

Summary
International Titisee Conference on Alzheimer's and Parkinson's Disease: From Basic Science to Therapeutic Treatment
Introduction
At this meeting, recent breakthrough findings on the molecular mechanisms, animal models and, in particular, the therapy of Alzheimer's disease (AD), and the second most common chronic neurodegenerative disorder Parkinson's disease (PD), were discussed. Both illnesses are paradigmatic for an expanding class of late-onset diseases that are characterized by brain deposits of misfolded proteins. The cross-β-sheet conformation of these pathologically misfolded proteins is a common biophysical feature of aggregation diseases. Specific dyes (such as thioflavin S) selectively bind to such so-called amyloid structures, irrespective of the individual protein that aggregates in each 'amyloidosis'. In the case of AD, the hallmark lesions are extracellular plaques composed of amyloid-β peptides (Aβ) that are derived from a larger Aβ precursor protein (APP; Fig. 1) and neurofibrillary tangles (NFTs) formed by the microtubule-associated protein tau. Furthermore, the pre-synaptic protein α-synuclein (α-SYN) fibrillizes into Lewy bodies (LBs), which are diagnostic for PD but also occur in some dementias including certain variants of AD. Although the underlying pathogenic cascades and the areas of the brain most affected are different for each disease, it is becoming increasingly apparent that the amyloidoses in the brain mutually influence each other, and experimental approaches used in one field have stimulated research in the other. Obviously, the amount of information and the broad area of research that is touched on at meetings such as this cannot all be incorporated into a brief meeting report. Here, we summarize some of the highlights.
Figure 1.
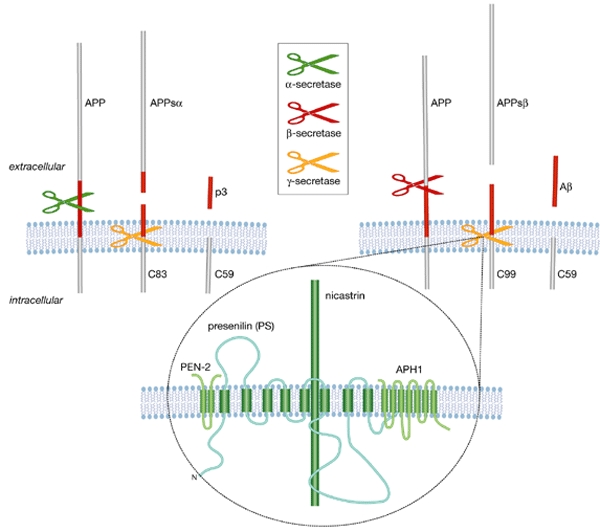
Cleavage of amyloid-β precursor protein. APP can be cleaved by α-secretase (upper, left) or by β-secretase (upper right), resulting in the release of the soluble ectodomains. The APP carboxy-terminal fragments (C83 and C99, respectively) are substrates for γ-secretase. This yields the p3 or Aβ peptides, which are secreted into the extracellular space, and the APP intracellular domain fragment C59, which is released into the cytoplasm. The γ-secretase is a multiprotein complex and presenilin (PS), nicastrin, PEN2 and APH1 are required for its full activity (lower part of the figure). Aβ, amyloid-β; APP, Aβ precursor protein; APPsα and APPsβ, soluble APP, cleaved by α- and β-secretase, respectively.

Almost 100 years after Alois Alzheimer saw his first patient with the complaint of “having lost herself” and his subsequent neuropathological description of what is now known as Alzheimer's disease, Christian Haass and Roger Nitsch invited a panel of international opinion leaders to the romantic Lake Titisee in the German Black Forest. This meeting, which took place during 19–23 March 2003, was the 87th International Titisee Conference, sponsored by the Boehringer Ingelheim Fonds, and the beauty of the surrounding scenery conferred a peaceful yet spirited environment to exchange thoughts and ideas.
Intramembrane cleavage
Three years ago, Brown and colleagues defined the concept of 'regulated intramembrane proteolysis' (Brown et al., 2000), a novel type of intracellular signalling that has been conserved in prokaryotes and eukaryotes. The two prototypes of this process are cleavage of the sterol-responsive-element binding protein (SREBP), which regulates cholesterol metabolism, and Notch, which regulates cell fate. Cleavage occurs first in the lumenal domain or ectodomain of the protein by a protease whose activity is regulated. A second cleavage occurs in the transmembrane domain of the substrate, resulting in the release of the cytoplasmic domain, which ultimately regulates gene transcription. The processing of APP that is relevant to the development of AD is initiated by β-secretase followed by intramembrane cleavage mediated by γ-secretase (Fig. 1). The latter liberates both a cytoplasmic domain that might participate in transcriptional control and at the same time produces the abundant 40-amino-acid soluble Aβ40, as well as the amyloidogenic 42-amino-acid Aβ42 (Haass & Steiner, 2002). Most familial AD mutations lead to a pathogenic increase of the Aβ42/Aβ40 ratio, explaining the particular interest in regulated intramembrane proteolysis at this meeting on neurodegenerative diseases.
The molecular composition of the γ-secretase complex that is responsible for the intramembrane proteolysis of APP was one of the hot topics. In addition to presenilin (PS), which constitutes the catalytic subunit of the complex, three other proteins—nicastrin, APH1 and PEN2—have been implicated in the γ-secretase cleavage process by genetic studies. T. Iwatsubo (Tokyo, Japan), H. Steiner (Munich, Germany) and D. Selkoe (Boston, MA, USA) reported that these three proteins, together with PS, form a stable complex (Edbauer et al., 2003; Kimberly et al., 2003; Takasugi et al., 2003). When they are expressed together in vivo, the cleavage rate of APP increases. Downregulation of any one of the components by RNA interference results in a strong decrease in γ-secretase activity, indicating that all four proteins are necessary to fully constitute activity. In yeast cells that do not have endogenous γ-secretase activity, expression of the four proteins together is sufficient and necessary to give cleavage of APP. The sum of the molecular weights of the different components is 200–250 kDa, but estimates of this total based on the electrophoretic motility of the complex in non-denaturing gels vary between 200 and 550 kDa. Furthermore, considering that APH1 and PS are both encoded by two different genes—APH1a and APH1b, and PS1 and PS2, respectively—several combinations of these proteins can be envisaged (De Strooper, 2003). This could explain why so many diverse substrates are cleaved by what was, until recently, considered to be a single γ-secretase complex. B. Martoglio (Zurich, Switzerland) broadened the discussion by examining the biology of signal-peptide peptidase (SPP), which is a presenilin-related protease that cleaves intramembrane proteins (Weihofen & Martoglio, 2003). SPP generates histocompatibility leukocyte antigen E (HLA-E) epitopes through the cleavage of signal peptides. Although SPP is a GXGD-type aspartyl protease like PS (Haass & Steiner, 2002), it is apparently not part of a multiprotein complex. Also, unlike PS, SPP does not undergo endoproteolysis, and has an opposite membrane orientation. This is probably the reason why it cleaves type II membrane proteins, whereas PS cleaves type I proteins. Nevertheless, several inhibitors that inhibit PS also inhibit SPP, pointing to potential side effects of using γ-secretase inhibitors to treat AD.
Therapeutic approaches against extracellular amyloid
One of the most fascinating and innovative concepts for clearing the extracellular lesions in the AD brain is immunotherapy. D. Schenk from ELAN Pharmaceuticals (San Francisco, CA, USA) explained the concept of AD immunotherapy, which involves the injection of pre-aggregated synthetic Aβ into patients to elicit an immune response against misfolded Aβ (Dodel et al., 2003). This approach has been validated in preclinical studies involving transgenic mouse models. ELAN's phase IIa trial unfortunately had to be stopped because of severe inflammatory side effects in several patients treated with AN-1792 (the Aβ42 formulation used in the clinical trials). However, the immunized patients are under further clinical monitoring and the definitive data will be disclosed at the end of this year. C. Hock (Zurich, Switzerland) provided some positive spin to the discussion by presenting the preliminary results from the Zurich cohort of the study. Twenty out of the 30 individuals who participated in this randomized trial responded to the Aβ immunization by generating antibodies that recognized the pathological Aβ species but not soluble Aβ or APP (Hock et al., 2002). In fact, the patients who developed an immune response against plaques showed a significantly decreased cognitive decline compared with the rapidly deteriorating control cohort (Hock et al., 2003). So far, 6% of the patients have developed post-vaccination complications (meningoencephalitis), but this grave side effect was treatable with corticosteroids. A recent case report on one of the patients from the British cohort of the ELAN study, who developed side effects and later died (Nicoll et al., 2003), suggests that the vaccine has 'clearing' capacities: the temporal cortex of this patient was almost entirely free of plaques and the other pathologies normally found in their immediate surroundings, such as plaque-associated dystrophic neurites and astrocytosis. However, plaque load in other brain regions (such as the frontal cortex) remained high, and vascular amyloidosis was unaffected even in the brain sections that were devoid of plaques. NFT pathology was also not suppressed within the time frame of this patient's immunization. Not surprisingly given the inflammatory complications, massive lymphocyte invasion into the brain was persistent even 12 months after the last AN-1792 injection. What was probably benign recruitment of helper T cells was found in the brain, but an alarming infiltration of white matter by macrophages was also detected. It remains to be determined whether or not any benefits gained by immunotherapy outweigh the risk of potential side effects.
In any event, Schenk suggested dissociating the epitopes in the Aβ peptide, as the amino-terminal region of Aβ is responsible for the B-cell response, whereas the middle region generates the T-cell response (Schenk, 2002). IgG2a subtype immunoglobulins that recognize N-terminal Aβ epitopes seem to be optimal. M. Jucker (Basel, Switzerland) pointed out that the cerebral amyloidosis present in up to 80% of AD patients might be involved in the development of vascular side effects. In a transgenic mouse model that has a pronounced cerebral amyloid angiopathy, passive Aβ immunization caused cerebral haemorrhage (Pfeifer et al., 2002). Thus, efforts should be undertaken to develop methods to identify patients at risk with high cerebral amyloid angiopathy scores, possibly by using functional magnetic resonance imaging.
Another approach to dissolving amyloid plaques is to disrupt their structure chemically. C. Soto (Plan-les-Ouates, Switzerland) has developed so-called β-sheet breakers, which are peptides that intercalate with the amyloid-seeding peptide sequence of aggregation-prone proteins and are spiked with conformation-breaking proline residues. A five-residue β-sheet-breaker peptide (iAβ5p) specifically prevented Aβ fibrillization in vitro. Administration of this β-sheet breaker reduced the plaque load and ameliorated astrogliosis and microglial activation as well as neurodegeneration in a transgenic mouse model. This treatment also improved water maze performance in a rat model of amyloidosis despite only partial reduction of plaque burden. Soto reported that the toxicological effects so far are acceptable and that no immunological responses to β-sheet breaking peptides have been detected. Current optimization strategies for this approach include the development of small peptidomimetics and non-hydrolysable peptide derivatives (Adessi et al., 2003).
Finally, another approach to the treatment of AD would be to prevent the formation of Aβ and thus inhibit plaque formation altogether. The three secretases involved in processing APP are obvious targets for amyloid-modulating approaches (Fig. 1), and proof-of-concept for β- and γ-secretase inhibition was provided at the meeting. M. Citron (Thousand Oaks, CA, USA) reviewed the properties of β-site APP-cleaving enzyme 1 (BACE1). The elimination of this β-secretase by knocking out the Bace1 gene in mice abrogated Aβ formation. More importantly, Citron reported that knocking out the Bace1 gene in a transgenic mouse model for plaque formation suppressed pathology, with no adverse effects, due to the elimination of Bace1. Thus, BACE1 inhibitors should have no side effects, but developing a drug to inhibit this enzyme may not be straightforward. So far, only peptides have been used to block the wide and complex active cleft of the BACE1 protease, and the development of drug-like small-molecule inhibitors of BACE1 remains a challenge (Citron, 2002).
The complex nature of the membrane-embedded γ-secretase (see above) and its many biological functions also make this a challenging drug target. E.H. Koo (La Jolla, CA, USA) has found that a subset of non-steroidal anti-inflammatory drugs (NSAIDs) are allosteric inhibitors of γ-secretase (Weggen et al., 2001). This effect is independent of the intended effect of NSAIDs, namely cyclooxygenase inhibition. The therapeutic value of NSAIDs against AD had been suspected from retrospective studies of patients who had been prescribed NSAIDs for rheumatoid arthritis. It was found that the incidence of AD in these patients was significantly reduced, and the anti-AD potential of NSAIDs was ascribed to the suppression of the inflammation around plaques. Koo then explained that the NSAID indomethacin was the only cyclooxygenase inhibitor investigated clinically for AD that showed efficacy in clinical trials and that inhibited γ-secretase. Thus, it may be possible to optimize NSAID derivatives for maximal γ-secretase inhibition and minimal cyclooxygenase inhibition. While the exact molecular mechanism of allosteric γ-secretase inhibition awaits elucidation, Koo has begun a prospective, two-centre phase I clinical trial with R-Flurbiprofen in 48 healthy elderly (55–80-year-old) subjects to assess its safety, tolerability and pharmacokinetics, as well as blood and cerebrospinal fluid levels of the biomarker Aβ.
Intracellular amyloid: tau and α-synuclein
A model of how the tau protein could interfere with neuronal viability before fibril maturation was presented by E.-M. Mandelkow (Hamburg, Germany). The motor protein kinesin moves towards the plus ends of microtubules, and another motor protein, dynein, moves towards the minus ends. However, when kinesin slips off the microtubule, tau physically blocks its reassembly, whereas dynein reassembly is not affected. Thus, dynein-mediated microtubular transport prevails in the presence of elevated tau levels, and plus-end transport is inhibited. This leads to the accumulation of axonal transport cargoes (synaptic vesicles, mitochondria, peroxisomes, and so on) in the cell soma, which deprives the synapse of vital support and antioxidative defence systems and, ultimately, causes a 'dying back' of the axon (Stamer et al., 2002). Microtubule affinity regulating kinases (MARKs) can alleviate this process by phosphorylating tau at sites that cause its dissociation from microtubules, which de-represses mitochondrial transport and thereby restores synaptic energy production. It remains to be shown how the AD-specific phosphorylation of tau influences this process.
An emerging topic in amyloid research is the reciprocal influence of the aggregating proteins Aβ, tau and α-SYN on each other. E. Masliah (La Jolla, CA, USA) provided experimental evidence that elevated levels of Aβ42 enhanced α-SYN inclusion body formation in a bigenic mouse model (Masliah et al., 2001). Similarly, crossbreeding between plaque mice or the direct injection of Aβ into tau transgenic mice promoted tau fibrillization (Lee, 2001). It is not yet clear how extracellular Aβ42 impinges on intracellular α-SYN and tau. The synergistic fibrillization of the axonally transported proteins tau and α-SYN, as described by V.M.-Y. Lee (Philadelphia, PA, USA), seems more straightforward, at least from a spatial point of view. In vitro experiments suggested that all isoforms of tau and α-SYN reciprocally seed each other to form separate homopolymers (Giasson et al., 2003). Transgenic mice were engineered to express tau or α-SYN in oligodendrocytes, and amyloid fibrils formed only on crossbreeding the two lines. Indeed, NFTs and LBs were occasionally observed in the same neuron.
J.Q. Trojanowski (Philadelphia, PA, USA) and P.J. Kahle (Munich, Germany) reported on the recent achievements in recapitulating LB pathology in transgenic mouse models that express human mutant α-SYN (Giasson et al., 2002; Neumann et al., 2002). In an age- and gene-dose-dependent manner, these animals developed fibrillar α-SYN deposits within neurites and neuronal perikarya and showed all of the traits of human pathology that are concomitant with lethal locomotor deterioration. Remarkably, the dopaminergic neurons in the midbrain, the degeneration of which accounts for parkinsonian symptoms in human patients, were consistently unaffected in the transgenic mouse models. By contrast, D. Kirik (Lund, Sweden) and P. Aebischer (Lausanne, Switzerland) reported that viral delivery of high gene doses of α-SYN into the substantia nigra did result in dopaminergic neurodegeneration (Kirik et al., 2002; Lo Bianco et al., 2002). This illustrates that a combination of transgenic technology and viral gene transfer considerably expands our experimental toolkit for the study of neurological disease. Aebischer went on to give an overview of the potential of lentiviral gene transfer to generate animal models and therapeutic approaches for neurodegenerative diseases. One approach would be to downregulate dominant genes or pathologically active enzymes (such as APP, PS, BACE1 and α-SYN) through the introduction of small interfering RNAs using lentiviral vectors. A second approach would be to restore the expression of recessive genes (such as parkin and DJ1). Third, neuroprotective and neurotrophic genes could be delivered to affected tissue. It might even become possible to optimize the therapeutic doses for individual patients by the use of tetracycline-regulatable lentiviral constructs.
Conclusions and perspectives
This was an exciting meeting that showed the enormous research efforts in the fields related to amyloid diseases. It is remarkable that amyloidogenic proteins such as tau, α-SYN and Aβ mutually influence each other's aggregation properties, which leaves one wondering whether common molecular mechanisms or common molecular principles underlie the two major neurodegenerative disorders AD and PD. At least, the fact that so many researchers cross the borders between these two pathological entities is remarkable. A great deal has been learned in recent years about the pathogenesis of both disorders, and a general feeling of optimism spread throughout the meeting when looking at the spectacular results obtained with Aβ vaccination in AD patients or with viral vectors that express growth factors in PD models. We look forward to future meetings focusing on drugs and therapy, and to evaluating the clinical progress in these fields.
Acknowledgments
This meeting was sponsored by the Boehringer Ingelheim Fonds (Foundation for Basic Research in Medicine). We apologize to the participants whose work could not be cited here due to space limitations.
References
- Adessi C. et al. ( 2003) Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer's disease. J. Biol. Chem., 278, 13905–13911. [DOI] [PubMed] [Google Scholar]
- Brown M.S., Ye J., Rawson R.B. & Goldstein J.L. ( 2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell, 100, 391–398. [DOI] [PubMed] [Google Scholar]
- Citron M. ( 2002) Emerging Alzheimer's disease therapies: inhibition of β-secretase. Neurobiol. Aging, 23, 1017–1022. [DOI] [PubMed] [Google Scholar]
- De Strooper B. ( 2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron, 38, 9–12. [DOI] [PubMed] [Google Scholar]
- Dodel R.C., Hampel H. & Du Y. ( 2003) Immunotherapy for Alzheimer's disease. Lancet Neurol., 2, 215–220. [DOI] [PubMed] [Google Scholar]
- Edbauer D., Winkler E., Regula J.T., Pesold B., Steiner H. & Haass C. ( 2003) Reconstitution of γ-secretase activity. Nature Cell Biol., 5, 486–488. [DOI] [PubMed] [Google Scholar]
- Giasson B.I., Duda J.E., Quinn S.M., Zhang B., Trojanowski J.Q. & Lee V.M.-Y. ( 2002) Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron, 34, 521–533. [DOI] [PubMed] [Google Scholar]
- Giasson B.I., Forman M.S., Higuchi M., Golbe L.I., Graves C.L., Kotzbauer P.T., Trojanowski J.Q. & Lee V.M.-Y. ( 2003) Initiation and synergistic fibrillization of tau and α-synuclein. Science, 300, 636–640. [DOI] [PubMed] [Google Scholar]
- Haass C. & Steiner H. ( 2002) Alzheimer disease γ-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol., 12, 556–562. [DOI] [PubMed] [Google Scholar]
- Hock C., Konietzko U., Papassotiropoulos A., Wollmer A., Streffer J., von Rotz R.C., Davey G., Moritz E. & Nitsch R.M. ( 2002) Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nature Med., 8, 1270–1275. [DOI] [PubMed] [Google Scholar]
- Hock C. et al. ( 2003) Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron, 38, 547–554. [DOI] [PubMed] [Google Scholar]
- Kimberly W.T., Ostaszewski B.L., Ye W., LaVoie M.J., Wolfe M.S. & Selkoe D.J. ( 2003) γ-Secretase is a membrane protein complex of presenilin, nicastrin, aph-1 and pen-2. Proc. Natl Acad. Sci. USA, 100, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D., Rosenblad C., Burger C., Lundberg C., Johansen T.E., Muzyczka N., Mandel R.J. & Björklund A. ( 2002) Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J. Neurosci., 22, 2780–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V.M. ( 2001) Tauists and βaptists united—well almost! Science, 293, 1446–1447. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C., Ridet J.L., Schneider B.L., Deglon N. & Aebischer P. ( 2002) α-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc. Natl Acad. Sci. USA, 99, 10813–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E., Rockenstein E., Veinbergs I., Sagara Y., Mallory M., Hashimoto M. & Mucke L. ( 2001) β-amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc. Natl Acad. Sci. USA, 98, 12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M. et al. ( 2002) Misfolded proteinase K-resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J. Clin. Invest., 110, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll J.A.R., Wilkinson D., Holmes C., Steart P., Markham H. & Weller R.O. ( 2003) Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med., 9, 448–452. [DOI] [PubMed] [Google Scholar]
- Pfeifer M., Boncristiano S., Bondolfi L., Stalder A., Deller T., Staufenbiel M., Mathews P.M. & Jucker M. ( 2002) Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science, 298, 1379. [DOI] [PubMed] [Google Scholar]
- Schenk D. ( 2002) Amyloid-β immunotherapy for Alzheimer's disease: the end of the beginning. Nature Rev. Neurosci., 3, 824–828. [DOI] [PubMed] [Google Scholar]
- Stamer K., Vogel R., Thies E., Mandelkow E. & Mandelkow E.-M. ( 2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol., 156, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N., Tomita T., Hayashi I., Tsuruoka M., Niimura M., Takahashi Y., Thinakaran G. & Iwatsubo T. ( 2003) The role of presenilin cofactors in the γ-secretase complex. Nature, 422, 438–441. [DOI] [PubMed] [Google Scholar]
- Weggen S. et al. ( 2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature, 414, 212–216. [DOI] [PubMed] [Google Scholar]
- Weihofen A. & Martoglio B. ( 2003) Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides. Trends Cell Biol., 13, 71–78. [DOI] [PubMed] [Google Scholar]