


Abstract
Serine/arginine-rich (SR) splicing factors play an important role in constitutive and alternative splicing as well as during several steps of RNA metabolism. Despite the wealth of functional information about SR proteins accumulated to-date, structural knowledge about the members of this family is very limited. To gain a better insight into structure-function relationships of SR proteins, we performed extensive sequence analysis of SR protein family members and combined it with ordered/disordered structure predictions. We found that SR proteins have properties characteristic of intrinsically disordered (ID) proteins. The amino acid composition and sequence complexity of SR proteins were very similar to those of the disordered protein regions. More detailed analysis showed that the SR proteins, and their RS domains in particular, are enriched in the disorder-promoting residues and are depleted in the order-promoting residues as compared to the entire human proteome. Moreover, disorder predictions indicated that RS domains of SR proteins were completely unstructured. Two different classification methods, the charge-hydropathy measure and the cumulative distribution function (CDF) of the disorder scores, were in agreement with each other, and they both strongly predicted members of the SR protein family to be disordered. This study emphasizes the importance of the disordered structure for several functions of SR proteins, such as for spliceosome assembly and for interaction with multiple partners. In addition, it demonstrates the usefulness of order/disorder predictions for inferring protein structure from sequence.
INTRODUCTION
Serine/arginine-rich (SR) proteins constitute a family of metazoan splicing factors that are essential for both constitutive and alternative splicing of pre-mRNAs (1). In constitutive splicing, they are known to promote cross-intron and cross-exon interactions, and to influence the recruitment of the U1 snRNP and U2AF splicing factor into the spliceosome (2). In alternative splicing, SR proteins are known to interact with exonic splicing enhancers (ESEs) and to stimulate the splicing of adjacent introns (3). Recent studies suggested several additional functions for SR proteins in mRNA metabolism [reviewed in (4)].
It is generally accepted that there are 10 canonical SR proteins in mammals, with sizes ranging from 20 to 75 kDa (5). These proteins were initially identified and grouped into a family based on common biochemical and immunological properties (1). SR proteins belong to a larger superfamily of SR-like proteins that are characterized by the presence of RS or RS-like domains (6). A bioinformatic approach identified about 50 proteins with RS domains in Homo sapiens, 80 in Caenorhabditis elegans and 110 in Drosophila melanogaster (7).
All SR proteins have a modular organization and consist of one or two RNA recognition motifs (RRMs), located on their N-terminus, and one arginine-serine-rich (RS) domain, located on the C-terminus. The RRM domains generally recognize specific RNA sequences through a wide range of interactions (8), and they can also participate in protein–protein interactions (9). Likewise, RS domains can engage in homotypical protein–protein interactions (2), and it was shown recently that they could also contact the pre-mRNA branchpoint (10). Thus, both RRM and RS domains have a broad binding specificity.
RS domains are required for all essential functions of SR proteins. It has been shown that RS domains function as splicing activation domains (11), and that they harbor signals for nuclear localization and nucleocytoplasmic shuttling (12,13). Besides these important functions, recent studies demonstrated that the RS domain of the ASF/SF2 splicing factor is also required for the nonsense-mediated mRNA decay (14). The RS domains are heavily phosphorylated on the serines by two families of kinases (15). Phosphorylation and dephosphorylation of RS domains modulates their interactions with other proteins and RNA (16).
Despite the fact that SR proteins have been a topic of intense investigation for the last fifteen years, only limited structural knowledge about this protein family is available to-date. Structural and functional studies of SR proteins are largely impeded by the difficulty of their purification. These proteins are very prone to inclusion bodies formation and aggregation during the purification procedure (A. Krainer, personal communication). Possibly for this reason, structural knowledge about SR proteins is currently limited to the RRM domains. The structures of the RRM domains from several RNA-binding proteins (but not from the canonical SR proteins) have been determined [reviewed in (17)]. In addition, there is only one circular dichroism (CD) study that investigates the structures of the full-length ASF/SF2 protein as well as the structures of its deletion mutants, delta-RS and the RS domain itself (18). The CD spectrum of the RS domain is characteristic of the random coil conformation, whereas full-length ASF/SF2 and the delta-RS construct have some α-helical content (18).
The aim of this study is to expand structural knowledge about the SR protein family using sequence analysis combined with the prediction of ordered and disordered protein regions. Intrinsically disordered (ID) proteins represent a new class of proteins that lack a folded structure under physiological conditions and that exist in the ensemble of conformations (19–21). The growing list of ID proteins currently consists of over 200 proteins [(22), see also http://www.disprot.org/]. It has been shown that ID proteins and regions are involved in numerous important biological functions (23–25), including signaling (26), protein–protein interactions with multiple partners (27) and post-translational modifications (28).
Here, we show that the RS domains of SR proteins are predicted to be completely disordered, and that SR proteins belong to the growing class of intrinsically unstructured proteins. These findings emphasize the importance of disorder for determining broad binding specificity of SR proteins and for spliceosome assembly. In addition, they add splicing to the growing list of biological functions in which disordered proteins and protein regions are involved.
MATERIALS AND METHODS
Datasets
Sequences of 10 human SR proteins (Table 1) were extracted from the SWISS-PROT database release 46.3. The dataset of the disordered protein regions was extracted from the DisProt Database (22). The dataset of ordered protein regions (O_PDB_S25) was constructed as described (29) and represents a non-redundant subset of well-ordered globular proteins extracted from the PDB Select 25 database (30). The disorder predictions on this dataset served as a control for estimating the false-positive prediction error rate in Figure 3. The Globular-3D dataset consisted of ordered protein regions extracted from PDB (31); fibrous sequences such as coiled coils, collagen and silk fibroins were removed from this dataset (29). The sequences of human proteins for the human proteome dataset were extracted from the NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/protein). The datasets of completely ordered and completely disordered proteins used in Figure 6 were constructed as described (32).
Table 1.
Frequencies of disorder- and order-promoting residues in SR proteins and RS domains as compared with the human proteome
| Protein name | SWISS-PROT/TrEMBL ID | RS domain location | Disorder-promoting residues (A,R,S,Q,E,G,K,P) | Order-promoting residues (N,C,I,L,F,W,Y,V) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Full-lenth protein | RS domain | Full-lenth protein | RS domain | |||||||
| n | % | n | % | n | % | n | % | |||
| ASF/SF2 | SFRS1 | 197–246 | 153 | 61.7 | 45 | 90.0 | 62 | 25.0 | 4 | 8.0 |
| SC35 | SFRS2 | 116–220 | 163 | 73.8 | 99 | 94.3 | 31 | 14.0 | 4 | 3.8 |
| SRp20 | SFRS3 | 86–164 | 108 | 65.9 | 66 | 83.5 | 42 | 25.6 | 9 | 11.4 |
| SRp75 | SFRS4 | 179–494 | 380 | 76.9 | 284 | 89.9 | 72 | 14.6 | 15 | 4.7 |
| SRp40 | SFRS5 | 182–267 | 185 | 68.0 | 79 | 91.9 | 62 | 22.8 | 4 | 4.6 |
| SRp55 | SFRS6 | 184–343 | 241 | 70.1 | 143 | 89.4 | 68 | 19.8 | 9 | 5.6 |
| 9G8 | SFRS7 | 121–238 | 173 | 72.7 | 106 | 89.8 | 46 | 19.3 | 9 | 7.6 |
| SRp30c | SFRS9 | 188–200 | 124 | 56.1 | 10 | 76.9 | 64 | 29.0 | 2 | 15.4 |
| SRp54 | SFR11 | 247–353 | 327 | 67.6 | 94 | 87.8 | 87 | 18.0 | 1 | 0.9 |
| SRp46 | Q9BRL6 | 98–274 | 209 | 74.1 | 151 | 85.3 | 44 | 15.6 | 18 | 10.2 |
| Average | 206.3 | 68.7 | 107.7 | 87.9 | 58 | 20.4 | 7.5 | 7.2 | ||
| Human proteome | 7388852 | 51 | 4754771 | 32.9 | ||||||
The RS domain boundaries correspond to the SWISS-PROT (42) database annotations.
Disorder predictions
Predictions of intrinsic disorder in SR proteins were carried out using a well-characterized disorder predictor PONDR® VL-XT (29,33). This predictor was trained on the experimentally (X-ray and NMR) confirmed disordered protein regions of a length of at least 30 residues, while the ordered training set included completely ordered proteins extracted from the non-redundant set of proteins from PDB Select 25 (30). The accuracy of this predictor, benchmarked on the 42 CASP5 targets, reached 72.8% (34). PONDR® VL-XT is currently being used successfully to guide the removal of disordered regions that interfere with crystallization of ‘problematic’ proteins for high-throughput structure determination (35). Access to PONDR® VL-XT was provided by Molecular Kinetics (Indianapolis, IN). VL-XT is copyright© 1999 by the WSU Research Foundation, all rights reserved. PONDR® is copyright© 2004 by Molecular Kinetics, all rights reserved.
Sequence complexity
Shannon's entropy (36), first applied to amino acid sequences as a measure of sequence complexity by Wootton (37), was calculated for each dataset using a window of 45 residues.
Cumulative distribution function (CDF)
The CDF represents a cumulative histogram of the PONDR® VL-XT prediction scores for each residue in a given protein. This histogram allows the separation of ordered and disordered proteins based on the distribution of the disorder scores (38). The boundary points on the CDF plot were calculated as previously described (32).
Charge-hydropathy classification
The charge-hydropathy method developed by Uversky et al. (20) has been used to classify SR proteins as ordered or disordered. The mean net charge and the mean normalized Kyte–Doolittle hydropathy (39) were calculated for each protein and their values were plotted against each other. The boundary between ordered and disordered proteins was determined using a linear discriminant function as previously described (32).
RESULTS AND DISCUSSION
Amino acid composition of SR proteins
It was previously established that ordered and disordered protein regions are characterized by significantly different amino acid compositions, with the prevalence of hydrophilic and charged amino acids and the depletion of hydrophobic and aromatic amino acids amongst the disordered regions (21,40). To determine whether sequence attributes of SR proteins are similar to those of the disordered protein regions, we calculated the amino acid frequencies for each of these two datasets (Figure 1). The plot represents the difference in the frequencies between the two studied datasets and a completely ordered set of proteins, Globular-3D (Materials and Methods).
Figure 1.

Amino acid composition of SR proteins. Amino acid compositions of SR and disordered proteins are shown relative to the composition of completely ordered globular proteins Globular-3D. Amino acids are arranged from left to right in order of increasing flexibility as defined by Vihinen et al. (41). The error bars represent 95% confidence intervals.
Due to the presence of RS domains, the frequencies of arginine and serine in the SR protein dataset are significantly higher than in the dataset of disordered proteins. The frequencies of aromatic residues, aliphatic residues and cysteine (the left side of the graph, Figure 1) are very similar for both the SR and disordered protein datasets, with the exception of one residue, tyrosine. Whereas disordered proteins are depleted in tyrosine, the SR proteins are slightly enriched in this residue. Since tyrosine has several distinct properties (such as partial hydrophobicity, aromatic side chain and a reactive hydroxyl group), it is tempting to speculate that it could participate in stacking interactions with the RNA bases. Another interesting difference between the two datasets is the frequency of the negatively charged glutamic acid: SR proteins are depleted in E while the disordered proteins are significantly enriched in E (Figure 1). The depletion of SR proteins in the negatively charged D and E and their enrichment in the positively charged K and R may be essential for interaction with the negatively charged RNA. In spite of a few compositional differences between SR and disordered proteins, the overall trend for the two datasets is very similar, with their overall depletion in the hydrophobic residues and enrichment in some hydrophilic, charged and flexible residues (such as proline, lysine and arginine).
The analysis of ID proteins that were characterized by various experimental techniques (such as NMR, X-ray and CD) have indicated that independent of the characterization method, all disordered proteins have similar amino acid frequencies (21). Based on this analysis, it was proposed that the residues found to be enriched in all disordered proteins, be called disorder-promoting residues (A,R,S,Q,E,G,K,P), and the residues found to be depleted in all disordered proteins, be called order-promoting residues (N,C,I,L,F,W,Y,V) (21). We calculated the frequencies of disorder- and order-promoting residues for each of the SR proteins as well as for each of the RS domains (Table 1). This analysis shows that the SR proteins and their RS domains in particular, are enriched in disorder-promoting residues and are depleted in order-promoting residues as compared to the entire human proteome. The average percentages of disorder promoters reach 68.7% for SR proteins and 87.9% for RS domains, while the average for the human proteome is only 51%. At the same time, the average for order promoters are 20.4%, 7.2% and 32.9% for SR proteins, RS domains and the human proteome, respectively. Given the high proportion of disorder-promoting residues within RS domains, it is very likely that these domains would be unable to adopt a stable 3D structure in solution without binding partners.
Sequence complexity distributions
Shannon's entropy (36) could be used as a measure of the sequence complexity of a protein (29), when applied to protein sequence. Previously, it has been shown that disordered sequences have an overall lower sequence complexity than ordered sequences (29). Furthermore, an independent analysis of 126 intrinsically unstructured sequences indicated that they are characterized by a higher frequency of short repetitive regions (43), thereby confirming the prevalence of low complexity segments among this protein class. SR proteins represent a perfect example of ID proteins that carry low complexity regions corresponding to the RS domains.
Here, we determined the overall sequence complexity of the SR protein family and compared it to the complexity of ordered and disordered protein regions (Figure 2). As expected, SR proteins and disordered protein regions have similar complexity distributions that differ from the complexity distribution of ordered regions. The analysis shows that SR proteins have an even higher proportion of extremely low complexity segments than the disordered proteins (compare the complexity values from ∼1.5 to 3.0). In addition, the peaks for SR and disordered regions overlap and are shifted towards lower complexity values as compared to the ordered regions (Figure 2). Thus, a sequence complexity analysis of SR proteins suggests that, similar to the disordered proteins, they are enriched in low complexity segments.
Figure 2.

Sequence complexity distributions of three datasets. Sequence complexity was calculated as described in Materials and Methods. SR proteins and disordered proteins have similar sequence complexity distributions.
Disorder analysis of SR proteins
We then applied a well-characterized disorder predictor PONDR® VL-XT (Materials and Methods) to SR proteins to predict the location of ordered and disordered regions. Results of the prediction agreed with the sequence analysis and further confirmed the high disorder content within this protein family. The disorder predictions, when analyzed based on the percentages of predicted disordered residues, clearly indicate an extremely high disorder content for SR proteins as compared to human proteome (Figure 3). For example, for regions of ≥40 consecutive disorder predictions (where the false-positive disorder prediction error rate is <1%) SR proteins have ∼4.9-fold more predicted disordered residues than human proteins (∼44% for the SR proteins versus ∼9% for the human proteins), whereas for regions of ≥100 consecutive disorder predictions SR proteins have ∼11-fold more predicted disordered residues (∼30% for the SR proteins versus ∼2.7% for the human proteins).
Figure 3.

Prediction of intrinsic disorder for three datasets. Human proteome dataset was constructed as described in Materials and Methods. The O_PDB_S25 dataset serves as a control for estimating the false-positive disorder prediction error rate.
Other disorder attributes (such as overall percentage of predicted disordered residues, average disorder score and longest disordered region), calculated on a per protein basis, also indicate that SR proteins belong to a class of intrinsically unstructured proteins (Table 2). With the exception of two proteins, ASF/SF2 and SRp30c, overall percentages of predicted disordered residues exceed 50% for all remaining SR proteins. Furthermore, the average disorder score for all but one SR protein (SRp30c) is greater than 0.5, where 0.5 is a boundary score between order and disorder.
Table 2.
Predicted disorder attributes for SR proteins calculated on a per protein basis
| Protein name | Protein length | % Disordered residues | Disorder average score | Longest DR |
|---|---|---|---|---|
| ASF/SF2 | 248 | 47.2 | 0.52 | 65 |
| SC35 | 221 | 83.3 | 0.82 | 130 |
| SRp20 | 164 | 58.5 | 0.64 | 89 |
| SRp75 | 494 | 76.3 | 0.78 | 339 |
| SRp40 | 272 | 66.5 | 0.67 | 104 |
| SRp55 | 344 | 73.5 | 0.72 | 183 |
| 9G8 | 238 | 60.9 | 0.68 | 121 |
| SRp30c | 221 | 33.0 | 0.38 | 25 |
| SRp54 | 484 | 74.2 | 0.74 | 214 |
| SRp46 | 282 | 78.0 | 0.80 | 195 |
The analysis of individual predictions shows that the disorder predictions for SR proteins highly correlate with their domain organization (Figure 4). In general, RRM domains are predicted to be ordered, while Gly-rich regions and RS domains are predicted to be disordered. Although the boundaries of these predictions do not always correspond exactly to the domain boundaries, for the most part, they agree with each other fairly well (Figure 4).
Figure 4.
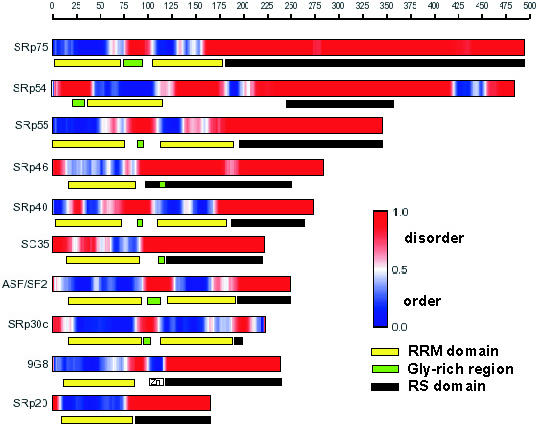
Domain organization and disorder predictions for SR proteins. The upper bar for each protein represents PONDR® VL-XT predictions with the red color signifying disorder and the blue color signifying order (see the gradient representation of the disorder scores shown on the vertical bar of the legend). The lower bar represents the location of the domains: RRM domain in yellow, Gly-rich region in green and RS domain in black. The Zn knuckle of the 9G8 protein is marked ‘Zn’. The RS domain boundaries correspond to the SWISS-PROT (42) database annotations.
Remarkably, our predictions highly correlate with the limited structural information that is available for individual domains constituting SR proteins. For example, we predict that the RRM domains of SR proteins are ordered (Figure 4). This prediction is supported by the experimental data: the structures of RRM domains from several RNA-binding proteins are solved, and RRMs indeed are ordered. They consist of four antiparallel β-strands packed against two α-helices, forming a β-sheet that makes multiple contacts with RNA (44). In addition, disorder predictions for the RS domains also agree with experimental observations. The CD spectra of the isolated recombinant RS domain of the SF2/ASF splicing factor is typical of a completely unstructured protein, with maximum negative ellipticity ∼200–202 nm and the isodichroic point around 212 nm, suggestive of random coil conformation (18). The flexibility and disorder of the glycine-rich protein regions in solution have also been previously observed (45).
Classification of SR proteins using the CDF analysis
The binary classification of proteins as either ordered or disordered is an oversimplification of a real biological situation since most proteins consist of a mixture of ordered and disordered regions. At the same time, it has been proven useful for estimating the disorder content of genomes (32). Such binary classification can also be used to estimate the disorder content of protein families or protein functional categories.
One of the methods for classifying protein as ordered or disordered is the CDF of disorder scores (38). This method separates ordered and disordered sequences based on the per-residue disorder score, and the optimal boundary, determined using the univariate normal probability density function, could be drawn between these two protein classes (32). The CDF curves for ordered proteins are located above the boundary, while the CDF curves for disordered proteins are located below the boundary.
Here, we applied the CDF method to classify SR proteins (Figure 5). According to the CDF classification, 9 out of 10 SR proteins belong to the class of ID proteins. The CDF curve for only one SR protein, SRp30c, is located slightly above the order-disorder boundary. This protein could be considered marginally ordered (or marginally disordered) using this classification method. Indeed, SRp30c carries the shortest RS domain (only 13 residues), and all other disorder attributes (Table 2) for this protein are suggestive of its marginally ordered structure (Figure 4). Thus, CDF analysis classifies the majority of SR proteins as disordered.
Figure 5.

CDF analysis of SR proteins. Ten CDF curves are shown, each corresponding to one SR protein. The order-disorder boundary is represented by the line with open circles.
Charge-hydropathy classification of SR proteins
Another method that has previously been developed for binary classification of proteins (20) is based on the calculated mean net charge and mean normalized Kyte–Doolittle hydropathy (39). When plotted against each other, these measures are known to separate ordered and disordered proteins by a boundary that could be determined using a linear discriminant function (32). Disordered proteins are generally clustered above the boundary, and therefore are characterized by a combination of high net charge and low hydropathy. In contrast, ordered proteins are generally clustered below the boundary and are characterized by lower net charge and higher hydropathy than disordered proteins.
When we applied the charge-hydropathy classification to the SR protein family, all 10 members of this family fell into the disordered protein category (Figure 6). As expected from the previous analysis, the SRp30c protein is located closer to the order-disorder boundary than the remaining SR proteins. The SRp75 protein has the lowest hydrophobicity and one of the highest values for the net charge (the leftmost red diamond in Figure 6), in agreement with the highest content of predicted disorder and the longest RS domain in comparison to other SR proteins (Figure 4).
Figure 6.
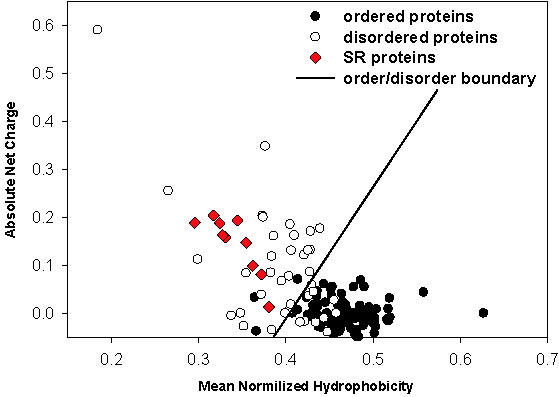
Charge-hydropathy analysis of SR proteins. The ordered proteins are represented as solid circles, the disordered proteins as open circles and the SR proteins as red diamonds. The order-disorder boundary is shown as a solid line.
In summary, two classification methods applied to the SR protein family are in agreement with each other, and they both predict that SR proteins belong to the class of intrinsically unstructured proteins. To a large degree, this classification is attributable to the disordered nature of the RS domains that comprise a significant portion (up to 64% of protein length in the extreme case of SRp75) of the SR proteins. The potential importance of disorder for RS domain functions is discussed in the section below.
Importance of disorder for RS domain functions
It is widely accepted that the RS domains of SR proteins participate in homotypical protein–protein interactions with the RS domains of numerous other SR and non-SR proteins (2,46). Moreover, it was recently suggested that RS domains could also specifically contact the pre-mRNA branchpoint (10). Thus, the RS domains seem to have dual specificity, e.g. they participate in interactions with both proteins and RNA.
It is difficult to understand how such a broad binding specificity is achieved assuming that the RS domain has a folded globular structure. In contrast, a broad specificity is in perfect agreement with the disordered structure because intrinsic structural disorder could facilitate accommodation of structurally diverse partners. Indeed, numerous examples from the literature indicate that ID proteins and protein regions are involved in protein–protein and protein–nucleic acids interactions (23,24), and that such interactions may also include folding upon binding (47). Structural plasticity is especially important for interactions with multiple partners (27,48). Thus, we suggest that disorder of the RS domain of SR proteins is crucial in determining the broad binding specificity of these factors.
Another important function of disorder within RS domains arises from their dispensability in splicing reaction. The fact that the RS domains from non-SR proteins as well as synthetic RS domains, consisting of only RS dipeptide repeats, are sufficient to activate splicing (49) argues against the requirement for a particular 3D structure for this function. Rather, it supports the requirement for a disordered structure, because the RS domains of other proteins, as well as the sequences of RS dipeptides, are also predicted to be unfolded (data not shown).
The RS domains of SR proteins are extensively phosphorylated by two families of kinases, the SR protein-specific kinases (SRPKs) and Clk/Sty protein kinases (15). Phosphorylation is required for translocation of SR proteins from the cytoplasm to the nucleus (50), and it is also known to regulate the activity of SR proteins during early development (51). Interestingly, we have previously shown that the phosphorylation sites of numerous other proteins are preferentially located in the disordered regions (28). Furthermore, other modifications such as methylation (52) and ubiquitination (P. Radivojac and L. M. Iakoucheva, manuscript in preparation) are also predicted to occur in disordered protein regions. Consistent with these observations, extensive phosphorylation of the RS domain supports the prediction of its disordered structure. Moreover, methylation of three arginines (R93, R97 and R109) has recently been observed in the ASF/SF2 splicing factor, as well as in several other hnRNPs and SR-like proteins (53). Thus, disorder could potentially facilitate numerous post-translational modifications of the RS domains.
ID proteins are often involved in the assembly of macromolecular complexes (54). The building of such complexes usually proceeds in a step-by-step manner and requires conformational flexibility and adaptability of constituting components during the assembly process. One example of a macromolecular complex that depends on the flexibility of its components is a ribosome. CD studies of individual ribosomal proteins from Escherichia coli showed that they are substantially disordered when separated from the ribosome (55). Moreover, some of the ribosomal proteins remain in the largely extended conformation even within the ribosome; they are filling ‘gaps and cracks’ between rRNA loops (56). Recent investigation of the biophysical properties of proteins comprising another macromolecular assembly, a nuclear pore complex, showed that they also exhibit structural characteristics typical of natively unfolded proteins (57).
The spliceosome represents another example of a large macromolecular complex, for which only limited structural knowledge is currently available (58). The spliceosome resembles a ribosomal subunit with respect to composition (RNA and proteins), complexity (large number of proteins) and size. SR proteins play key roles in the spliceosome assembly by facilitating recruitment of components of the spliceosome via protein–protein interactions that are potentially mediated by the RS domains (59). It is logical to suggest that the disordered structure of the RS domains would play an important role in facilitating interactions of spliceosome components during the assembly process.
As shown above, numerous functions performed by SR proteins in general, and their RS domains in particular, seem to rely on the disordered structure. Our predictions of disorder for RS domains, together with the classification of SR proteins as a disordered family, strongly suggest that unstructured conformation may be essential for the activity of SR splicing factors. Furthermore, our findings add splicing to the growing list of biological functions (23) performed by the disordered proteins and protein regions.
Acknowledgments
We would like to thank Adrian Krainer and Keith Dunker for helpful discussions and valuable comments and suggestions. We thank Katherine Montague for proofreading the manuscript. This study was supported by NSF grant MCB 0444818 to L.M.I. Funding to pay the Open Access publication charges for this article was provided by the National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zahler A.M., Lane W.S., Stolk J.A., Roth M.B. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 2.Wu J.Y., Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 3.Graveley B.R. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y., Steitz J.A. SRprises along a messenger's journey. Mol. Cell. 2005;17:613–615. doi: 10.1016/j.molcel.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Bourgeois C.F., Lejeune F., Stevenin J. Broad specificity of SR (serine/arginine) proteins in the regulation of alternative splicing of pre-messenger RNA. Prog. Nucleic Acid Res. Mol. Biol. 2004;78:37–88. doi: 10.1016/S0079-6603(04)78002-2. [DOI] [PubMed] [Google Scholar]
- 6.Fu X.D. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 7.Boucher L., Ouzounis C.A., Enright A.J., Blencowe B.J. A genome-wide survey of RS domain proteins. RNA. 2001;7:1693–1701. [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Canadillas J.M., Varani G. Recent advances in RNA–protein recognition. Curr. Opin. Struct. Biol. 2001;11:53–58. doi: 10.1016/s0959-440x(00)00164-0. [DOI] [PubMed] [Google Scholar]
- 9.Kielkopf C.L., Rodionova N.A., Green M.R., Burley S.K. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. doi: 10.1016/s0092-8674(01)00480-9. [DOI] [PubMed] [Google Scholar]
- 10.Shen H., Kan J.L., Green M.R. Arginine-serine-rich domains bound at splicing enhancers contact the branchpoint to promote prespliceosome assembly. Mol. Cell. 2004;13:367–376. doi: 10.1016/s1097-2765(04)00025-5. [DOI] [PubMed] [Google Scholar]
- 11.Graveley B.R., Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol. Cell. 1998;1:765–771. doi: 10.1016/s1097-2765(00)80076-3. [DOI] [PubMed] [Google Scholar]
- 12.Caceres J.F., Screaton G.R., Krainer A.R. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998;12:55–66. doi: 10.1101/gad.12.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cazalla D., Zhu J., Manche L., Huber E., Krainer A.R., Caceres J.F. Nuclear export and retention signals in the RS domain of SR proteins. Mol. Cell Biol. 2002;22:6871–6882. doi: 10.1128/MCB.22.19.6871-6882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z., Krainer A.R. Involvement of SR proteins in mRNA surveillance. Mol. Cell. 2004;16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 15.Stojdl D.F., Bell J.C. SR protein kinases: the splice of life. Biochem. Cell Biol. 1999;77:293–298. [PubMed] [Google Scholar]
- 16.Soret J., Tazi J. Phosphorylation-dependent control of the pre-mRNA splicing machinery. Prog. Mol. Subcell. Biol. 2003;31:89–126. doi: 10.1007/978-3-662-09728-1_4. [DOI] [PubMed] [Google Scholar]
- 17.Singh R., Valcarcel J. Building specificity with nonspecific RNA-binding proteins. Nature Struct. Mol. Biol. 2005;12:645–653. doi: 10.1038/nsmb961. [DOI] [PubMed] [Google Scholar]
- 18.Labourier E., Rossi F., Gallouzi I.E., Allemand E., Divita G., Tazi J. Interaction between the N-terminal domain of human DNA topoisomerase I and the arginine-serine domain of its substrate determines phosphorylation of SF2/ASF splicing factor. Nucleic Acids Res. 1998;26:2955–2962. doi: 10.1093/nar/26.12.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 20.Uversky V.N., Gillespie J.R., Fink A.L. Why are ‘natively unfolded’ proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Dunker A.K., Lawson J.D., Brown C.J., Williams R.M., Romero P., Oh J.S., Oldfield C.J., Campen A.M., Ratliff C.M., Hipps K.W., et al. Intrinsically disordered protein. J. Mol. Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 22.Vucetic S., Obradovic Z., Vacic V., Radivojac P., Peng K., Iakoucheva L.M., Cortese M.S., Lawson J.D., Brown C.J., Sikes J.G., et al. DisProt: a database of protein disorder. Bioinformatics. 2005;21:137–140. doi: 10.1093/bioinformatics/bth476. [DOI] [PubMed] [Google Scholar]
- 23.Dunker A.K., Brown C.J., Lawson J.D., Iakoucheva L.M., Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 24.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nature Rev. Mol. Cell. Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 25.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 26.Iakoucheva L.M., Brown C.J., Lawson J.D., Obradovic Z., Dunker A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002;323:573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 27.Dunker A.K., Cortese M.S., Romero P., Iakoucheva L.M., Uversky V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 28.Iakoucheva L.M., Radivojac P., Brown C.J., O'Connor T.R., Sikes J.G., Obradovic Z., Dunker A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero P., Obradovic Z., Li X., Garner E.C., Brown C.J., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Hobohm U., Sander C. Enlarged representative set of protein structures. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oldfield C.J., Cheng Y., Cortese M.S., Brown C.J., Uversky V.N., Dunker A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 33.Li X., Romero P., Rani M., Dunker A.K., Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 34.Obradovic Z., Peng K., Vucetic S., Radivojac P., Brown C.J., Dunker A.K. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53:566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 35.Oldfield C.J., Ulrich E.L., Cheng Y., Dunker A.K., Markley J.L. Addressing the intrinsic disorder bottleneck in structural proteomics. Proteins. 2005;59:444–453. doi: 10.1002/prot.20446. [DOI] [PubMed] [Google Scholar]
- 36.Shannon C.E. The mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423. and 623–656. [Google Scholar]
- 37.Wootton J.C. Statistic of local complexity in amino acid sequences and sequence databases. Comput. Chem. 1993;17:149–163. [Google Scholar]
- 38.Dunker A.K., Obradovic Z., Romero P., Garner E.C., Brown C.J. Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 39.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 40.Uversky V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 41.Vihinen M., Torkkila E., Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19:141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 42.Bairoch A., Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tompa P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays. 2003;25:847–855. doi: 10.1002/bies.10324. [DOI] [PubMed] [Google Scholar]
- 44.Varani G., Nagai K. RNA recognition by RNP proteins during RNA processing. Annu. Rev. Biophys Biomol. Struct. 1998;27:407–445. doi: 10.1146/annurev.biophys.27.1.407. [DOI] [PubMed] [Google Scholar]
- 45.Konrat R., Krautler B., Weiskirchen R., Bister K. Structure of cysteine- and glycine-rich protein CRP2. Backbone dynamics reveal motional freedom and independent spatial orientation of the lim domains. J. Biol. Chem. 1998;273:23233–23240. doi: 10.1074/jbc.273.36.23233. [DOI] [PubMed] [Google Scholar]
- 46.Kohtz J.D., Jamison S.F., Will C.L., Zuo P., Luhrmann R., Garcia-Blanco M.A., Manley J.L. Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 47.Dyson H.J., Wright P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 48.Tompa P., Szasz C., Buday L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Philipps D., Celotto A.M., Wang Q.Q., Tarng R.S., Graveley B.R. Arginine/serine repeats are sufficient to constitute a splicing activation domain. Nucleic Acids Res. 2003;31:6502–6508. doi: 10.1093/nar/gkg845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai M.C., Lin R.I., Tarn W.Y. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl Acad. Sci. USA. 2001;98:10154–10159. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanford J.R., Bruzik J.P. Developmental regulation of SR protein phosphorylation and activity. Genes Dev. 1999;13:1513–1518. doi: 10.1101/gad.13.12.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daily K.M., Radivojac P., Dunker A.K. Intrinsic disorder and protein modifications: building an SVM predictor for methylation. IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology; San Diego, CA: CIBCB; 2005. pp. 475–481. [Google Scholar]
- 53.Ong S.E., Mittler G., Mann M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nature Methods. 2004;1:119–126. doi: 10.1038/nmeth715. [DOI] [PubMed] [Google Scholar]
- 54.Namba K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells. 2001;6:1–12. doi: 10.1046/j.1365-2443.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 55.Venyaminov S., Gudkov A., Gogia Z., Tumanova L. Absorption and Circular Dichroism Spectra of Individual Proteins from Escherichia coli Ribosomes. Pushchino, USSR: Biological Research Center, Institute of Protein Research; 1981. [Google Scholar]
- 56.Ban N., Nissen P., Hansen J., Moore P.B., Steitz T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 57.Denning D.P., Patel S.S., Uversky V., Fink A.L., Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc. Natl Acad. Sci. USA. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurica M.S., Moore M.J. Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 59.Roscigno R.F., Garcia-Blanco M.A. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA. 1995;1:692–706. [PMC free article] [PubMed] [Google Scholar]