

Abstract
The cycle of the photoactive yellow protein (PYP) has been extensively studied, but the dynamics of the isolated chromophore responsible for transduction is unknown. Here, we present real-time observation of the dynamics of the negatively charged chromophore and detection of intermediates along the path of trans-to-cis isomerization using femtosecond mass selection/electron detachment techniques. The results show that the role of the protein environment is not in the first step of double-bond twisting (barrier crossing) but in directing efficient conversion to the cis-structure and in impeding radical formation within the protein.
Keywords: femtobiology, transduction, molecular dynamics, photoelectron spectroscopy
Photoactive yellow protein (PYP) is a water-soluble photoreceptor found in Halorhodospira halophila and related halophilic bacterial species (1). The chromophore responsible for perception of light and phototactic response is a deprotonated trans-4-hydroxycinnamic acid (also known as p-coumaric acid) covalently linked to a cysteine residue of the protein via a thioester bond (Fig. 1). Light absorption of the PYP chromophore leads to the initiation of a complex photocycle involving several intermediates. With a variety of biophysical techniques (2–5), it is concluded that the first intermediate (I0)inthe photocycle is formed within a few picoseconds, with the chromophore changing to the cis configuration, in a trans-to-cis isomerization process (7–11). The intermediates at longer times have been trapped and identified by x-ray crystallography (12, 13).
Fig. 1.

Structure of the PYP and its chromophore. (a) Structure of the PYP, from refs. 5 and 6. The chromophore structure is indicated. (b) Chemical structure of the PYP chromophore and P-chromophore used in this study.
The critical change of the chromophore in the primary process of isomerization is accompanied or followed by several other processes: proton transfer, protein conformational change, solvation, and disruption of hydrogen-bonding. This complexity can be reduced if the chromophore in its true anionic, not neutral, structure can be studied free of perturbations. With model compounds, the solvent effect can be assessed by studying them in the solution phase (14–18). However, the nature of intramolecular change and the timescales involved are still unknown. It is, therefore, desirable to study the primary isomerization dynamics of the PYP chromophore in isolation, especially in view of the fact that the mechanism for forming the first intermediate (I0) is debatable (2). A gas-phase spectroscopic study has already been reported (19), but it was for the neutral molecule, not the anion structure in the protein.
In this work, we report direct observation of the dynamics of the isolated PYP anionic chromophore. To disentangle the role of protein binding and distant torsions, we preserved the central structure involved in the isomerization about the double bond but use the CH3 group for termination (see Fig. 1). The chromophore, hereafter called P, is excited with a femtosecond pulse from its ground state, the dark state in the protein, to the trans configuration using the mass-selected ions. For probing we use another femtosecond pulse to photodetach and resolve the photoelectrons at different kinetic energies. This resolution in time and energy provides the transient behavior of the intermediates involved. Remarkably, the P-chromophore shows primary step dynamics of twisting in 1 ps, which is essentially the same as that in the protein, indicating that the efficient, almost frictionless, transduction process by isomerization is central to the double bond. The meaning of pure double- vs. single-bond twisting is convoluted because of nuclear couplings and electronic/resonance delocalization (see conclusion). The role of the protein environment is primarily in directing the crossing to the ground-state cis structure and suppressing radical formation.
Results and Discussion
Fig. 2 shows the photoelectron spectra of the P-chromophore with and without the probe pulse. With the pump pulse alone (blue trace), a major peak was observed at ≈0.2 eV (1 eV = 1.602 × 10-19 J) together with a broad and much less intense peak at ≈2 eV; no signal was observed with the probe pulse alone. The sharp major peak at ≈0.2 eV is identified as the photoelectron signal from single-photon excitation; its energy matches well with the difference between the photon energy of the pump pulse (3.1 eV) and the vertical detachment energy of the anion to the radical ground state (≈2.9 eV).‡ With both the probe and pump pulses overlapping at 200 fs, a new peak appears at ≈0.9 eV in the electron kinetic energy, whereas the peak at ≈0.2 eV is depleted (the red trace in Fig. 2). As shown below, these temporal changes are present because of the evolution from the trans to cis configuration with the formation of intermediates. Because of the clear separation in energy, low and high kinetic energy, it is possible to map out the dynamics of different species.
Fig. 2.

Photoelectron spectra of the P-chromophore obtained with pump femtosecond pulse (blue trace) and pump-plus-probe femtosecond pluses (red trace) at 200-fs delay. (Inset) Blowup of the energy region of 0–1.5 eV, in which energy windows for time-dependent transients are marked as I, II, and III. The windows allow for probing of  ,
, 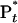 , and
, and 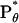 (see text).
(see text).
The temporal behavior at three windows of kinetic energies (I–III in Fig. 3) provides a stringent test of the nature of intermediates involved and the consistency of the rates. In Fig. 3, we display 15 such transients over different, short, and long timescales. Transients in the same row show temporal behaviors of a specific energy window, on different timescales, whereas those in the same column show the temporal behavior of different energy windows for a specific timescale. The clear trends in Fig. 3, depletion of the population in region I, decays (and coherence) in region II, and build-up and decay in region III, indicate that the dynamics involve three states of the chromophore: the initially launched population, the twisted intermediate, and the hot ground-state population of the cis form. To obtain the timescales involved and quantify the fraction of population for each structure, we invoked the following elementary steps:
 |
where Pdark is the structure in the ground state,  is the initially excited state,
is the initially excited state,  is the twisted state, and Pc is the hot-ground state of the cis form. All of the transients were fit with the same parameter set in a global search. The results of the global fit are shown as solid lines overlaid on the experimental transients in Fig. 3.
is the twisted state, and Pc is the hot-ground state of the cis form. All of the transients were fit with the same parameter set in a global search. The results of the global fit are shown as solid lines overlaid on the experimental transients in Fig. 3.
Fig. 3.

Time-resolved dynamics of the P-chromophore. Shown are the photoelectron spectra at three different kinetic energies and different timescales. Transients in each row are from the same energy window, and those in the same column have the same timescale. The theoretical fits are the solid lines superimposed on the experimental transients. The damped coherent oscillations are shown in region II on the short timescale. Note the clear depletion and recovery in window I and prompt response and decay in window II. In window III, the buildup and decay is evident; some contribution (prompt response) for the overlap of windows also is observed.
The agreement between the theoretical analysis and experimental results at different energies and the self-consistency among all transients are impressive and reflect the validity of the above picture with well defined rates for the elementary steps involved. In detail, the population of the initially excited state, P*, bifurcates on the femtosecond timescale to produce the trans population 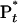 (80%); the remaining fraction (20%,
(80%); the remaining fraction (20%, 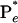 ) autodetach to form the corresponding radicals. This finding is evidenced in the decay of the population in region II (1 ps; 4 ps) and the recovery of population in region I (4 ps; 52 ps). Monitoring population in region III, we observed a clear buildup (1 ps) and a decay (52 ps). Accordingly, the high-energy region (II) results from the initial population (prompt response), and the low-energy region (III) results from the trapped, in a well, twisted intermediate. The isomerization by twisting occurs in 1 ps, whereas the twisted state (
) autodetach to form the corresponding radicals. This finding is evidenced in the decay of the population in region II (1 ps; 4 ps) and the recovery of population in region I (4 ps; 52 ps). Monitoring population in region III, we observed a clear buildup (1 ps) and a decay (52 ps). Accordingly, the high-energy region (II) results from the initial population (prompt response), and the low-energy region (III) results from the trapped, in a well, twisted intermediate. The isomerization by twisting occurs in 1 ps, whereas the twisted state ( ) decays in 52ps. A damped coherent oscillation was observed in the decay of the population of region II (see Fig. 3), and it has a period of ≈800 fs, corresponding to a vibrational frequency of ≈40 cm-1.
) decays in 52ps. A damped coherent oscillation was observed in the decay of the population of region II (see Fig. 3), and it has a period of ≈800 fs, corresponding to a vibrational frequency of ≈40 cm-1.
The above results are surprising for several reasons. First, isomerization time for the isolated chromophore is similar to that observed for the same chromophore in the solution phase (1.3 ps) (D. H. Paik, A. Espagne, M. M. Martin, and A.H.Z., unpublished data). Second, unlike the protein case, the chromophore is free from backbone bonding and can undergo other torsions and rotations, and yet the isomerization time is similar to that of the protein (within a few picoseconds) (7–11). Third, the coherence, which reflects the involvement of low-frequency motion(s) in isomerization (20), is preserved on going from the isolated molecule to the protein. For the chromophore in PYP, these oscillations were reported to be ≈50 and ≈135 cm-1 (21). One then must ask the following. Why is this nearly a frictionless dynamics? What is its significance to the function?
The structural change of the chromophore is a precursor to other dynamical events in the PYP function. After isomerization, protonation in the protein environment occurs, and the protein itself changes on a longer timescale. From theoretical calculations (22, 23), it was proposed that the trans-to-cis isomerization proceeds by, first, the passage through a small barrier for twisting around the double bond (coordinate θ) and then a search for a conical intersection (CI), which facilitates the internal conversion to the ground state. The same calculations (22) suggested that the protein promotes the isomerization of the chromophore by restricting the rotation of its p-hydroxyphenyl moiety.
Our experimental results given above indicate that the primary role of the chromophore is determined by the twisting around the central double bond (C2=C3) and that other motions involving the hydroxyphenyl or the sulfur linkage are relatively unimportant. In fact, careful examination of the trajectories in the molecular dynamics simulations (22) indicate that the C2=C3 motion occurs on the timescale of ≈1 ps, significantly faster (factor of 2) than the other bonds involved in the linkage to the protein. Thus, the chromophore twisting is nearly insensitive to the protein environment in that it does not occur by a typical barrier-crossing process with multiple “collisions” to cross over to the cis configuration. The ultrafast nature of the reaction ensures the high efficiency of the initial “molecular impulse,” which then triggers subsequent changes in the protein.
This initial step is facilitated by the electronic excitation, which weakens the double bond (more antibonding character) and by (coherent) low-frequency motion(s) that brings the atoms into the needed configuration for efficient twisting.
The barrier height estimates range from 5 to 20 kcal/mol, and the effect of the environment on the height is debatable (22, 23). It is clear that at our wavelength the system is above the barrier energy. The degree of frictionless crossing is also clear from comparison with results for the same chromophore in solution. Upon varying the viscosity from 1 to 60 centipoise (D. H. Paik, A. Espagne, M. M. Martin, and A.H.Z., unpublished data), the fluorescence decay changes from 1.3 to 16 ps, indicating the effect of friction. The quantum yield of fluorescence in solution (24) and in the protein (25) is ≈10-3 and increases by 2 orders of magnitude when the chromophore is locked around the double bond (26). Again, this behavior suggests an ultrafast nonradiative crossing, but we now know that it is an intrinsic property of the isolated chromophore around its central double bond. The analogy with trans-stilbene, diphenyl ethylene, is relevant because the barrier is similarly low (≈3 kcal/mol) (27).
In Fig. 4, we depict the potential energy along the twisting coordinate θ and the effect of a second coordinate(s) describing the coherent motion, for example, that of skeletal deformation(s). After barrier crossing, the population of the twisted state in the isolated chromophore can decay only by searching for the CI and/or by redistributing the energy by intramolecular vibrational-energy redistribution. The twisted configuration is in a well with a depth of ≈1 eV.§ For this intermediate, we measured 52 ps for its decay, but in the protein the formation of the ground-state cis isomer (I0, the first intermediate of the PYP photocycle) is within a few picoseconds (8–11). This large discrepancy suggests that the position of CI on the potential energy surface is dependent on the environment. Groenhof et al. (22) proposed that the CI of the PYP chromophore in vacuo is located far from the reaction coordinate plane on the skeletal deformation coordinate(s), whereas it is near the reaction coordinate in the protein (see Fig. 4 Inset).
Fig. 4.

Conceptual potential energy surface for the primary dynamics of the P-chromophore. The rotation angle around the double bond is indicated by θ and the other coordinate(s) are denoted by a perpendicular plane (γ coordinate) with the CI. The initial excitation prepares a wave packet localized at the trans configuration, after bifurcation (see text). The 3D potential in Inset shows the energy surface of the chromophore in the protein environment (adapted from ref. 22). A cut in this surface displays a similar θ-dependence but with a much closer, to the reaction coordinate, CI.
Nuclear motions, particularly those of low frequency, can promote intramolecular vibrational-energy redistribution to coordinates other than θ, and hence internal conversion at the CI would be delayed, explaining the slow decay of  population in the isolated chromophore; once the CI is reached, the crossing occurs in <1 ps. In the protein the twisted state can easily reach the CI near the reaction coordinate plane without the assistance of intramolecular vibrational-energy redistribution. We show this difference in Fig. 4 (perpendicular plane). The momentum carried by the twisted state along the reaction coordinate is conserved better in the protein than in the isolated system because of the ultrashort interaction time in the CI region, and therefore more cis isomers can be formed with high reaction quantum yield (0.24–0.64) (8, 11, 28, 29). That is a critical design by the protein environment to enhance trans-to-cis isomerization in the PYP photocycle.
population in the isolated chromophore; once the CI is reached, the crossing occurs in <1 ps. In the protein the twisted state can easily reach the CI near the reaction coordinate plane without the assistance of intramolecular vibrational-energy redistribution. We show this difference in Fig. 4 (perpendicular plane). The momentum carried by the twisted state along the reaction coordinate is conserved better in the protein than in the isolated system because of the ultrashort interaction time in the CI region, and therefore more cis isomers can be formed with high reaction quantum yield (0.24–0.64) (8, 11, 28, 29). That is a critical design by the protein environment to enhance trans-to-cis isomerization in the PYP photocycle.
There is another influence for the protein. In the isolated chromophore, the internal conversion of the twisted state near the CI creates vibrationally hot ground-state cis molecules. The excess energy is released by ejecting an electron (autodetachment), and some of the anions convert to radicals, because of the lack of competing relaxation processes. In the protein, however, the energy can be released efficiently because of the presence of many degrees of freedom, and creating a radical by autodetachment is not a dominant process; the radical state, from our autodetachment experiments, lies near 2.9 eV above the ground state. Vibrational relaxation in the protein is highly efficient, as indeed measured in the protein; the absorption of the formed cis molecules is that of an equilibrated species within a few picoseconds (7–11). Consistent with our findings is the fact that the radical state was not observed in the PYP photocycle with single-photon absorption; only with multiphoton excitation of high-energy photons were radicals detected (7).
The studies reported here on the anionic, isolated chromophore of PYP provide direct evidence of the ultrafast, nearly frictionless dynamics of the first step of isomerization. The key primary step involves structural change including the central double bond. As pointed out earlier, double-bond twisting must include the effect of nuclear couplings to other bonds and electronic/resonance effects. In a very recent report (30), the gas-phase absorption spectrum of the PYP chromophore indicates that the excitation energy is lower than calculated value (22), indicating that an excess vibrational energy is sufficient for double-bond twisting. Moreover, recent molecular dynamics calculations for the protein and in vacuo support the involvement of the double-bond twisting (31). The protein environment is not significant in changing the low-energy barrier for twisting, but it is critical for the subsequent conversion to the cis form and for the impedance of radical formation during the cycle. The design by the protein is for an efficient ultrafast phototransduction and for directing the dynamics by reducing the phase space of many nuclear motions into that of the reaction coordinate, as observed here in the direct comparison of the behavior of isolated-chromophore and protein dynamics.
Materials and Methods
The P-chromophore, 4-(3-oxobut-1-enyl)-phenolate anion, was prepared from 4-hydroxybenzylideneacetone (HBA; Alfa Aesar, Ward Hill, MA; 97%) by using pulsed electron impact (1 keV, 1.0 ms) and expansion with oxygen at ≈200 kPa in a molecular beam. The temperature was ≈80°C, and under these conditions we obtained the P-chromophore mass, which confirms the deprotonation of HBA. The anions were directed into a field-free time-of-flight region by applying a -2.0-kV electric pulse in a two-stage accelerator. The beam of the P-chromophore, which was separated from other ions by its mass, was interrogated by the two beams of femtosecond laser pulses. The laser pulse (110 fs) at 800 nm was generated from a Ti:sapphire oscillator and amplified by a regenerative and multipass amplifier. The amplified laser pulse was split into two parts. The first was frequency doubled by a type-I beta barium borate crystal, generating a 400-nm pulse, and was used as the pump pulse to excite the P-chromophore. The second part was used as the probe pulse. The polarization was set at the magic angle (54.7°), and the power of the pulse was 0.35 mJ for the pump and 3.6 mJ for the probe, collimated to ≈3-mm diameter. The resulting photoelectrons were analyzed in our apparatus (32) by using a magnetic bottle photoelectron spectrometer. The powerful methodology of mass selection/electron detachment provided the needed resolution of species and state (33–36), whereas the pump-probe femtosecond chemistry configuration allowed for the temporal resolution.
Ab initio calculations for geometry optimization and energy minimization of the P-chromophore and single-point energy calculation of its radical form were performed by using the gaussian 98 program (37) at the B3LYP/cc-pVTZ level of theory.
Acknowledgments
We thank Prof. Monique M. Martin and Drs. Agathe Espagne and D. Hern Paik for stimulating discussion; the collaborative work on solution-phase studies will be published elsewhere. This work was supported by the National Science Foundation.
Author contributions: A.H.Z. designed research; I-R.L. and W.L. performed research; I-R.L. and W.L. analyzed data; and I-R.L., W.L., and A.H.Z. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: PYP, photoactive yellow protein; CI, conical intersection.
Footnotes
Based on energetics obtained by ab initio calculation (see Materials and Methods), the peak at ≈0.9 eV (pump-plus-probe) corresponds to probe detachment with the formed radical having 0.8 eV of excitation. The broad peak at ≈2 eV (pump only), similarly, is a two-photon excitation.
When the population of  is promoted by the probe to form the corresponding radical, the radical would carry similar amount of vibrational energy (Evib) as
is promoted by the probe to form the corresponding radical, the radical would carry similar amount of vibrational energy (Evib) as  , because of Franck–Condon consideration. This energy is the sum of δ, the energy difference between the trans and twisted form of the radical, and Δ, the difference in their electron-kinetic-energy observed here to be ≈0.5 eV. Small δ values are improbable, and if δ is ≈ 0.5 eV, Evib would be ≈1 eV, which matches well with the theoretical calculation (23).
, because of Franck–Condon consideration. This energy is the sum of δ, the energy difference between the trans and twisted form of the radical, and Δ, the difference in their electron-kinetic-energy observed here to be ≈0.5 eV. Small δ values are improbable, and if δ is ≈ 0.5 eV, Evib would be ≈1 eV, which matches well with the theoretical calculation (23).
References
- 1.Sprenger, W. W., Hoff, W. D., Armitage, J. P. & Hellingwerf, K. J. (1993) J. Bacteriol. 175, 3096-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larsen, D. S. & van Grondelle, R. (2005) Chemphyschem. 6, 828-837. [DOI] [PubMed] [Google Scholar]
- 3.Hellingwerf, K. J., Hendriks, J. & Gensch, T. (2003) J. Phys. Chem. A 107, 1082-1094. [Google Scholar]
- 4.Cusanovich, M. A. & Meyer, T. E. (2003) Biochemistry 42, 4759-4770. [DOI] [PubMed] [Google Scholar]
- 5.Borgstahl, G. E. O., Williams, D. R. & Getzoff, E. D. (1995) Biochemistry 34, 6278-6287. [DOI] [PubMed] [Google Scholar]
- 6.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33-38. [DOI] [PubMed] [Google Scholar]
- 7.Larsen, D. S., van Stokkum, I. H. M., Vengris, M., van der Horst, M. A., de Weerd, F. L., Hellingwerf, K. J. & van Grondelle, R. (2004) Biophys. J. 87, 1858-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groot, M. L., van Wilderen, L. J. G. W., Larsen, D. S., van der Horst, M. A., van Stokkum, I. H. M., Hellingwerf, K. J. & van Grondelle, R. (2003) Biochemistry 42, 10054-10059. [DOI] [PubMed] [Google Scholar]
- 9.Gensch, T., Gradinaru, C. C., van Stokkum, I. H. M., Hendriks, J., Hellingwerf, K. J. & van Grondelle, R. (2002) Chem. Phys. Lett. 356, 347-354. [Google Scholar]
- 10.Imamoto, Y., Kataoka, M., Tokunaga, F., Asahi, T. & Masuhara, H. (2001) Biochemistry 40, 6047-6052. [DOI] [PubMed] [Google Scholar]
- 11.Devanathan, S., Pacheco, A., Ujj, L., Cusanovich, M., Tollin, G., Lin, S. & Woodbury, N. (1999) Biophys. J. 77, 1017-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genick, U. K., Soltis, S. M., Kuhn, P., Canestrelli, I. L. & Getzoff, E. D. (1998) Nature 392, 206-209. [DOI] [PubMed] [Google Scholar]
- 13.Genick, U. K., Borgstahl, G. E. O., Ng, K., Ren, Z., Pradervand, C., Burke, P. M., Šrajer, V., Teng, T. Y., Schildkamp, W., McRee, D. E., et al. (1997) Science 275, 1471-1475. [DOI] [PubMed] [Google Scholar]
- 14.Usman, A., Mohammed, O. F., Heyne, K., Dreyer, J. & Nibbering, E. T. J. (2005) Chem. Phys. Lett. 401, 157-163. [Google Scholar]
- 15.Changenet-Barret, P., Espagne, A., Plaza, P., Hellingwerf, K. J. & Martin, M. M. (2005) New J. Chem. 29, 527-534. [Google Scholar]
- 16.Changenet-Barret, P., Espagne, A., Charier, S., Baudin, J. B., Jullien, L., Plaza, P., Hellingwerf, K. J. & Martin, M. M. (2004) Photochem. Photobiol. Sci. 3, 823-829. [DOI] [PubMed] [Google Scholar]
- 17.Vengris, M., van der Horst, M. A., Zgrablic, G., van Stokkum, I. H. M., Haacke, S., Chergui, M., Hellingwerf, K. J., van Grondelle, R. & Larsen, D. S. (2004) Biophys. J. 87, 1848-1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen, D. S., Vengris, M., van Stokkum, I. H. M., van der Horst, M. A., Cordfunke, R. A., Hellingwerf, K. J. & van Grondelle, R. (2003) Chem. Phys. Lett. 369, 563-569. [Google Scholar]
- 19.Ryan, W. L., Gordon, D. J. & Levy, D. H. (2002) J. Am. Chem. Soc. 124, 6194-6201. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen, S., Bañares, L. & Zewail, A. H. (1992) J. Chem. Phys. 97, 8801-8804. [Google Scholar]
- 21.Mataga, N., Chosrowjan, H. & Taniguchi, S. (2004) J. Photochem. Photobiol. C 5, 155-168. [DOI] [PubMed] [Google Scholar]
- 22.Groenhof, G., Bouxin-Cademartory, M., Hess, B., de Visser, S. P., Berendsen, H. J. C., Olivucci, M., Mark, A. E. & Robb, M. A. (2004) J. Am. Chem. Soc. 126, 4228-4233. [DOI] [PubMed] [Google Scholar]
- 23.Thompson, M. J., Bashford, D., Noodleman, L. & Getzoff, E. D. (2003) J. Am. Chem. Soc. 125, 8186-8194. [DOI] [PubMed] [Google Scholar]
- 24.Changenet-Barret, P., Plaza, P. & Martin, M. M. (2001) Chem. Phys. Lett. 336, 439-444. [Google Scholar]
- 25.Meyer, T. E., Tollin, G., Causgrove, T. P., Cheng, P. & Blankenship, R. E. (1991) Biophys. J. 59, 988-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chosrowjan, H., Taniguchi, S., Mataga, N., Unno, M., Yamauchi, S., Hamada, N., Kumauchi, M. & Tokunago, F. (2004) J. Phys. Chem. B 108, 2686-2698. [Google Scholar]
- 27.Baskin, J. S., Bañares, L., Pedersen, S. & Zewail, A. H. (1996) J. Phys. Chem. 100, 11920-11933. [Google Scholar]
- 28.van Brederode, M. E., Gensch, T., Hoff, W. D., Hellingwerf, K. J. & Braslavsky, S. E. (1995) Biophys. J. 68, 1101-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer, T. E., Tollin, G., Hazzard, J. H. & Cusanovich, M. A. (1989) Biophys. J. 56, 559-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen, I. B., Boye-Peronne, S., El Ghazaly, M. O. A., Kristensen, M. B., Nielsen, S. B. & Andersen, L. H. (2005) Biophys. J. 89, 2597-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada, A., Ishikura, T. & Yamato, T. (2004) Proteins 55, 1063-1069. [DOI] [PubMed] [Google Scholar]
- 32.Paik, D. H., Kim, N. J. & Zewail, A. H. (2003) J. Chem. Phys. 118, 6923-6929. [Google Scholar]
- 33.Leopold, D. G., Murray, K. K., Miller, A. E. S. & Lineberger, W. C. (1985) J. Chem. Phys. 83, 4849-4865. [Google Scholar]
- 34.Posey, L. A., Deluca, M. J. & Johnson, M. A. (1986) Chem. Phys. Lett. 131, 170-174. [Google Scholar]
- 35.Metz, R. B., Weaver, A., Bradforth, S. E., Kitsopoulos, T. N. & Neumark, D. M. (1990) J. Phys. Chem. 94, 1377-1388. [Google Scholar]
- 36.Castleman, A. W. & Bowen, K. H. (1996) J. Phys. Chem. 100, 12911-12944. [Google Scholar]
- 37.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery, J. A., Stratmann, R. E., Burant, J. C., et al. (1998) gaussian 98 (Gaussian, Pittsburgh), Revision A.9.