ferritin mRNA (3). A challenge in such a strategy is identifying a small molecule that selectively interacts with the ferritin IRE compared to mRNA with other IRE isoforms, such as the transferrin receptor (TfR) IRE. Members of the IRE family share the hairpin loop structure but vary in the structure of the helical stem. The ferritin IRE has a distinctive internal loop structure (U6-C8 with a G7-C25 base pair) that is absent in other IRE isoforms (31–34). Therefore, compounds that bind to the internal loop of the ferritin IRE should be selective for the ferritin mRNA over other IREs.
Fig. 1.

RNA secondary structures of human ferritin IRE, mutant TfR IRE, and the structure of the small molecule yohimbine.
A number of chemical reagents have been used to oxidize and cleave RNA, which revealed a variety of structural features and interactions (17–19). We have focused on complexes based on Ru(tpy)(bpy)O2+ (tpy, 2,2′,2″-terpyridine; bpy, 2,2′-bipyridine), which oxidizes guanines in RNA based on their solvent accessibility (35, 36). Reaction of the ferritin IRE with Ru(tpy)(bpy)O2+ produces cleavage at guanine with intensities that track the solvent accessibilities determined from NMR structures (16, 37). Based on the differences in structures for the ferritin and TfR IREs, we reasoned that small molecules that blocked sites in the region of the ferritin internal loop (i.e., G23, G26, G27) but not guanines in the hairpin loop (i.e., G16, G18) would be promising candidates for selective binding to the ferritin IRE. In this article, a small molecule that blocks oxidation of guanines by Ru(tpy)(bpy)O2+ adjacent to the internal loop is shown to bind to the IRE and change or disrupt the IRE–IRP complex to increase the rate of ferritin synthesis in cell-free extracts.
Materials and Methods
Cloning, Transcription, and Ru(tpy)(bpy)O2+ Assay. Desired sequences along with the T7 promoter sequence were cloned by PCR into pUC19K vectors. Plasmids were digested with a restriction enzyme to allow for run-off transcription by using a MEGAshortscript T7 Kit (Ambion, Austin, TX). RNA was 3′-end-labeled with 5′-[32P]pCp (PerkinElmer) by using T4 RNA ligase overnight at 4°C (38). Ru-OH2 was synthesized according to published procedures (39). The RuO2+ oxidant was generated by bulk electrolysis, holding the aqueous solution at 0.85 V for 10 min. [32P]RNA was folded (40 mM Tris/100 mM KCl, pH 7.2) by heating to 95°C for 5 min followed by slow cooling to room temperature. [32P]RNA and yohimbine HCL (Sigma) were incubated at room temperature for 15 min; RuO2+ (100 μM) was added and reacted for 5 min. The reaction was quenched with ethanol, speed-vacuumed to dryness, aniline-treated, and washed twice with water. Samples were separated on 20% (7 M urea) denaturing gels, which then were exposed to a phosphorimaging screen overnight and analyzed by using a phosphorimager. Bands were quantitated by using imagequant software.
Gel Mobility-Shift Assay. [32P]RNA was folded (10 mM Hepes/40 mM KCl, pH 7.2); 5 μl of folded [32P]RNA, binding buffer (10 mM Hepes/40 mM KCl/0.3% Nonidet P-40/5% glycerol/3 mM MgCl2), yohimbine, and heparin (5 mg/ml) were incubated at 30°C for 20 min. Human IRP1 (65 nM; MBI Fermentas, Amherst, NY) and DTT (1 mM) were added, and incubation continued at 37°C for 20 min. The reaction was terminated by the addition of 80% glycerol/bromophenol blue dye. RNA–protein complexes were separated on 6% nondenaturing gels (29:1 acrylamide/bisacrylamide) at 4°C. Gels were dried and phosphorimaged. Bands were quantitated by using imagequant software. For shift assays on full-length mRNA, the conditions were the same as described above with the following exceptions: [α-32P]UTP was added to the transcription reaction as a trace label, 220 nM RNA and 660 nM IRP1 or IRP2 was used, and 4% nondenaturing gels were run for at least 16 h. For these studies, the human IRP1 and IRP2 used were isolated as described by Allerson et al. (40).
Small-Molecule Translation Assay. Full-length human ferritin mRNA (human H-chain; the 5′ UTR is 268 bases and contains the IRE) was transcribed from a linearized pCRII-TOPO vector by using the SP6 promoter. For the G18A ferritin mutant, site-directed mutagenesis was performed on the human ferritin sequence [from pCRII-TOPO vector, subcloned into pBluescript IISK(+)] by using a QuikChange mutagenesis kit (Stratagene), and RNA was transcribed from the linearized vector by using the T7 promoter. For IRE-luc, a 1,739-bp fragment containing the gene coding for Luciferase from a pGEM-luc vector (Promega) was amplified by PCR and blunt-end ligated into a vector containing the human IRE sequence. The resulting plasmid contained the IRE 18 bases upstream of the luciferase start codon, and RNA was synthesized off of the T7 promotor.
Translation of mRNA (133 nM), after preincubation with or without yohimbine for 20 min at 30°C, used reticulocyte lysates (Promega) and [35S]methionine (PerkinElmer; final volume, 25 μl) incubated at 30°C for 15 min. Newly synthesized protein was analyzed by electrophoresis on 16% polyacrylamide gels in Tris-glycine SDS buffer (pH 8.3), which was run for 1.75 h, fixed in 50% methanol/7% acetic acid for 10 min, dried, and exposed to a phosphorimaging screen overnight. Protein bands (based on incorporation of [35S]methionine) were quantitated by using imagequant software.
Fluorescence Binding Assay. Steady-state fluorescence measurements were performed on a Cary Eclipse fluorimeter (Varian). All measurements were made at room temperature in 50 mM Tris (pH 7.4)/100 mM NaCl/1 mM MgCl2 on RNA that had been melted and reannealed. The excitation wavelength was 290 nm, and the emission wavelength was 359 nm. The Kd of the yohimbine–RNA complex was calculated by curve fitting of the fluorescence intensity as a function of [RNA] at a concentration of 10 μM yohimbine by using the following equation (assuming 1:1 binding):
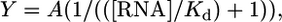 |
[1] |
where A is the difference in fluorescence intensity at 0 μM RNA and an infinite RNA concentration. Binding assays were repeated at least three times on two different preparations of RNA.
Results
Identification of IRE Ligands. Several commercially available small molecules have been studied for binding to RNA (6, 41). A subset of these molecules was selected and screened for selective binding near the internal loop IRE, as revealed by oxidation with Ru(tpy)(bpy)O2+. We primarily focused on compounds based on the parent structures of indole, phenothiazine, and diazepam. In this experiment, radiolabeled IRE oligoribonucleotide (50-mer) was subjected to oxidation with Ru(tpy)(bpy)O2+ in the presence of increasing amounts of the selected small molecule. The majority of the compounds screened blocked oxidation of G16 but did not affect oxidation of the guanines in the region of the internal loop. One compound, the natural product yohimbine (42) (Fig. 1) selectively inhibited cleavage at G23 and G27 by >50% with little effect on G16 (Fig. 2), indicating selective binding of yohimbine in the region of the ferritin IRE-specific internal loop. The internal loop region confers selectivity to IRP binding among IRE isoforms (3). NMR studies on ferritin IRE structures used either the frog ferritin IRE sequence (33), which has the same internal loop sequence as the human L-ferritin, or a consensus sequence that has a C bulge, rather than the internal loop (34); the results were very similar except for the size of the helix distortion. In the human ferritin-H IRE used here (Fig. 1), which differs from human ferritin-L and frog ferritin IRE only by having a C-G closing base pair rather than a U-G, the G26 residue, also in the internal loop region of the IRE, was not protected by yohimbine from oxidation. This result is consistent with the observation that G26 and G27 exhibit different environments when probed by NMR and metal-binding studies (43).
Fig. 2.

Effect of yohimbine on guanine oxidation in human IRE RNA. (Left) Lanes 1–6, [RuO2+] = 100 μM. [yohimbine]: lane 1, 0 μM; lane 2, 1 μM; lane 3, 5 μM; lane 4, 10 μM; lane 5, 20 μM; lane 6, aniline-treated control; lane 7, RNA only control. (Right) Quantitations of [32P]RNA bands. Cleavage intensity is in arbitrary units normalized to cleavage at G0.
The native fluorescence of yohimbine was also used to study binding to the IRE. Titration of yohimbine with increasing quantities of IRE quenched the yohimbine fluorescence, and subsequent fitting of the emission profile, gave a binding constant of 3.9 ± 1.2 μM. The affinity is comparable in magnitude to values determined for a number of RNA–ligand interactions identified by systematic evolution of ligands by exponential enrichment (SELEX) (44, 45) and reflects the overall binding of yohimbine to the RNA, not necessarily that for the specific internal loop site. Although the data fit well for the formation of a 1:1 yohimbine–RNA complex, the binding of a second yohimbine cannot be completely excluded.
The effect of yohimbine binding to the ferritin IRE was also analyzed as the shift in electrophoretic mobility by using radio-labeled IRE and the IRP. Addition of yohimbine to the ferritin IRE inhibited formation of the IRE–IRP complex and concomitantly increased the quantity of free RNA (Fig. 3). As shown in Fig. 3, the increase in the quantity of free RNA was 8.0 ± 2.7% in the presence of 40 μM yohimbine. Given the dissociation constant of the IRE–IRP interaction of 90 pM (46) and the concentrations of IRP and yohimbine, the calculated displacement is 8%, which is in good agreement with the observed displacement.
Fig. 3.

Gel mobility-shift assay. (Left) Upper band is the shifted ferritin IRE RNA–IRP1 complex, and the lower band is unshifted RNA. [IRP1] = 65 nM; [RNA] = 32.5 nM. [yohimbine]: lane 1, 0 μM; lane 2, 10 μM; lane 3, 20 μM; lane 4, 40 μM; lane 5, 0 μM, no IRP1. (Right) Quantitations of [32P]RNA bands. Legend values indicate the final concentration of yohimbine in the assay.
Selectivity of the yohimbine–ferritin IRE interactions was evaluated by comparing the interaction of yohimbine with ferritin to an IRE isoform, a single TfR IRE (Fig. 1), which has a C bulge instead on an internal loop using Ru(tpy)(bpy)O2+ oxidation as a probe. The G selectivity of the probe and the decrease in potential sites caused by the relatively A-U-rich base pairing in TfR-IRE helices, required conversion of a G-C base pair to a G-U base pair (C24 → U24 substitution) (Fig. 1) to provide a reporter near the C bulge. The mutation did not alter the predicted folding of the structure as determined by mfold analysis (47, 48). When the mutant TfR IRE was oxidized by Ru(tpy)(bpy)O2+ with increasing amounts of yohimbine, oxidation at G16 was inhibited but the guanines near the C bulge were not blocked (Fig. 4), in contrast to the ferritin IRE, which emphasizes the selectivity of yohimbine binding to the ferritin IRE. The dissociation constants of yohimbine for both the native TfR IRE and the mutant were detectably weaker than that for the ferritin IRE by fluorescence titration (see supporting information, which is published on the PNAS web site). Another measure of the selectivity of the yohimbine–ferritin IRE interaction is the fact that the majority of the other compounds tested with the ferritin IRE gave results similar to those of the yohimbine–TfR IRE interaction.
Fig. 4.

Effect of yohimbine on guanine oxidation in the C24 → U24 mutant TfR IRE. (Left) Lanes 1–6, [RuO2+] = 100 μM. [yohimbine]: Lane 1, 0 μM; lane 2, 1 μM; lane 3, 5 μM; lane 4, 10 μM; lane 5, 20 μM; lane 6, 40 μM; lane 7, RNA-only control; lane 8, aniline-treated control. (Right) Quantitations of [32P]RNA bands. Cleavage intensity is in arbitrary units normalized to the intensity at G0.
Acceleration of Ferritin Synthesis by Yohimbine. To determine whether binding of yohimbine to the internal loop of the ferritin IRE would increase the rate of ferritin translation, we analyzed ferritin synthesis directed by full-length ferritin mRNA in a eukaryotic cell-free expression system, generated by lysing rabbit reticulocytes. Addition of yohimbine to ferritin mRNA before translation increased ferritin synthesis by >40% (Fig. 5). In contrast, addition of Hoechst 33258, which is well known to bind nucleic acids (49), inhibited ferritin synthesis by >50% (data not shown). As shown in this (see supporting information) and earlier work (16, 43), a G18A mutation in the ferritin IRE (G18 forms a base pair with C14 in the IRE terminal loop) (33, 34) displays a reduced affinity for IRP (16, 43), which increases ferritin mRNA translation. Translation of mRNA with the G18A mutation in the IRE was unaffected by yohimbine (Fig. 5), again indicating the selectivity of the yohimbine effect for the native ferritin-IRE structure.
Fig. 5.

Effects of yohimbine on IRE-dependent mRNA translation in cell-free extracts as measured by incorporation of [35S]methionine (see Materials and Methods). Data are representative of at least three different trials on at least two separate preparations or batches of RNA. The results are expressed as a percentage of the control (no yohimbine added), and the error bars represent the SD.
To rule out any possible contributions of the mRNA coding sequence on yohimbine-enhanced ferritin mRNA translation, the ferritin IRE was cloned upstream of the sequence coding for the luciferase protein (IRE-luc). The chimeric mRNA was shown to bind both IRP1 and IRP2; luciferase synthesis directed by the IRE-luciferase chimeric mRNA was repressed comparably to ferritin (see supporting information). Addition of yohimbine to IRE-luc produced a similar effect to ferritin with an increase of >40% in the levels of luciferase synthesis (Fig. 5). Taken together, these data indicate that yohimbine selectively alters mRNA repression by the ferritin IRE–IRP interaction.
Discussion
The three conclusions to be drawn from these experiments are that (i) differential binding of small molecules to specific sites in mRNA can be identified through chemical oxidation reactions, and the targeting apparent in the footprinting experiment is confirmed through examining alternative sequences and compounds and by comparison of dissociation constants; (ii) differential binding of small molecules to specific three-dimensional structures in mRNA can alter mRNA translation in a cell-free expression system; and (iii) small molecules can effectively distinguish between the different members of the IRE family of regulatory structures to enhance RNA function. These strategies should prove useful in developing therapeutic approaches to increasing iron-storage capacity during iron overload and to targeting related mRNA elements.
When yohimbine selectively bound the internal loop of the ferritin IRE, the IRE/IRP repression was changed as indicated by up-regulation of ferritin mRNA (Fig. 5). The increase in translation could result simply from the increase in free RNA generated by yohimbine binding, as seen in the gel-shift assay. However, the 40% increase in synthesized protein is greater than what would be expected solely from free RNA, because the ferritin mRNA (133 nM) saturates the endogenous IRP (9–12 nM) (50) in the cell-free expression system. The yohimbine effect, therefore, is more than simply displacing the IRP. One explanation could be that unwinding of the IRE structure by yohimbine is more complete than by endogenous initiation factors, a conjecture supported by the observations that during iron overload, when the IRP is presumed to be displaced (51), 40–50% of endogenous ferritin mRNA remains untranslated (52, 53). The complexity of the interactions among the IRE RNA, IRP repressor, and initiation factor proteins is illustrated by the binding of the IRP repressor to both the IRE and the large initiation scaffold protein, eIF-4G (54). It likely is a combination of decreased IRP binding coupled with unwinding of the IRE structure by yohimbine that results in the increased protein-synthesis levels observed.
Positive regulation of mRNA translation by a small molecule, illustrated here for the binding of yohimbine to a natural RNA helix distortion in the ferritin IRE, is an example of a process that can find use where increased synthesis would be efficacious. Earlier examples of small molecules that altered translation acted through dimerization (22) or intercalation (21) and inhibited translation. In addition, until now, manipulating mRNA function with small molecules has relied on in vitro aptamer selection of the RNA followed by insertion of the aptamer into the UTR of mRNA (20, 22) or the use of engineered RNA–protein interactions to control protein synthesis (22). The demonstration that natural, noncoding RNA regulatory structures in mRNAs for normal proteins such as ferritin can be recognized and targeted by small molecules indicates the importance of searching for other targets in mRNAs encoding proteins in which increased synthesis is needed (e.g., in insulin deficiency).
Supplementary Material
Acknowledgments
We are grateful to Dr. Joanne C. Long for construction of the mRNA template used in the small-molecule translation assay and to Dr. Tracey A. Rouault for the clones of human iron-regulatory protein. This research was supported by National Institutes of Health Grant DK020251.
Author contributions: J.D.T., P.M.F., and P.A.R. performed research; and E.C.T. and H.H.T. designed research.
Conflict of interest statement: No conflicts declared.
Abbreviations: IRE, iron-responsive element; IRP, iron-regulatory protein; TfR, transferrin receptor; tpy, 2,2′,2″-terpyridine; bpy, 2,2′-bipyridine.
References
- 1.Pearson, N. D. & Prescott, C. D. (1997) Chem. Biol. 4, 409-414. [DOI] [PubMed] [Google Scholar]
- 2.Ecker, D. J. & Griffey, R. H. (1999) Drug Discov. Today 4, 420-429. [DOI] [PubMed] [Google Scholar]
- 3.Theil, E. C. & Eisenstein, R. S. (2000) J. Biol. Chem. 275, 40659-40662. [DOI] [PubMed] [Google Scholar]
- 4.Xavier, K. A., Eder, P. S. & Giordano, T. (2000) Trends Biotechnol. 18, 349-356. [DOI] [PubMed] [Google Scholar]
- 5.Dassonneville, L., Hamyl, F., Colson, P., Houssier, C. & Bailly, C. (1997) Nucleic Acids Res. 25, 4487-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lind, K. E., Du, Z., Fujinaga, K., Peterlin, B. M. & James, T. L. (2002) Chem. Biol. 9, 185-193. [DOI] [PubMed] [Google Scholar]
- 7.Chapman, R. L., Stanley, T. B., Hazen, R. & Garvey, E. P. (2002) Antiviral Res. 54, 149-162. [DOI] [PubMed] [Google Scholar]
- 8.Tor, Y. (2003) Chembiochem. 4, 998-1007. [DOI] [PubMed] [Google Scholar]
- 9.Leeper, T. C., Athanassiou, Z., Dias, R. L., Robinson, J. A. & Varani, G. (2005) Biochemistry 44, 12362-12372. [DOI] [PubMed] [Google Scholar]
- 10.Mandal, M., Lee, M., Barrick, J. E., Weinberg, Z., Emilsson, G. M., Ruzzo, W. L. & Breaker, R. R. (2004) Science 306, 275-279, and erratum (2004) 306, 1477. [DOI] [PubMed] [Google Scholar]
- 11.Nudler, E. & Mironov, A. S. (2004) Trends Biochem. Sci. 29, 11-17. [DOI] [PubMed] [Google Scholar]
- 12.Famulok, M. (2004) Science 306, 233-234. [DOI] [PubMed] [Google Scholar]
- 13.Rocak, S. & Linder, P. (2004) Nat. Rev. Mol. Cell Biol. 5, 232-241. [DOI] [PubMed] [Google Scholar]
- 14.Alen, C. & Sonenshein, A. L. (1999) Proc. Natl. Acad. Sci. USA 96, 10412-10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stormo, G. D. & Ji, Y. (2001) Proc. Natl. Acad. Sci. USA 98, 9465-9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorp, H. H., McKenzie, R. A., Lin, P.-N., Walden, W. E. & Theil, E. C. (1996) Inorg. Chem. 35, 2773-2779. [Google Scholar]
- 17.Theil, E. C. (1994) New J. Chem. 18, 435-441. [Google Scholar]
- 18.Chow, C. S. & Barton, J. K. (1992) Methods Enzymol. 212, 219-242. [DOI] [PubMed] [Google Scholar]
- 19.Burrows, C. J. & Rokita, S. E. (1994) Acc. Chem. Res. 27, 295-301. [Google Scholar]
- 20.Werstuck, G. & Green, M. R. (1998) Science 282, 296-298. [DOI] [PubMed] [Google Scholar]
- 21.Malina, A., Khan, S., Carlson, C. B., Svitkin, Y., Harvey, I., Sonenberg, N., Beal, P. & Pelletier, J. (2005) FEBS Lett. 579, 79-89. [DOI] [PubMed] [Google Scholar]
- 22.Harvey, I., Garneau, P. & Pelletier, H. (2002) Proc. Natl. Acad. Sci. USA 99, 1882-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theil, E. C. (1994) Biochem. J. 304, 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aziz, N. & Munro, H. N. (1987) Proc. Natl. Acad. Sci. USA 84, 8478-8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrell, C. M., McKenzie, A. R., Patino, M. M., Walden, W. E. & Theil, E. C. (1991) Proc. Natl. Acad. Sci. USA 88, 4166-4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickey, L. F., Wang, Y. H., Shull, G. E., Wortman, I. A., III, & Theil, E. C. (1988) J. Biol. Chem. 263, 3071-3074. [PubMed] [Google Scholar]
- 27.Walden, W. E., Daniels-McQueen, S., Brown, P. H., Gaffield, L., Russell, D. A., Bielser, D., Bailey, L. C. & Thach, R. E. (1988) Proc. Natl. Acad. Sci. USA 85, 9503-9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ke, Y. & Theil, E. C. (2002) J. Biol. Chem. 277, 2373-2376. [DOI] [PubMed] [Google Scholar]
- 29.Zahringer, J., Baliga, B. S. & Munro, H. N. (1976) Biochem. Biophys. Res. Commun. 68, 1088-1093. [DOI] [PubMed] [Google Scholar]
- 30.Dandekar, T., Stripecke, R., Gary, N. K., Goossen, B., Constable, A., Johansson, H. E. & Hentze, M. W. (1991) EMBO J. 10, 1903-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ke, Y., Wu, J., Leibold, E. A., Walden, W. E. & Theil, E. C. (1998) J. Biol. Chem. 273, 23637-23640. [DOI] [PubMed] [Google Scholar]
- 32.Koeller, D. M., Casey, J. L., Hentze, M. W., Gerhardt, E. M., Chan, L.-N. L., Klausner, R. D. & Harford, J. B. (1989) Proc. Natl. Acad. Sci. USA 86, 3574-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gdaniec, Z., Sierzputowska-Gracz, H. & Theil, E. C. (1998) Biochemistry 37, 1505-1513, and erratum (1999) 38, 5676. [DOI] [PubMed] [Google Scholar]
- 34.Addess, K. J., Basilion, J. P., Klausner, R. D., Rouault, T. A. & Pardi, A. (1997) J. Mol. Biol. 274, 72-83. [DOI] [PubMed] [Google Scholar]
- 35.Carter, P. J., Cheng, C.-C. & Thorp, H. H. (1996) Inorg. Chem. 35, 3348-3354. [DOI] [PubMed] [Google Scholar]
- 36.Carter, P. J., Cheng, C.-C. & Thorp, H. H. (1998) J. Am. Chem. Soc. 120, 632-642. [Google Scholar]
- 37.Ciftan, S. A., Theil, E. C. & Thorp, H. H. (1998) Chem. Biol. 5, 679-687. [DOI] [PubMed] [Google Scholar]
- 38.Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Woodbury, NY), 2nd Ed.
- 39.Moyer, B. A. & Meyer, T. J. (1981) Inorg. Chem. 20, 436-444. [Google Scholar]
- 40.Allerson, C. R., Martinez, A., Yikilmaz, E. & Rouault, T. A. (2003) RNA 9, 364-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu, M.-C., Schutt, A. D., Holly, M., Slice, L. W., Sherman, M. I., Richman, D. D., Potash, M. J. & Volsky, D. J. (1991) Science 254, 1799-1802. [DOI] [PubMed] [Google Scholar]
- 42.Tam, S. W., Worcel, M. & Wyllie, M. (2001) Pharmacol. Ther. 91, 215-243. [DOI] [PubMed] [Google Scholar]
- 43.Ke, Y., Sierzputowska-Gracz, H., Gdaniec, Z. & Theil, E. C. (2000) Biochemistry 39, 6235-6242. [DOI] [PubMed] [Google Scholar]
- 44.Wang, Y. & Rando, R. R. (1995) Chem. Biol. 2, 281-290. [DOI] [PubMed] [Google Scholar]
- 45.Sassanfar, M. & Szostak, J. W. (1993) Nature 364, 550-553. [DOI] [PubMed] [Google Scholar]
- 46.Barton, H. A., Eisenstein, R. S., Bomford, A. & Munro, H. N. (1990) J. Biol. Chem. 265, 7000-7008. [PubMed] [Google Scholar]
- 47.Mathews, D. H., Sabina, J., Zuker, M. & Turner, D. H. (1999) J. Mol. Biol. 288, 911-940. [DOI] [PubMed] [Google Scholar]
- 48.Zuker, M. (2003) Nucleic Acids Res. 31, 3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blackburn, G. M. & Gait, M. J. (1996) Nucleic Acids in Chemistry and Biology (Oxford Univ. Press, New York), p. 528.
- 50.Dix, D. J., Lin, P.-N., Kimata, Y. & Theil, E. C. (1992) Biochemistry 31, 2818-2822. [DOI] [PubMed] [Google Scholar]
- 51.Hentze, M. W., Muckenthaler, M. U. & Andrews, N. C. (2004) Cell 117, 285-297. [DOI] [PubMed] [Google Scholar]
- 52.Melefors, O., Goossen, B., Johansson, H. E., Stripecke, R., Gray, N. K. & Hentze, M. W. (1993) J. Biol. Chem. 268, 5974-5978. [PubMed] [Google Scholar]
- 53.Zahringer, J., Baliga, B. S. & Munron, H. N. (1976) Proc. Natl. Acad. Sci. USA 73, 857-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Gregorio, E., Preiss, T. & Hentze, M. W. (1999) EMBO J. 18, 4865-4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.