

Abstract
The monomeric RAL (RAS-like) GTPases have been indirectly implicated in mitogenic regulation and cell transformation. Here, we show that RALA and RALB collaborate to maintain tumorigenicity through regulation of both proliferation and survival. Remarkably, this task is divided between these highly homologous isoforms. RALB is specifically required for survival of tumour cells but not normal cells. RALA is dispensable for survival, but is required for anchorage-independent proliferation. Reducing the 'oncogenic burden' in human tumour cells relieves the sensitivity to loss of RALB. These observations establish RAL GTPases as crucial components of the cellular machinery that are exploited by factors that drive oncogenic transformation.
Introduction
RAL (RAS-like) GTPases, as the name implies, were originally identified on the basis of their sequence similarity to the RAS family of small GTPases (Chardin, 1988). Two RAL genes, RALA and RALB, are ubiquitously expressed in humans and produce proteins that are 80% identical. Interest in the function of RAL proteins in cell regulation was sparked by the observation that RAL proteins lie in a RAS effector pathway. RAL GTPases are activated in response to several mitogenic regulatory cascades, and RAL activation has been implicated as a contributing factor to oncogenic RAS-induced cellular transformation (Feig et al., 1996; Reuther & Der, 2000). The mechanistic contribution of RAL proteins to cell proliferation and transformation is unclear at present; however, expression of gain-of-function RAL variants suggests that RAL can affect several mitogenic regulatory cascades, including Src (Goi et al., 2000), phospholipase D1 (PLD1; Jiang et al., 1995) and nuclear factor-κB (Henry et al., 2000). As is generally the case with RAS-family isoforms, it is unknown at present whether RALA and RALB have non-overlapping, fully overlapping or partially overlapping functions.
Here, we use loss-of-function analysis to define explicitly the roles of RALA and RALB in the proliferation and transformation of human cells. We show that RALA is dispensable for the proliferation of human epithelial cells and tumour-derived cell lines in adherent cultures; however, RALA is required for the anchorage-independent proliferation of transformed cells. By contrast, RALB is required to prevent transformed cells from initiating programmed cell death. We propose that RAL isoforms collaborate in the maintenance of oncogenic transformation, mediating both oncogenic proliferation and survival signals.
Cell-autonomous molecular events that can drive the genesis of cancers are multifarious and complex. However, the activation of proliferation coupled with suppression of apoptosis has been aptly described as a “minimal platform” that supports oncogenic transformation (Evan & Vousden, 2001; Green & Evan, 2002). The observations described here show that RAL GTPases have the ability to support both 'legs' of this oncogenic platform. The selective sensitivity of tumour cells versus normal cells to loss of RALB reveals an Achilles' heel with potential for exploitation by targeted therapy approaches.
Results
To assess directly the contribution of RAL GTPases to cell regulation, we selectively inhibited the expression of RALA and RALB using small-interfering-RNA (siRNA)-mediated RNA interference (RNAi; Elbashir et al., 2001; Fig. 1). Inhibiting RALA expression had no noticeable effect on cell proliferation under standard in vitro culture conditions. By contrast, inhibition of RALB expression with either of two siRNA duplexes was toxic to HeLa cells, resulting in the appearance of condensed picnotic nuclei and marked cell loss by 150 h post-transfection (Fig. 2A). Annexin-V staining and TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP–biotin nick-end labelling) 72 h post-transfection showed that inhibition of RALB activates programmed cell death (Fig. 2B). This was partially reversed by the broad-specificity caspase inhibitor zVAD-FMK. Previous work suggested that inhibition of RAL function, by the expression of dominant inhibitory RAL variants to block RAL activation, or by expression of a minimal RAL-binding domain (RBD) to block RAL–effector interactions, is not toxic in a variety of cell types (Goi et al., 1999; Henry et al., 2000; Jullien-Flores et al., 2000; Moskalenko et al., 2002; Rosario et al., 2001). These apparently conflicting observations may be a consequence of selective inhibition of RALB function by siRNA versus inhibition of both RALA and RALB by the expression of dominant interfering molecules. Consistent with this, we found that siRNA-mediated inhibition of RALA and RALB together reversed the cell-death phenotype seen on loss of RALB alone (Figs 2A,3). This result suggests that RALA and RALB have antagonistic functions in the regulation of cell survival.
Figure 1.
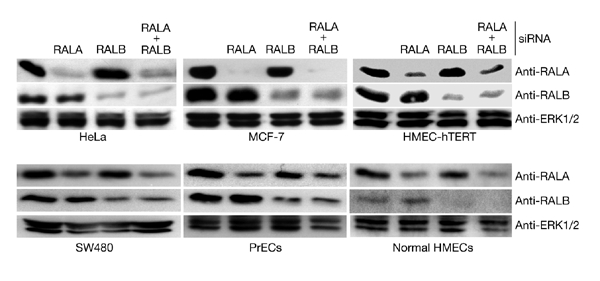
Small-interfering-RNA-mediated inhibition of RAL isoform expression. The indicated cell lines were transfected with small interfering RNAs (siRNAs) that were designed to selectively target RALA or RALB. Whole-cell lysates were prepared 72 h post-transfection and equivalent amounts of total protein were analysed by SDS–polyacrylamide gel electrophoresis for the indicated proteins. Extracellular-signal-regulated protein kinase 1/2 (ERK1/2) was used as a loading control. Similar results were obtained using two independent siRNA sequences for both RALA and RALB. HMECs, human-mammary epithelial cells; HMEC-hTERT, human diploid mammary epithelial cell line immortalized by hTERT expression; PrECs, primary human-prostate epithelial cells; RAL, RAS-like.
Figure 2.
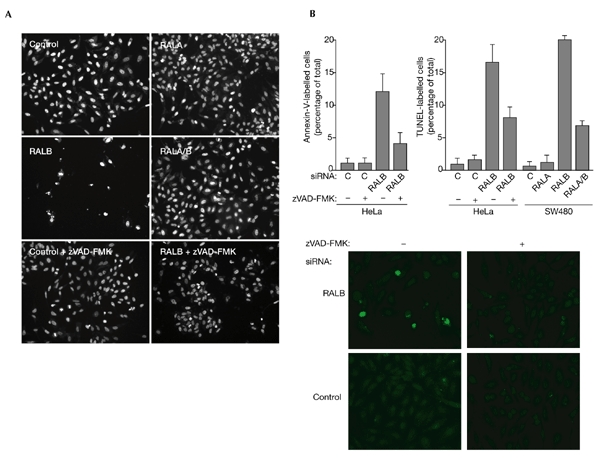
RALB is required for cell survival. (A) HeLa cells were transfected with the indicated small interfering RNAs (siRNAs) and incubated in the presence or absence of 50 μM zVAD-FMK. Ninety-six hours post-transfection, cells were fixed and stained with DAPI (4′,6-diamidino-2-phenylindole) to visualize nuclei. Representative fields of view are shown for each treatment. (B) HeLa and SW480 cells were transfected as described above. Seventy-two hours post-transfection, cells were labelled by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP–biotin nick-end labelling) to detect fragmented DNA, or with annexin V to detect surface phosphatidyl serine. The percentages of annexin-V-positive and TUNEL-positive cells were quantified by microscopic observation. Bars indicate s.e.m.s for three independent experiments. Panels showing TUNEL labelling from a representative experiment are shown below the graphs. RAL, RAS-like.
Figure 3.
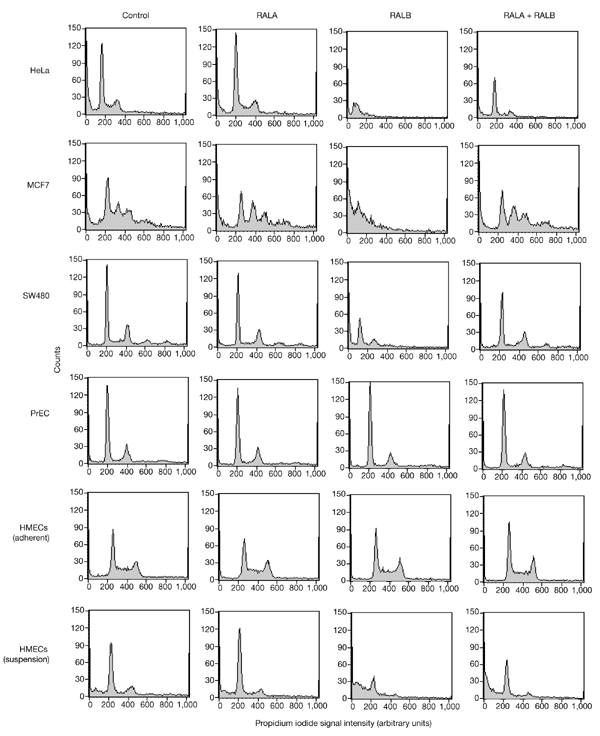
Tumour-derived cell lines are sensitized to RALB-dependent survival pathways. DNA content in propidium-iodide-treated cells was analysed by fluorescence-activated cell-sorting (FACS) 96 h after transfection with the indicated siRNAs. Asynchronous, proliferating, adherent cell cultures were used, except where indicated. MCF7 and SW480 cell lines are aneuploid and give multiple peaks. DNA fragmentation results in a shift of the population of events towards reduced signal intensities (hypodiploid). Inhibition of RALA and/or RALB expression was verified by western blotting. The data shown are representative of three independent experiments. HMECs, human-mammary epithelial cells; PrECs, primary human-prostate epithelial cells; RAL, RAS-like.
To explore the broad-spectrum contribution of RALB to cell survival, we inhibited RALB expression in two other tumour-derived cell lines, MCF7 (human breast adenoma) and SW480 (human colorectal carcinoma), as well as in non-cancerous primary human-prostate epithelial cells (PrECs), non-cancerous primary human-mammary epithelial cells (HMECs), and in non-cancerous, telomerase-immortalized normal HMECs (HMEC-hTERT; Herbert et al., 2002). Similar to HeLa cells, SW480 cells responded to inhibition of RALB expression by the induction of programmed cell death, as observed by microscopic examination (not shown), by TUNEL (Fig. 2B), and by a marked increase in the number of hypodiploid apoptotic bodies (Fig. 3). Similarly, MCF7 cells were acutely sensitive to loss of RALB expression. Also consistent with observations in HeLa cells, MCF7 and SW480 sensitivity to loss of RALB was relieved by co-inhibition of RALA expression. In contrast with the behaviour of cancer cell lines, loss of RALB expression did not induce apoptosis in 'normal' human prostate or mammary epithelial cells (Fig. 3). For both HMECs and PrECs, less than 1% of the cells were apoptotic in control cultures, and no differences were seen on inhibition of RALA or RALB alone or together. This suggests that tumour cells may develop an increased dependency on RALB-mediated survival pathways relative to non-cancerous, proliferating epithelial cells. Whereas RALB is dispensable for the survival of HMECs proliferating on tissue-culture plates, loss of RALB sensitized HMECs to induction of apoptosis on release from the extracellular matrix (Fig. 3). At least 48 h of incubation in suspension culture is typically required for most HMECs to induce anoikis (data not shown). Inhibition of RALB accelerated this process such that most cells were apoptotic within 16 h (Fig. 3). This was partially rescued by co-inhibition of RALA.
Several reports suggest that RAL GTPases can promote cell proliferation and oncogenic RAS-dependent transformation (Lu et al., 2000; Miller et al., 1997; Urano et al., 1996; White et al., 1996) and are required for serum-independent tumour-cell proliferation (Rosario et al., 2001). The observations described above suggest that RAL proteins contribute primarily to the regulation of cell survival in human cell lines and are not limiting for serum-dependent proliferation under standard culture conditions. To examine the contribution of endogenous RAL GTPases to oncogenic transformation, we tested the consequences of RAL inhibition on the anchorage-independent proliferation of human tumour cell lines. Expression of the minimal RBD of RAL-binding protein 1 (RALBP1), a candidate RAL effector, inhibits RAL function in cells, presumably through inhibition of the association of endogenous RAL effectors with activated RALA and RALB (De Ruiter et al., 2001; Jullien-Flores et al., 2000; Moskalenko et al., 2002; Rosario et al., 2001). As shown in Fig. 4A, and consistent with observations using RALA and RALB siRNAs, RBD expression does not significantly interfere with proliferation of these cell lines in adherent cultures. By contrast, the proliferative ability of RBD-expressing cells was severely impaired in suspension cultures. BrdU (5-bromo-2-deoxyuridine)-negative RBD-expressing cells showed no signs of blebbing or of condensed or picnotic nuclei (as visualized by DAPI (4′,6-diamidino-2-phenylindole) staining), suggesting that the proliferative defect was a consequence of cell-cycle arrest rather than apoptosis. Selective inhibition of RALA expression by RNAi resulted in a similar inhibition of anchorage-independent proliferation to that seen on RBD expression (Fig. 4B). This last observation suggests that RALA is responsible for transmitting a positive proliferative signal in tumour cells that is selectively required in the absence of matrix association.
Figure 4.
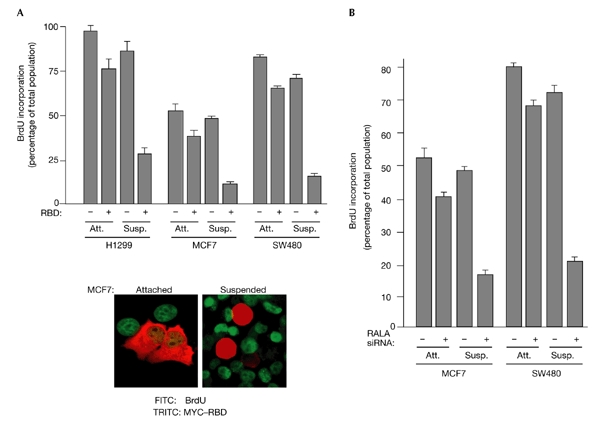
RALA is required for anchorage-independent proliferation of transformed cells. (A) The indicated cell lines were transiently transfected with MYC–RBD (a fusion of a MYC epitope to the RAL-binding domain (RBD) of RAL-binding protein 1) or empty vector. Twenty-four hours after transfection, cells were incubated with BrdU (5-bromo-2-deoxyuridine) for another 24 h in adherent (attached (Att.)) or suspension (Susp.) cultures. The percentages of RBD-expressing cells that incorporated BrdU are shown. More than 100 transfected cells were analysed for each experimental group. Transfection efficiencies ranged from 30% to 50%. Bars represent s.e.m.s for three independent experiments. An overlay image showing the detection of RBD expression and BrdU incorporation from a representative experiment is shown. (B) MCF7 and SW480 cells were transfected with control small interfering RNAs (siRNAs) that targeted mouse caveolin 1, or with RALA siRNAs. Seventy-two hours after transfection, cells were incubated with BrdU as described above. Quantitation of BrdU incorporation was carried out as described in (A). RAL, RAS-like.
Telomerase-immortalized HMECs that stably express H-RAS-G12V gain the ability to proliferate in the absence of matrix association (K. Kaur, J. Shay and M.W., unpublished data; Fig. 5), and this phenotype is RAL-dependent (Fig. 5A). We therefore tested the ability of oncogenic RAS to render cells sensitive to RALB-dependent survival pathways. As shown in Fig. 5, HMEC–hTERT cells are resistant to loss of RALB expression, whereas the partially transformed HMEC–hTERT:H-RAS-G12V cells are sensitive to the loss of RALB, mimicking the observations from tumour-derived cell lines.
Figure 5.
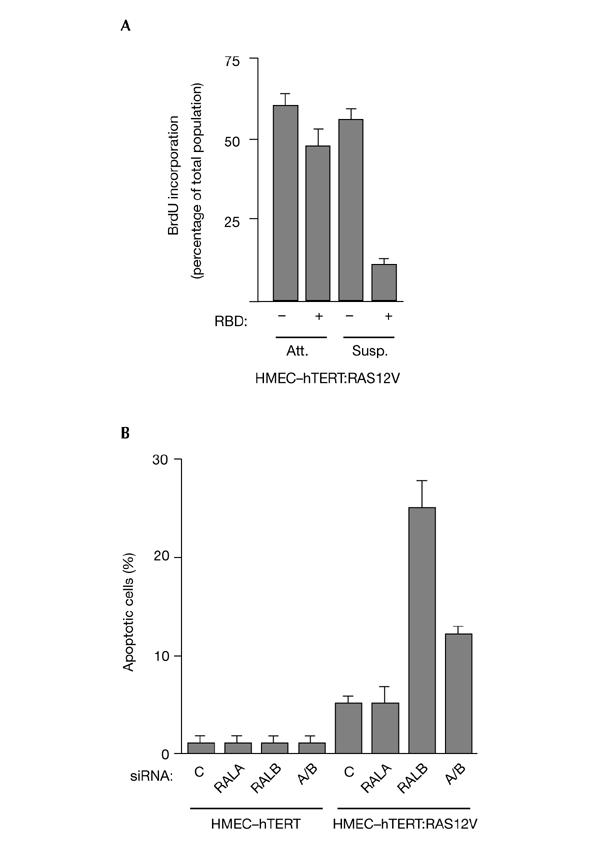
Oncogenic RAS sensitizes mammary epithelial cells to loss of RALB. (A) HMEC–hTERT:H-RAS-G12V cells were transiently transfected with MYC–RBD (a fusion of a MYC epitope to the RAL-binding domain (RBD) of RAL-binding protein 1) or empty vector. Proliferation assays were carried out as described in Fig. 4B. HMEC-hTERT and HMEC-hTERT:H-RAS-G12V cells were transfected with the indiciated small interfering RNAs (siRNAs). Seventy-two hours post-transfection, cells were stained with DAPI (4′,6-diamidino-2-phenylindole) to visualize chromatin structure. Cells with condensed, picnotic nuclei were scored as apoptotic. HMEC–hTERT, human diploid mammary epithelial cell line immortalized by hTERT expression; RAL, RAS-like.
Discussion
Through selective inhibition of RALA and RALB expression, we have revealed surprisingly discrete but interlocking contributions of these highly similar GTPases to the regulation of cell proliferation and survival. We have shown that RALB is essential for the survival of a variety of tumour-derived cell lines in culture. However, RALB is not limiting for the survival of non-cancerous, proliferating epithelial cells. By contrast, RALA seems to be dispensable for cell proliferation in adherent cultures but is required for tumour cells to maintain the ability to proliferate in the absence of matrix association. The coupling of RALA and RALB regulatory function was revealed by the observation that inhibition of RALA can relieve the sensitivity of tumour cells to loss of RALB.
In 'normal' cells, there is a tight coupling of the proliferative and apoptotic machinery, such that increasing the propensity to proliferate increases sensitivity to apoptosis (Evan & Littlewood, 1998). This observation has led to the hypothesis that oncogenic transformation minimally requires the acquisition of enhanced proliferative ability, together with suppression of apoptosis (Evan & Vousden, 2001; Green & Evan, 2002). The observations described here suggest that RAL GTPases are crucial components of oncogenic regulatory pathways, mediating both mitogenic and survival signals in tumour cells. This task seems to be split between the two isoforms, as RALA is required for anchorage-independent proliferation, whereas RALB is required for suppression of apoptosis. This division of responsibility may be the explanation for the apparent yin and yang relationship between RAL isoforms in the context of survival signalling. Inhibition of RALA seems to relieve mitogenic pressure, at least partially reversing oncogenic transformation, and therefore reducing tumour-cell dependency on RALB survival pathways.
We have not yet characterized the molecular basis of the divergent contributions of RALA and RALB to cell regulation. Several RAL-interacting proteins have been identified that may mediate RAL function in cells. These include phospholipase D1 (PLD1; Jiang et al., 1995), filamin (Ohta et al., 1999), the RAC/CDC42 GTPase-activating protein RALBP1 (Cantor et al., 1995; Jullien-Flores et al., 1995; Park & Weinberg, 1995), and the exocyst subunit Sec5 (Moskalenko et al., 2002; Sugihara et al., 2002). There is no indication that any of these proteins can selectively associate with either RAL isoform. However, as endogenous RAL–effector complexes have not been identified, the possibility remains that isoform-specific RAL–effector interactions may occur in cells. Similar to RAS isoforms, most of the sequence variation between RALA and RALB is in the carboxy-terminal hypervariable domains. These domains are required for appropriate lipid modification and membrane association (Reuther & Der, 2000). In the case of RAS GTPases, the C-terminal hypervariable domains seem to direct compartmentalization of RAS isoforms to different membrane microdomains (Roy et al., 1999). This compartmentalization may contribute to selective effector association among RAS isoforms by restricting partner availability (Jaumot et al., 2002; Yan et al., 1998). A similar phenomenon may contribute to the discrete biological actions of RALA versus RALB. In addition, partial loss-of-function RAL variants suggest that there are effectors still to be identified, which may display selective isoform association.
In summary, the loss-of-function analysis described here directly implicates RAL GTPases as crucial components in the maintenance of oncogenic transformation. The selective dependency of tumour cells on RALB expression suggests that this GTPase may induce survival pathways that are crucial for counteracting oncogene-driven apoptotic propensities. Therefore, RALB-dependent regulatory cascades may represent an Achilles' heel against which to target future therapeutic strategies.
Methods
Cell culture.
HeLa cells were maintained in DMEM supplemented with 10% FBS (Life Technologies). MCF7 cells were maintained in RPMI medium 1640 (Invitrogen) supplemented with 10% FBS. NCI-H1299 cells were cultured in DMEM supplemented with 5% FBS. PrECs, HMECs and the HMEC–hTERT cell line (a human diploid mammary epithelial cell line immortalized by hTERT expression; a gift from J. Shay) were grown in MCDB serum-free medium (Invitrogen) supplemented with 0.4% bovine pituitary extract (Hammond Cell Tech), 10 ng ml−1 epidermal growth factor (EGF), 5 μg ml−1 insulin, 0.5 μg ml−1 hydrocortisone, 5 μg ml−1 transferrin and 50 μg ml−1 gentamicin (Sigma). MCF7 and NCI-H1299 cells were transfected using Lipofectin and Lipofectin Plus reagents (Life Technologies) in accordance with the manufacturer's instructions. HME50–hTERT cells were transfected with Lipofectamine (Invitrogen). Before transfection, cells were seeded into 35-mm culture dishes and grown to 80% confluence.
Materials.
pRK5myc and pRK5myc–RBD have been described previously (Moskalenko et al., 2002). Synthetic siRNAs against RALA and RALB were designed by standard methods using the following sense sequences: 5′-GACAGGUUUCUGUAGAAGAdTdT-3′ (RALA), 5′-CAGAGCUGAGCAGUGGAAUTdTdT-3′ (RALA), 5′-GGUGAUCAUGGUUGGCAGCdTdT-3′ (RALB) and 5′-GACUAUGAACCUACCAAAGdTdT-3′ (RALB). The following antibodies' were used: mouse monoclonal anti-RALA and rabbit polyclonal anti-RALB (Becton Dickinson, Transduction Laboratories); mouse monoclonal anti-BrdU (Becton Dickinson); and rabbit polyclonal anti-MYC A14 (Santa Cruz Biotechnology). zVAD-FMK (Enzyme Systems Products) was used at a final concentration of 50 μM.
Transfections.
siRNAs (200 pM) were transfected using Oligofectamine (Life Technologies) in all cell lines. To obtain high transfection efficiencies (>90%) in HMEC–hTERT cells, cultures were briefly exposed to trypsin to induce macropinocytosis on apical and lateral surfaces (Hodges et al., 1973). Immediately before transfection, cultures were incubated in 0.05% trypsin, 0.5 mM EDTA for 60 s. The reaction was stopped with trypsin inhibitor, followed by a standard Oligofectamine transfection protocol. Plasmids were transfected using Lipofectin Plus reagents (Life Technologies) for MCF7 and NCI-H1299, and Lipofectamine 2000 (Life Technologies) for HME50–hTERT: H-RAS-G12V cells.
Apoptosis assays.
TUNEL and Annexin-V binding were carried out in accordance with the manufacturer's instructions (Becton Dickinson, Biosciences). For fluorescence-activated cell sorting (FACS), cells were collected, fixed with 50% ethanol at 4 °C for 1 h, and stained with propidium iodide at 37 °C for 30 min. Approximately 10,000 cells were collected for each assay, and were analysed using Cell Quest software (Becton Dickinson).
Proliferation assays.
Forty-eight hours post-transfection, cells were split and replated onto glass coverslips and 1%-agarose-coated dishes. BrdU was then added to a final concentration of 30 μM. After another 24-h incubation, cells were fixed with 3.7% formaldehyde, permeabilized with acetone at −20 °C for 5 min, and treated with 2 M HCl for 10 min. BrdU incorporation was visualized using mouse monoclonal anti-BrdU and FITC-conjugated anti-mouse IgG. Expression of the MYC-tagged RBD was visualized using rabbit anti-MYC A14 polyclonal antibodies and rhodamine-red X-conjugated anti-rabbit IgG.
Acknowledgments
This work was supported by the National Cancer Institute (grant number CA71443), the Welch Foundation (grant number I-1414) and a Lung Spore Grant development award (P50 CA70907).
References
- Cantor S.B., Urano T. & Feig L.A. ( 1995) Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol. Cell. Biol., 15, 4578–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chardin P. ( 1988) The ras superfamily proteins. Biochimie, 70, 865–868. [DOI] [PubMed] [Google Scholar]
- De Ruiter N.D., Burgering B.M. & Bos J.L. ( 2001) Regulation of the Forkhead transcription factor AFX by Ral-dependent phosphorylation of threonines 447 and 451. Mol. Cell. Biol., 21, 8225–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K. & Tuschl T. ( 2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Evan G. & Littlewood T. ( 1998) A matter of life and cell death. Science, 281, 1317–1322. [DOI] [PubMed] [Google Scholar]
- Evan G.I. & Vousden K.H. ( 2001) Proliferation, cell cycle and apoptosis in cancer. Nature, 411, 342–348. [DOI] [PubMed] [Google Scholar]
- Feig L.A., Urano T. & Cantor S. ( 1996) Evidence for a Ras/Ral signaling cascade. Trends Biochem. Sci., 21, 438–441. [DOI] [PubMed] [Google Scholar]
- Goi T., Rusanescu G., Urano T. & Feig L.A. ( 1999) Ral-specific guanine nucleotide exchange factor activity opposes other Ras effectors in PC12 cells by inhibiting neurite outgrowth. Mol. Cell. Biol., 19, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goi T., Shipitsin M., Lu Z., Foster D.A., Klinz S.G. & Feig L.A. ( 2000) An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J., 19, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D.R. & Evan G.I. ( 2002) A matter of life and death. Cancer Cell, 1, 19–30. [DOI] [PubMed] [Google Scholar]
- Henry D.O., Moskalenko S.A., Kaur K.J., Fu M., Pestell R.G., Camonis J.H. & White M.A. ( 2000) Ral GTPases contribute to regulation of cyclin D1 through activation of NF-κB. Mol. Cell. Biol., 20, 8084–8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert B.S., Wright W.E. & Shay J.W. ( 2002) p16(INK4a) inactivation is not required to immortalize human mammary epithelial cells. Oncogene, 21, 7897–7900. [DOI] [PubMed] [Google Scholar]
- Hodges G.M., Livingston D.C. & Franks L.M. ( 1973) The localization of trypsin in cultured mammalian cells. J. Cell Sci., 12, 887–902. [DOI] [PubMed] [Google Scholar]
- Jaumot M., Yan J., Clyde-Smith J., Sluimer J. & Hancock J.F. ( 2002) The linker domain of the Ha-Ras hypervariable region regulates interactions with exchange factors, Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem., 277, 272–278. [DOI] [PubMed] [Google Scholar]
- Jiang H., Luo J.Q., Urano T., Frankel P., Lu Z., Foster D.A. & Feig L.A. ( 1995) Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature, 378, 409–412. [DOI] [PubMed] [Google Scholar]
- Jullien-Flores V., Dorseuil O., Romero F., Letourneur F., Saragosti S., Berger R., Tavitian A., Gacon G. & Camonis J.H. ( 1995) Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem., 270, 22473–22477. [DOI] [PubMed] [Google Scholar]
- Jullien-Flores V., Mahe Y., Mirey G., Leprince C., Meunier-Bisceuil B., Sorkin A. & Camonis J.H. ( 2000) RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J. Cell Sci., 113, 2837–2844. [DOI] [PubMed] [Google Scholar]
- Lu Z., Hornia A., Joseph T., Sukezane T., Frankel P., Zhong M., Bychenok S., Xu L., Feig L.A. & Foster D.A. ( 2000) Phospholipase D and RalA cooperate with the epidermal growth factor receptor to transform 3Y1 rat fibroblasts. Mol. Cell. Biol., 20, 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.J., Prigent S., Kupperman E., Rioux L., Park S.H., Feramisco J.R., White M.A., Rutkowski J.L. & Meinkoth J.L. ( 1997) RalGDS functions in Ras- and cAMP-mediated growth stimulation. J. Biol. Chem., 272, 5600–5605. [DOI] [PubMed] [Google Scholar]
- Moskalenko S., Henry D.O., Rosse C., Mirey G., Camonis J.H. & White M.A. ( 2002) The exocyst is a Ral effector complex. Nature Cell Biol., 4, 66–72. [DOI] [PubMed] [Google Scholar]
- Ohta Y., Suzuki N., Nakamura S., Hartwig J.H. & Stossel T.P. ( 1999) The small GTPase RalA targets filamin to induce filopodia. Proc. Natl Acad. Sci. USA, 96, 2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.H. & Weinberg R.A. ( 1995) A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene, 11, 2349–2355. [PubMed] [Google Scholar]
- Reuther G.W. & Der C.J. ( 2000) The Ras branch of small GTPases: Ras family members don't fall far from the tree. Curr. Opin. Cell Biol., 12, 157–165. [DOI] [PubMed] [Google Scholar]
- Rosario M., Paterson H.F. & Marshall C.J. ( 2001) Activation of the Ral and phosphatidylinositol 3′ kinase signaling pathways by the ras-related protein TC21. Mol. Cell. Biol., 21, 3750–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Luetterforst R., Harding A., Apolloni A., Etheridge M., Stang E., Rolls B., Hancock J.F. & Parton R.G. ( 1999) Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nature Cell Biol., 1, 98–105. [DOI] [PubMed] [Google Scholar]
- Sugihara K., Asano S., Tanaka K., Iwamatsu A., Okawa K. & Ohta Y. ( 2002) The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nature Cell Biol., 4, 73–78. [DOI] [PubMed] [Google Scholar]
- Urano T., Emkey R. & Feig L.A. ( 1996) Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J., 15, 810–816. [PMC free article] [PubMed] [Google Scholar]
- White M.A., Vale T., Camonis J.H., Schaefer E. & Wigler M.H. ( 1996) A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem., 271, 16439–16442. [DOI] [PubMed] [Google Scholar]
- Yan J., Roy S., Apolloni A., Lane A. & Hancock J.F. ( 1998) Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem., 273, 24052–24056. [DOI] [PubMed] [Google Scholar]