

Abstract
Escherichia coli cells that lack the carboxy-terminal part of FtsK fail to segregate their chromosomes properly during cytokinesis and tend to form chains. These chains are possibly formed as a result of DNA being trapped in the division planes or a failure to fuse the membrane during septum formation. If so, small molecules might diffuse between the apparent cell compartments. To investigate this theory, we developed an optical workstation that allows simultaneous imaging of and surgical operations on cellular objects in the sub-micrometre range. By surgical incisions of E. coli cell poles, diffusion of propidium iodide (PI) can be followed in real time. This analysis showed that PI was unable to diffuse from one cell equivalent to another in chain-forming ftsK mutants. Thus, the cytoplasm of the cell compartments in the chains seems to be fully separated.
Introduction
The transmembrane protein FtsK is required in at least two processes during the proliferation of Escherichia coli cells: septum formation and chromosome segregation. In contrast with the amino-terminal domain of FtsK, the carboxy-terminal domain (FtsKC) is not essential (Diez et al., 1997) and takes part in dif-dependent chromosome-dimer resolution (Steiner et al., 1999; Barre et al., 2000; Boyle et al., 2000) by switching the catalytic state of the XerC and XerD recombinases (Aussel et al., 2002). In addition, it is suggested that FtsKC functions as a motor protein that pumps any trapped DNA through the closing septum during cell division (Aussel et al., 2002), which may explain why mutants lacking FtsKC tend to form chains rather than filaments during growth (Diez et al., 1997, 2000; Liu et al., 1998). Indeed, DNA staining of FtsKC− mutants has shown that the cells often have asymmetrically positioned nucleoids, large anucleate regions and regions where DNA seems to be trapped in some division zones (Yu et al., 1998; Liu et al., 1998). Thus, chain formation in FtsKC-deficient cells has been proposed to be due to the lack of translocase activity, leading to DNA remaining in the septum (Aussel et al., 2002). The FtsKC domain of E. coli is homologous to the SpoIIIE protein of Bacillus subtilis, which is required to translocate the forespore chromosome across the polar septum during sporulation (Wu & Errington, 1994). In addition, a second and distinct role of SpoIIIE is to allow fusion of the mother-cell membrane that migrates around the prespore during engulfment (Sharp & Pogliano, 1999). Thus, it is possible that chain formation in E. coli cells lacking FtsKC is a result of a failure to fuse the membrane during the final stages of septum formation.
In this study, we analysed whether the division zones of the FtsKC chains allow molecular transport between cells, that is, if cells of the chain exchange cytoplasmic material. If the chains are formed due to DNA being trapped or a failure to fuse the cytoplasmic membrane, small molecules should readily move between the apparent cell compartments. To investigate this, we developed a method for studying the diffusion of a small molecule, propidium iodide (PI), after laser-scalpel-induced incisions into cells in the chains. This high-precision surgery required the firm alignment of live cells, which was accomplished by the simultaneous use of non-disruptive laser trapping. We show that this technique can be used for spatial and temporal molecular movement analysis.
Results and Discussion
Multiphoton imaging of DNA aberrations in ftsK mutants
We used two chain-forming ftsK mutants, ftsK1::cat and ftsK15Δ1264–1329, in this work. The product of ftsK1::cat lacks the entire cytoplasmic SpoIIIE-like domain, whereas the dominant ftsK15Δ1264–1329 allele encodes a product that lacks only the proximal part of this domain (Fig. 1A; Diez et al., 1997, 2000). On average, chains of the mutant population consist of four cell equivalents, but long chains can also be detected, especially in cells carrying the ftsK15Δ1264–1329 allele (Fig. 1B). To analyse DNA localization in the chains, we used DAPI (4′, 6-diamidino-2-phenylindole), which preferentially stains double-stranded DNA, with little or no background from cytoplasmic material. By using multiphoton microscopy and DAPI staining, there is a probability of almost zero that chromophores outside the focal plane will absorb two photons, as the photon density is not high enough in this region. This provides a precise method for determining the spatial distribution of DNA within a cell. Multiphoton microscopy of DAPI-stained ftsK1::cat (data not shown) and ftsK15Δ1264–1329 mutants (Fig. 1B–D) confirmed that the cells often have asymmetrically positioned nucleoids, large anucleate regions and regions where DNA seems to be trapped in the division plane (Fig. 1B–D), as previously shown (Yu et al., 1998; Liu et al., 1998). However, it is also clear that some septa of the chains are located far from DNA stained regions (Fig. 1F,G), which raises the question of why cell separation does not occur in these regions. It is possible that these regions do contain DNA, but the quantity is too small to be detected by DAPI staining, or that the septum membrane fails to fuse properly. Next, we wanted to investigate whether cells in the ftsK chains exchange cytoplasmic material due to leakage of molecules through the aberrant septa.
Figure 1.
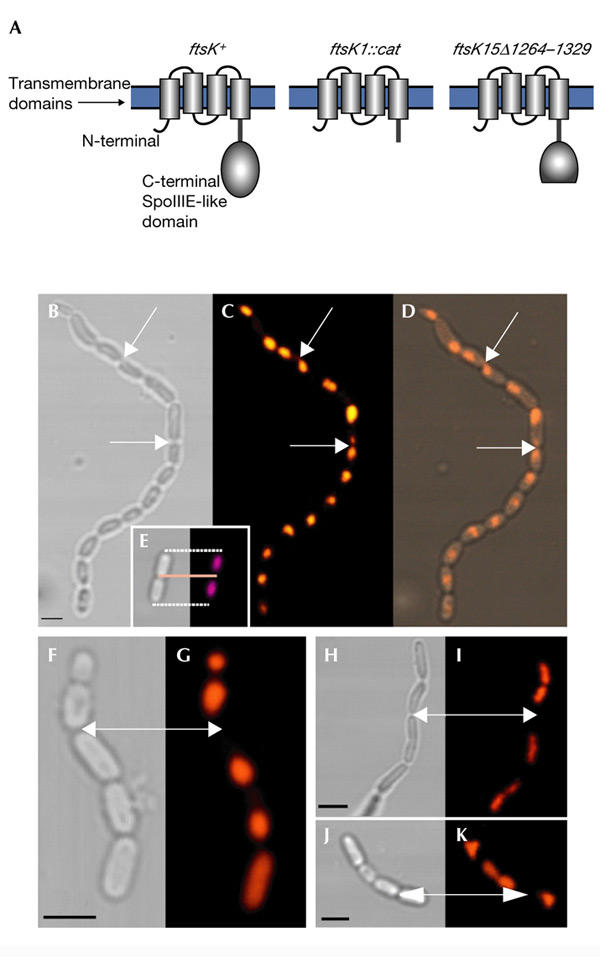
The FtsK protein, and DNA localization and chain formation in ftsK mutants. (A) The proteins encoded by the ftsK alleles used in this work. The transmembrane domains are located in the cytoplasmic membrane and the SpoIIIE-like domain is in the cytoplasm. ftsK+ denotes the wild-type allele. (B) Transmission micrograph of an ftsK15Δ1264–1329 chain. (C) Localization of DNA in the ftsK15Δ1264–1329 mutant chain shown in (B) analysed by DAPI (4′,6-diamidino-2-phenylindole) staining and multiphoton microscopy. (D) Merge of (B) and (C). The arrows indicate areas where DNA appears to be trapped in the division plane between two cell compartments. (E) The localization of DNA in dividing wild-type Escherichia coli is shown for comparison. (F,H,J) Transmission imaging and (G,I,K) multiphoton microscopy of DAPI-stained ftsK15Δ1264–1329 chains. Arrows indicate division zones, which appear to be far from and free of DNA-rich regions. Scale bars, 1 μm. C-terminal, carboxy-terminal; N-terminal, amino-terminal.
Molecular diffusion analysis of ftsK filaments
To study intracellular molecular movement and diffusion, we selected the probe PI, which does not penetrate intact cell membranes and emits red fluoresence when bound to nucleic acids. The experimental set-up was designed to combine optical tweezers for trapping, laser scalpels for dissecting, cutting and drilling, and confocal imaging (Fig. 2; and see the Methods section). Before analysing ftsK chains, we wanted to make sure that diffusion of PI in the bacterial cytoplasm could be followed for relatively long distances and times without bleaching problems and loss of signal. Therefore, we first analysed diffusion through long filaments of the ftsK mutant AD579 (Fig. 3) and an ftsZ temperaturesenstive (ts) strain (data not shown). This analysis was initiated by opening one pole of the filaments using an optical scalpel, which allowed the PI to penetrate into the cytoplasmic compartment of the bacteria and real-time diffusion analysis to be performed. The nucleic-acid distribution and the diffusion rate throughout the filamentous bacterium are shown in Fig. 3A. By assaying the changes in the pixel intensity of the image, we found that the time for PI to diffuse through the 20-μm-long filament was of the order of 10 min.
Figure 2.
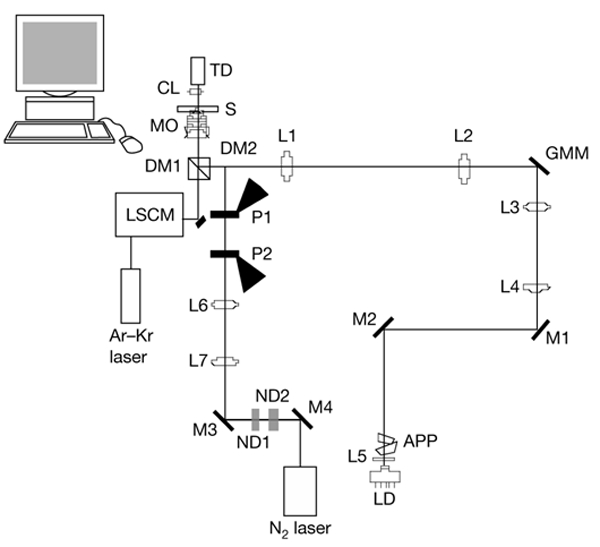
The optical set-up used in this study. The optical tweezers are formed using an 830-nm infrared laser diode (LD). The trap can be moved in the sample in all three dimensions, allowing it to individually position bacteria to a poly-L-lysine-coated coverslip. The scalpel is formed by a highly attenuated pulsed-ultraviolet-light laser. A laser scanning confocal microscope (LSCM) is used to simultaneously image the sample. A series of dichroic mirrors allows the excitation light from the Ar–Kr laser and the fluorescence from the sample to pass through, and the light forming both the optical tweezers and the laser scalpel is reflected through the objective (see the Methods section). APP, anamorphic prism pair; CL, condenser lens; DM, dichroic mirror; GMM, gimbal-mounted mirror; L, lens; M, mirror; MO, microscope objective; ND, neutral density filter; P, polarizer; S, sample; TD, transmission detector.
Figure 3.
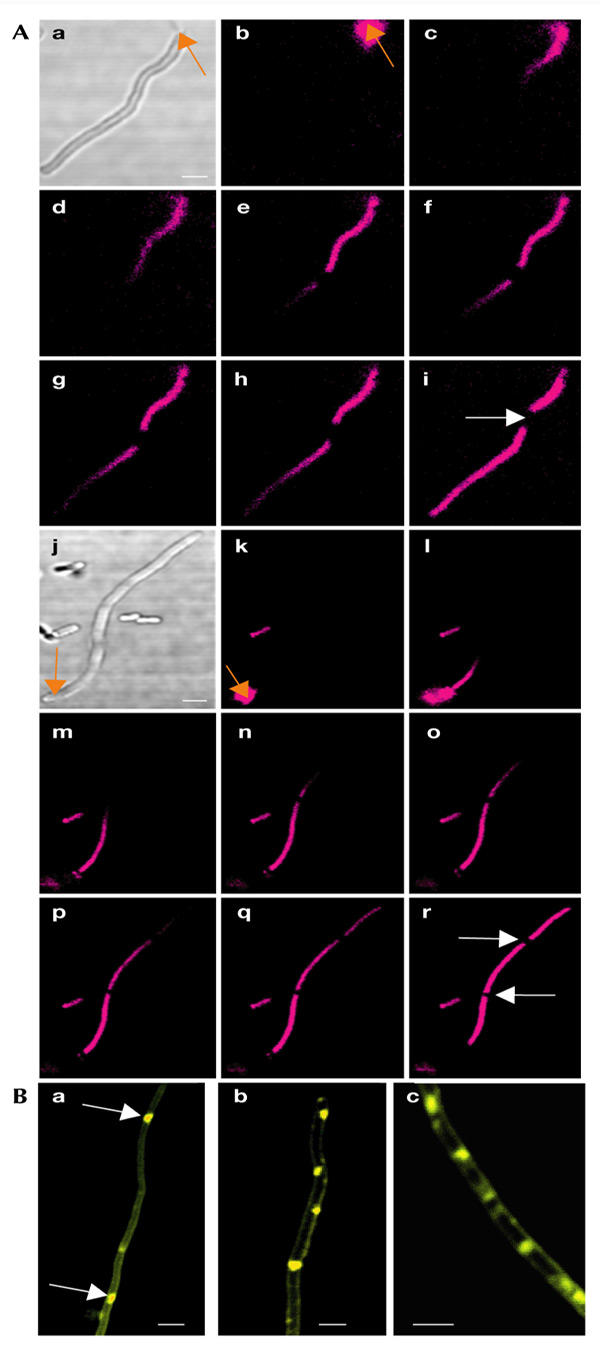
Molecular diffusion and membrane aberrancy in ftsK filaments. (A) Propidium iodide (PI) diffusion in filament-forming ftsK mutants (AD579). Diffusion was initiated by opening up the filaments with a pulsed ultraviolet light laser. PI fluorescence was detected using a confocal microscope. Orange arrows indicate the aiming points of the laser scalpel. The time elapsed between (Aa) to (Ah) was ∼10 min. The white arrows indicate areas devoid of fluorescent signal, which suggest that these compartments lack nucleic acids. (B) Membrane aberrancy in filament-forming ftsK mutants (AD579). The membranes were stained using FM4-64. Arrows indicate internal membrane regions. Scale bars, 1 μm.
As shown in Fig. 3A, filamentous ftsK1-derived mutants contain internal zones that do not fluoresce. These zones were not detected in ftsZ ts mutants (data not shown). After ablation of the cell pole, these zones move with the cytoplasmic contents towards the hole with a speed of ∼5 nm s−1. As PI binds to all forms of nucleic acids, we conclude that these zones are free from DNA and RNA but allow the PI to diffuse through them. At present, we do not know the exact nature of these nucleic-acid-free zones, but it is possible that the inner membrane forms internal pseudo-septa in the ftsK mutant. Consistent with this, membrane staining showed that the ftsK filaments contain aberrant equatorial membrane structures (Fig. 3B). It is not clear how these structures can move along the filaments, but it is possible that the entire cytoplasmic compartment, including surrounding membranes, moves towards the point of ablation in these experiments.
The diffusion analysis showed that the probe can be used for real-time spatial and temporal analysis of molecular movement, without significant bleaching problems or loss of signal.
Molecular movement analysis in ftsK chains
To allow molecular movement analysis in the chains of ftsK mutants, a small and discrete incision into the membrane was performed to avoid damage that might have spread along the cell to the division zones of the chains. Thus, low laser energy was used in this experiment, but the artificial pores created in the membrane had to be large enough to avoid immediate spontaneous self-repair. A theoretical minimum size of the laser focus, dmin, is the diffraction-limited focus described by the following equation:
where λ is the wavelength and NA is the numerical aperture of the objective. Using NA = 1.3, a minimal width of ∼316 nm is obtained. However, the images shown in Fig. 4Aa,Bb, which were taken just after the laser incision, indicate that the pore size is smaller than the diffraction limit of the focused laser beam; the fluorescent signal is concentrated at the central part of the bacterium, which has an approximate width of 300 nm. In addition, in contrast to the ablation experiment with ftsK filaments (Fig. 3), no fluorescent signal leaked out of cells in this experiment, which indicated that the pores produced may be even smaller than 100 nm (stable RNA on ribosomes is a major contributor to PI fluorescence in bacterial cells and ribosomes are ∼25 nm in diameter). Thus, the fluorescence signal seen in Fig. 4Aa,Bb, was probably a result of the creation of discrete pores, formed due to the breaking of molecular bonds in the membrane. The main conclusion is that we did not produce a single hole of the size of the focused laser beam. Instead, the laser pulses weakened the membrane integrity until one or many pores were created. It should be pointed out here that the ultraviolet (UV)-light laser beam is highly focused and is only destructive at the focal region.
Figure 4.
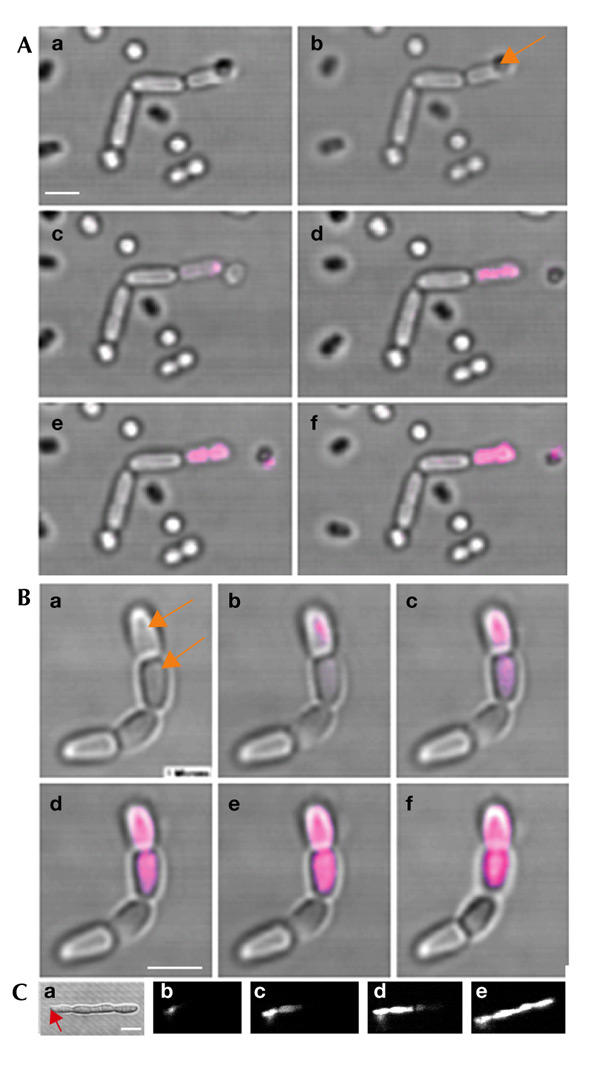
Molecular diffusion analysis in ftsK chains. (A) Propidium iodide (PI) diffusion in chain-forming ftsK mutants (AD158) initiated by opening up the pole of one cell in the chain with a pulsed ultraviolet-light laser. (B) Molecular diffusion analysis after opening up two neighbouring cells in the chain. The images shown are from the transmission detector, merged with the fluorescent response from the sample. The arrows indicate the aiming points of the laser scalpel. The time elapsed between the images in (Ba) and (Be) was ∼15 s. The image shown in (Bf) was taken ∼2 h later. (C) PI diffusion in an ftsI temperature sensitive (ts) mutant (after a shift from 30 °C to 42 °C), initiated by opening up the pole of one cell equivalent with the pulsed ultraviolet-light laser. Scale bars, 1 μm.
We did not see any diffusion between the cells in the ftsK mutant (both ftsK1::cat and ftsK15Δ1264–1329) chains (Fig. 4A). The experiment was repeated for 50 chains and gave the same results each time. In addition, optical drilling in cells at different locations within a chain gave the same result, that is, PI never entered the next neighbouring cell (Fig. 4B). Fluorescence was analysed continuously for 2 h after PI penetration, and during this time no signal was ever detected in the neighbouring cell compartments.
These data suggest that there is no free exchange of cytoplasmic material through a passage connecting the cells of the ftsK chains. Thus, it is unlikely that the ftsK chains of E. coli are formed because the edges of the membrane of the septum fail to fuse. However, the data do not rule out the possibility that small quantities of DNA are trapped in the division planes of these chains and that PI is too large to penetrate a small passage containing an unresolved terminus region of the chromosome. However, it should be noted that computer simulations, using the 1999 HyperChem, MM+ programme (Hypercube, Inc.), suggest that the diameter of the PI molecule is considerably less than that of DNA. We used ftsI ts mutants to show that diffusion of PI can occur in cells that are known to have an incomplete septal constriction. Constriction of the cell envelope requires FtsI, and electron microscopy studies have shown that filaments formed by ftsI mutants have blunt constrictions, indicating that constriction is initiated but not completed. As shown in Fig. 4C, PI was able to penetrate from one cell equivalent to another in the ftsI 'chains'.
Speculation
In this study, laser scalpel/drill technology was used to study diffusion processes within cells, and the holes achieved were smaller than the diffraction limited size of the focused laser beam. This set-up can therefore be used to manipulate subcellular structures, such as mitochondria, inside eukaryotic cells without affecting or harming the outer cell wall or other constituents of the cell. The question of whether mitochondria in some cell types form large interconnected networks is just one example of a problem that can be studied using this technique. We believe that the laser-induced molecular diffusion approach will be instrumental, together with standard bleaching and diffusion experiments using green fluorescent protein, in analysing molecular segregation, molecular barriers, vectorial transport phenomena and molecular filtering processes in a variety of model systems. In addition, the laser can be used as a source of heat rather than a scalpel, which allows spatial induction of any gene of interest that is linked to a heat shock promoter at subcellular, or even suborganellar, locations in specific model organisms or tissues.
Methods
Bacteria and growth conditions.
E. coli cells were grown aerobically (shaken at 220 r.p.m.) in LB media at 37 °C. The MC4100 E. coli background was used, as this strain lacks flagella, which facilitates optical trapping. AD10 (ftsK1::cat) and AD158 (carrying pAD15, which carries a truncated ftsK allele (Δ1264–1329)) have been described previously (Diez et al., 1997). One filament-forming suppressor mutant of ftsK1, AD579, was used in this study as it allowed long-distance analysis of intracellular molecular movement. The ftsI2158(ts) (AX655) and ftsZ844(ts) (PAT84) strains used were from the E. coli Genetic Stock Center.
The technical platform.
The experimental set-up was designed to combine optical tweezers for cell manipulation and trapping, laser scalpels for dissecting, cutting and drilling, and simultaneous confocal imaging (Fig. 2). The two manipulating laser beams enter the microscope through the empty fluorescence attachment port, and the laser scanning confocal unit is mounted on the side port of the microscope. The optical tweezers make use of an 830-nm infrared laser diode. A collimator lens (L5) and an anamorphic prism pair expand and form a well-collimated beam. Movement of the optical trap in the specimen plane (in the x–y direction) is accomplished by moving the gimbal-mounted mirror. The trap is also moveable in the z direction, perpendicular to the specimen plane. Moving the lens L4 and thus changing the divergence of the beam entering the microscope objective changes the z position of the focus in the sample. This technique was previously used by Fällman & Axner (1997). The power used to trap the bacteria ranged typically from 30 mW to 60 mW, measured at the entrance aperture of the objective.
The optical scalpel was formed by focusing a pulsed UV-light laser down the microscope objective. The nitrogen laser that was used had a wavelength of 337.1 nm, a pulse width of less than 4 ns and a maximum pulse energy of 300 μJ. Lenses L6 and L7 were used to ensure that the laser focus was formed in the image plane of the microscope objective. The pulse energy was optimized with respect to bacterial membrane damage by two neutral density filters (ND1 and ND2) combined with crossed polarizers (P1 and P2). The pulse energy needed to damage the membrane of a single bacterium ranged from 0.5 nJ to 5.0 nJ, measured at the entrance aperture of the microscope objective. Samples were imaged using a confocal microscope (MRC1024; Bio-Rad).
The three optical pathways (tweezers, scalpel and imaging) were combined in a filter block situated under the microscope objective. The filter block contains a dichroic mirror (DM1) and a blocking filter. The dichroic mirror and the blocking filter were coated in such a way that the excitation light from the Ar–Kr laser (488 nm) and the fluorescence light from the sample (500–635 nm) were able to pass through, whereas both the UV laser scalpel and the infrared laser tweezers were reflected into the microscope objective. The blocking filter was used to eliminate any scattered light from the laser beams. A dichroic mirror and variable emission filters in the scanning unit provided the possibility to separate the different emitted signals from the sample. In addition, a multiphoton microscope (Radiance 2000; Bio-Rad) was used to detect the DNA by using the blue fluorescent DAPI stain and a pulsed titanium sapphire laser (MaiTai), which was operated at a wavelength of 820 nm.
Laser-induced diffusion analysis.
PI was used at a concentration of 20 mM in anhydrous dimethyl sulphoxide. The chain-forming or filamentous bacteria were attached to a poly-L-lysine-coated cover glass using the optical tweezers. The bacteria were verified for viability using the fluorescent LIVE/DEAD BacLight (Molecular Probes) methodology in accordance with the protocol provided by the manufacturer (Ericsson et al., 2000; Haugland, 1996a).
Incision of the bacterial membrane required 1–50 pulses of the nitrogen laser, depending on the pulse energy and position of the laser focus relative to the bacterium studied. The pulse energy had to be attenuated to prevent bursting of the membrane and to leave the surroundings unaffected.
DNA and membrane staining.
Multiphoton microscopy (Radiance 2000; Bio-Rad) was used to detect DNA using the blue fluorescent DAPI stain (Haugland, 1996b). The pulsed femtosecond titanium sapphire laser was operated at a wavelength of 820 nm. Detection of membrane structures in ftsK filaments was carried out using the membrane-labelling stain FM4-64, as described previously (Haugland, 1996c).
Acknowledgments
This work was sponsored by grants form the Swedish Scientific Research Council VR (T.N. and D.H.) and by the Carl Tryggers Foundation for Scientific Research (D.H.).
References
- Aussel L., Barre F., Aroyo M., Stasiak A., Stasiak A. & Sherratt D. ( 2002) FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell, 108, 195–205. [DOI] [PubMed] [Google Scholar]
- Barre F.X., Aroyo M., Colloms S.D., Helfrich A., Cornet F. & Sherratt D.J. ( 2000) FtsK functions in the processing of a Holliday junction intermediate during bacterial chromosome segregation. Genes Dev., 14, 2976–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle D.S., Grant D., Draper G.C. & Donachie W.D. ( 2000) All major regions of FtsK are required for resolution of chromosome dimers. J. Bacteriol., 182, 4124–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez A., Farewell A., Nannmark U. & Nyström T. ( 1997) A mutation in the ftsK gene of Escherichia coli affects cell–cell separation, stationary phase survival, stress adaption and expression of the gene encoding the stress protein UspA. J. Bacteriol., 179, 5878–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez A., Gustavsson N. & Nyström T. ( 2000) The universal stress protein A of Escherichia coli is required for resistance to DNA damaging agents and is regulated by a RecA/FtsK-dependent regulatory pathway. Mol. Microbiol., 36, 1494–1503. [DOI] [PubMed] [Google Scholar]
- Ericsson M., Hanstorp D., Hagberg P., Enger J. & Nyström T. ( 2000) Sorting out bacterial viability with optical tweezers. J. Bacteriol., 182, 5551–5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fällman E. & Axner O. ( 1997) Design for fully steerable dual-trap optical tweezers. Appl. Opt., 36, 2107–2113. [DOI] [PubMed] [Google Scholar]
- Haugland R.P. ( 1996a) in Handbook of Fluorescent Probes and Research Chemicals (ed. Spence, M.), 406–407. Molecular Probes, Inc., Eugene, Oregon, USA. [Google Scholar]
- Haugland R.P. ( 1996b) in Handbook of Fluorescent Probes and Research Chemicals (ed. Spence, M.), 151. Molecular Probes, Inc., Eugene, Oregon, USA. [Google Scholar]
- Haugland R.P. ( 1996c) in Handbook of Fluorescent Probes and Research Chemicals (ed. Spence, M.), 372–374. Molecular Probes, Inc., Eugene, Oregon, USA. [Google Scholar]
- Liu G., Draper G.C. & Donachie W.D. ( 1998) FtsK is a bifunctional protein involved in cell division and chromosome localization in Escherichia coli. Mol. Microbiol., 29, 893–903. [DOI] [PubMed] [Google Scholar]
- Sharp M.D. & Pogliano K. ( 1999) An in vivo membrane fusion assay implicates SpoIIIE in the final stages of engulfment during Bacillus subtilis sporulation. Proc. Natl Acad. Sci. USA, 96, 14553–14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner W., Liu G., Donachie W.D. & Kuempel P. ( 1999) The cytoplasmic domain of ftsK protein is required for resolution of chromosome dimers. Mol. Microbiol., 31, 579–583. [DOI] [PubMed] [Google Scholar]
- Wu L.J. & Errington J. ( 1994) Bacillus subtilis SpoIIIE protein required for DNA segregation during asymmetric cell division. Science, 264, 572–575. [DOI] [PubMed] [Google Scholar]
- Yu X.C., Weihe E.K. & Margolin W. ( 1998) Role of the C terminus of FtsK in Escherichia coli chromosome segregation. J. Bacteriol., 180, 6424–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]