

Abstract
The smallest extrinsic polypeptide of the water-oxidizing complex (PsbQ) was extracted and purified from spinach (Spinacia oleracea) photosystem II (PSII) membranes. It was then crystallized in the presence of Zn2+ and its structure was determined by X-ray diffraction at 1.95-Å resolution using the multi-wavelength anomalous diffraction method, with the zinc as the anomalous scatterer. The crystal structure shows that the core of the protein is a four-helix bundle, whereas the amino-terminal portion, which possibly interacts with the photosystem core, is not visible in the crystal. The distribution of positive and negative charges on the protein surface might explain the ability of PsbQ to increase the binding of Cl− and Ca2+ and make them available to PSII.
Introduction
Oxygenic photosynthesis takes place in the thylakoid membrane of green plants, algae and cyanobacteria, in which sunlight is converted into chemical energy. Photosystem II (PSII) is one of the complexes of the thylakoid membrane and performs light-driven oxidation of water, with reduction of the plastoquinone pool and release of molecular dioxygen. PSII consists of a multi-component protein complex that comprises more than 25 subunits (Hankamer et al., 1997), most of which are embedded in the thylakoid membrane (Fig. 1). Several pigments (chlorophylls, carotenoids, pheophytin, heme and quinones), as well as inorganic cofactors (Mnn+, Ca2+, Cl− and Fe2+/3+) are bound to the protein matrix as essential or auxiliary parts of the electron-transport chain from water to plastoquinones. The reaction centre of PSII is composed of a heterodimer that consists of the two transmembrane proteins D1 and D2 and the heterodimeric cytochrome b559. All redox and photoredox cofactors that are involved in PSII activity are located in this 'core' of the complex (Nanba & Satoh, 1987). Water splitting, which gives rise to molecular dioxygen, is performed on the lumenal side of the membrane through a cluster of four Mn2+ ions that are coordinated with the D1 polypeptide. Several Ca (two or, more probably, one) and Cl ions are required for optimal activity of this 'water-oxidase complex', but the detailed molecular mechanism of their function is not yet fully understood (Ananyev et al., 2001; Ono, 2001). The function of Ca2+ and Cl− is modulated by the presence of three extrinsic proteins at the lumenal surface (Seidler, 1996), which are named photosystem b O (PsbO), PsbP and PsbQ in higher plants (formerly known as OEE1/33 kDa, OEE2/23 kDa and OEE3/16 kDa, respectively). PsbO is also thought to be involved in stabilization of the Mn cluster, whereas PsbP and PsbQ are more directly involved in Ca2+ and Cl− binding (Seidler, 1996). Although PsbO is also found in all oxygenic phototrophs, PsbP and PsbQ are not present in cyanobacteria, in which they are functionally substituted by the proteins PsbU and PsbV (Shen et al., 1995, 1988), which are also called cytochrome c550 and 12-kDa extrinsic protein, respectively. In the absence of the extrinsic polypeptides, oxygen evolution can still proceed, provided that high, non-physiological levels of Ca2+ and Cl− are present. This observation has suggested a function for the extrinsic proteins in creating the correct ionic environment during water oxidation (Ghanotakis et al., 1984).
Figure 1.
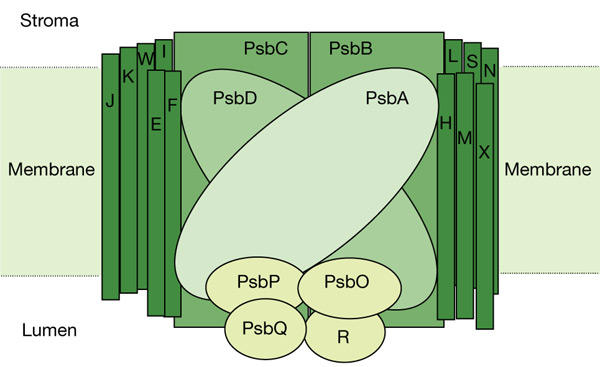
Proteins that form the photosystem II core complex. Light-harvesting complex components are not shown. Adapted from http://www.bio.ic.ac.uk/research/barber/psIIimages/psII.html. The letters E, F, H–N, R, S, W and X indicate photosystem b (Psb) components.
Recently, the structures of two reaction-centre complexes from cyanobacteria have been reported (Zouni et al., 2001; Kamiya & Shen, 2003). In the latter study, the location of all three of the extrinsic subunits was determined, giving a clear picture of the structure of the photosystem. However, only a relatively low-resolution structure (8 Å) of PSII from higher plants is available (Rhee et al., 1998), although this study has recently been extended and has allowed the identification of all transmembrane helices of the core proteins (Hankamer et al., 2001). However, as the preparation that was used lacked several PSII proteins, including PsbP and PsbQ, our knowledge of their structural organization relies on single-particle analysis of the intact oxygen-evolving PSII supercomplex (Nield et al., 2000a,b, 2002), with a resolution limited to ∼17 Å. We have isolated to purity and crystallized the PsbQ protein. Its crystal structure at a resolution of 1.95 Å is reported here.
Results and Discussion
Structure of PsbQ
The structure of the 16-kDa protein component of the oxygen-evolving enhancer complex (PsbQ) can be divided into two portions: a mobile amino-terminal chain (residues 1–45) and a well-folded domain that starts at around residue 48 and extends to the carboxy terminus. This latter portion has a typical four-helix bundle structure, which can be contained in a parallelepiped with dimensions of approximately 45 Å × 24 Å × 20 Å. The electron density of the main chain is well defined in all the residues of the bundle (Fig. 2). The same is true for the side chains, with the exception of a few positively charged residues (K53, K63, K90 and K123), which present partially mobile side chains. The four α-helices (I–IV) that form the bundle run approximately antiparallel and are connected by short turns (Fig. 3). The four helices span residues 48–67, 71–94, 98–123 and 127–146, respectively. α-Helix I shows some irregularities, as conformational torsion angles of residues 61 and 62 deviate from those of theoretical α-helix values. The other three α-helices do not show discontinuities. Residues 68–70, 95–97 and 124–126 connect α-helices I and II, II and III, and III and IV, respectively.
Figure 2.

Stereo view of part of an electron-density map. The map was calculated with the coefficients 2Fobs − Fcalc. Blue lines, contour level at 1.5σ. Red line, contoured to 15σ; represents the position of the Zn ion. Symmetry-related atoms are shown as green spheres. Red spheres represent solvent molecules.
Figure 3.

The PsbQ protein. Helices are numbered I–IV, from the amino terminus to the carboxy terminus. Red spheres indicate two zinc ions that are bound to the protein in the crystal state. Inside the crystal cell, only one zinc ion is bound per molecule but, as binding is asymmetric, each polypeptide chain interacts with two ions. The drawing was obtained using the Molscript programme (Kraulis, 1991) and rendered with Raster3D (Merrit & Bacon, 1997). PsbQ, photosystem b protein Q.
The four helices pack together mainly through hydrophobic interactions. In fact, all the potentially charged and hydrophilic residues point towards the solvent, with two exceptions: Thr 108 and Thr 137. The former residue is hydrogen bonded to carbonyl O 104, and the latter to carbonyl O 133. For the others, the internal surface of the bundle is highly hydrophobic, being occupied mainly by Leu, Ile, Ala and Val residues. Interestingly, aromatic amino acids lie on the surface of the bundle, partially exposed to the solvent, with the exception of residue 71, the only Trp present in the protein, which closes one of the extremities of the bundle.
Our model starts from residue 38, as no clearly interpretable density is present in the map for the N-terminal portion of the molecule. We were not able to state unequivocally whether the N-terminal part of the protein is disordered and cannot be seen in the crystal, or whether the protein was degraded during the crystallization process. SDS–polyacrylamide gel electrophoresis (PAGE) analysis of a drop containing the crystals revealed the presence, along with the intact protein, of a degradation band of ∼14 kDa. Moreover, the presence of several Pro and Gly residues at the N terminus suggests that this portion of the molecule could be highly flexible. From residues 38–44, the chain runs as a strand close to the surface of helix II. The electron density of the main chain is fairly clear, but it is rather poor for the side chains, so that some amino acids might not have been located correctly. Residues 44–47 form a β-turn, with the last residue starting the first α-helix. The overall structure is stabilized by 182 hydrogen bonds among protein atoms and four that are mediated by solvent molecules.
Zinc-ion coordination site
A zinc ion is bound to the surface of the protein and helps the molecules to stay together in the crystal lattice. Zn2+ forms a bridge between two symmetry-related molecules: it interacts with one residue, Gln 105, of one molecule, and two residues, His 120 and Glu 129, of a symmetry-related mate. The fourth position of the tetrahedral coordination is occupied by a solvent molecule. Zinc seems to be fundamental for crystallization and, in fact, crystals can only be grown in its presence, whereas several other divalent cations, such as Ca2+, Cd2+ and Mg2+, are unable to promote crystal growth. Because the residues involved in zinc binding are not completely conserved in other proteins of the family (see below), we suggest that the zinc ion is only necessary for crystal growth and that it does not have any function in vivo.
Structural comparisons
The four-helix bundle motif is fairly common in the Protein Data Bank (Berman et al., 2002). A search performed using the DALI server (Holm & Sander, 1993) showed that the closest structural homologues of spinach (Spinacia oleracea) PsbQ are α-catenin (1H6G; Z score, 11.8), cytochrome b562 (256B; Z score, 11.0), apolipoproptein e3 fragment (1NFN; Z score, 11.0) and aspartate receptor (1VLS; Z score, 10.4). All these structures include a four-helix bundle with up-and-down topology. Other homologous proteins in the list that have lower Z scores are some cytochrome c′ proteins, vinculin fragments, focal adhesion kinase 1 and myohemeritrin. No regions of the above molecules show any sequence similarity to PsbQ, and no functional similarity is apparent. Interestingly, PsbQ does not share any structural similarity with the PsbU or PsbV proteins of cyanobacteria (Kamiya & Shen, 2003).
Interaction with photosystem II
Electron crystallography and X-ray diffraction studies have provided a fairly detailed picture of the organization of oxygenic reaction-centre complexes of higher plants and cyanobacteria (Zouni et al., 2001; Kamiya & Shen, 2003; Rhee et al., 1998) at resolutions of 8 Å and 3.7 Å, respectively. Folding of the PsbO protein in particular has been shown clearly by Kamiya & Shen (2003), and this organization is probably shared by higher plants. However, PsbQ and PsbP proteins have no counterparts in cyanobacteria, in which they are functionally substituted by PsbU and PsbV. In addition, the best available model of PSII from higher plants was obtained with a relatively simple PSII core that lacks both PsbP and PsbQ (Rhee et al., 1998; Hankamer et al., 2001). At present, most of our knowledge about the structural organization of the oxygen-evolving PSII supercomplex is derived from single-particle reconstruction. This analysis assigns the three extrinsic proteins to a lumenal protrusion, on the basis of evidence that it is lost on washing with Tris. The size and shape of our model are comparable with those proposed on the basis of cryoelectron microscopy (Nield et al., 2002).
Biochemical data indicate that the N-terminal residues of PsbQ are involved in binding to the PSII complex, provided that the PsbP protein is already present (Seidler, 1996). This is consistent with the finding that this region is probably flexible in the isolated protein. One could speculate that this domain takes on its functional structure only when interacting with its docking site on the PSII complex.
Comparison of the available amino-acid sequences of the PsbQ components from plants and algae indicates that only 22 residues are fully conserved (Fig. 4A). The hydrophobic protein core is well preserved in terms of hydrophobicity. On the protein surface, 13 residues are fully conserved, and all but two (Ser 55 and Thr 91) are charged residues (Fig. 4B). In terms of charged side chains, there are ten positive and four negative charges that are conserved on the protein surface, which are located in two well-defined areas: one is positioned at the beginning of α-helix I and in the nearby region connecting α-helix II to α-helix III (region A in Fig. 4B). As this area is close to the N-terminal strand, which is thought to interact with the PSII core, it seems reasonable to conclude that this conserved region represents one of the areas that contacts the rest of the complex. The other conserved portion of the surface is at the opposite side of the molecule, around the end of helix III and the start of helix IV (region B in Fig. 4B). This second group of conserved charged residues might be involved in interactions with other extrinsic proteins or with a different site on the PSII core. Both would be consistent with experimental evidence that the extrinsic polypeptides can be released from the PSII core by an increase in ionic strength (Seidler, 1996). Alternatively, the latter region may be involved in interactions with ions.
Figure 4.

Conserved amino acids in PsbQ proteins. (A) Alignment of available amino-acid sequences of PsbQ proteins. In some species, more than one gene is present in the genome. The 'conserved' line shows fully conserved residues. Conserved positively charged residues are shown in blue, conserved negatively charged residues in red, and other conserved residues in green. Double-ended arrows indicate the positions of α-helices in the three-dimensional structure of Spinacia PsbQ. (B) Amino acids that are conserved on the surface of PsbQ proteins of known amino-acid sequence. Positively charged residues are shown in blue, negatively charged residues in red, and hydrophilic residues in green. Arabidopsis, A. thaliana; Chlamydomonas, C. reinhardtii; Onobrychis, O. viciifolia; Volvox, V. carteri; Zea, Z. mays. Psb, photosystem b.
The surface of the well-structured four-helix bundle domain is rich in potentially charged residues (18 Arg or Lys residues and 1 His residue, and 13 Glu or Asp residues). As shown by the electrostatic-potential surface, which was calculated using the GRASP programme (Nicholls et al., 1991), they are not uniformly distributed. The sides that include α-helices II and III are, in fact, more heavily charged (particularly positively), whereas negative charges are located more on the borders on the sides of α-helices I and IV. This distribution may explain one of the known properties of PsbQ: its electrostatic interaction with PsbP (De Vitry et al., 1989). Moreover, it may be responsible for the generation of an electric field that can increase the binding of Cl− and Ca2+ and make them available to PSII.
Methods
Purification and crystallization.
Extrinsic proteins were extracted by incubating spinach PSII membranes (Berthold et al., 1981) in 50 mM MES, pH 6.5, 1 M CaCl2 for 20 min in ice at a chlorophyll concentration of 1 mg ml−1 (Ono & Inoue, 1983). The suspension was centrifuged at 4 °C for 30 min at 40,000g and the supernatant, which contained the three extrinsic proteins (PsbO, PsbP and PsbQ), was dialysed against 20 mM Tris-HCl buffer, pH 7.2, in the presence of 0.15 M NaCl, using an Amicon system with a cut-off membrane of 3 kDa. The solution was then loaded onto a Superdex 75 HiLoad 26/60 column for gel filtration to separate the two small proteins (16 kDa and 23 kDa) from the larger one (33 kDa). The elution was performed isocratically, with the same buffer used for dialysis, with a flow rate of 2 ml min−1. The fraction of interest, which contained the 16- and 23-kDa subunits, was collected at an elution time of 88 min, whereas the 33-kDa subunit eluted at 78 min. The sample was then dialysed against 20 mM Tris-HCl, pH 7.5, and applied to a Mono Q HR 5/5 column for anionic exchange to achieve separation of the 16-kDa subunit from the 23-kDa subunit. Elution was carried out with a 0.0–0.5 M NaCl gradient in 20 mM Tris-HCl, pH 7.5. The elution was performed for 30 min, with a pre-gradient of 15 min, at a flow rate of 1 ml min−1. Consistent with its high theoretical pI (9.25), the 16-kDa subunit does not bind to the anionic exchanger and is collected as a highly purified protein during the pre-gradient. The 23-kDa subunit and minor impurities interact with the stationary phase and are eluted during the gradient. The PsbQ collected was finally dialysed against 50 mM Tris-HCl, pH 7.2, 50 mM NaCl, and concentrated to 24 mg ml−1 for crystallization trials. Purification was checked by SDS–PAGE, which showed that the intact protein of 16 kDa was recovered at the end of the purification.
For crystal growth, drops of 2–4 μl of the concentrated protein solution (20–24 mg ml−1) were mixed with an equivalent volume of the precipitant solution, which contained 25–30% (w/v) PEG 3350 or PEG 4000 as precipitant agents, 5 mM Zn2+, acetate buffer, pH 4.6, and was equilibrated against 0.5 ml of the same precipitant solution. PsbQ crystals grew in 4–5 days at 20 °C.
Data collection, processing, structure determination and refinement.
The crystals did not need a cryoprotectant solution and were frozen directly under a nitrogen stream at 100 K for the diffraction experiment. Diffraction data were collected using synchrotron radiation (ID14-4 and ID29, European Synchrotron Radiation Facility (ESRF); and XRD1, Elettra) with charge-coupled device detectors. The crystals belong to the P3221 space group, with a and b cell parameters of ∼50 Å, and c varying from 92 to 97 Å. One molecule is present in the asymmetric unit, which corresponds to a VM coefficient of 2.15 Å3/Da and a solvent content of ∼42% (Matthews, 1968).
A native data set was measured on a crystal that diffracted to 1.95-Å resolution (a = b = 50.282 Å; c = 94.067 Å). The multiwavelength anomalous diffraction experiment was performed on a crystal that diffracted to 2.15-Å resolution (a = b = 49.95 Å; c = 91.92 Å). The remote wavelength used was 0.93 Å, and the inflex and peak wavelengths were 1.284 Å and 1.282 Å, respectively. Table 1 shows statistics for the two data sets.
Table 1.
Statistics of X-ray data collection and refinement
| Native data set | Peak | Inflection point | Remote | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 0.9393 | 1.284 | 1.282 | 0.93 |
| Resolution range (Å) | 39–1.95 (2.06–1.95) | 45–2.15 | – | – |
| Unique reflections | 10,333 (1,340) | 6,475 (551) | 6,616 (446) | 7,538 (1,094) |
| Multiplicity | 8.9 (5.3) | 4.5 (2.5) | 4.3 (2.2) | 5 (5.0) |
| Completeness (%) | 98.1 (90.9) | 99.4 (96.8) | 98.8 (94.1) | 99.6 (99.6) |
| <I/σ(I)> | 3.3 (1.2) | 5.0 (1.7) | 5.9 (2.2) | 4.0 (1.6) |
| Rsymm | 0.11 (0.33) | 0.063 (0.44) | 0.05 (0.39) | 0.065 (0.45) |
| Phasing power | – | 2.33 (1.15) | 1.87 (0.80) | 1.74 (0.97) |
| FOM | – | 0.31 (0.13) | 0.38 (0.15) | 0.46 (0.28) |
| FOM (overall) | 0.66 (0.37) | – | – | |
| Refinement | ||||
| Reflections used | 10,307 | – | – | – |
| Reflections Rfree set | 1,062 | – | – | – |
| Rcryst | 0.227 (0.382) | – | – | – |
| Rfree | 0.268 (0.387) | – | – | – |
| r.m.s. deviation from ideal values | ||||
| Bond lengths (Å) | 0.01 | – | – | – |
| Bond angles (°) | 1.5 | – | – | – |
Values in brackets refer to the highest resolution shell. σ, standard deviation; FOM, figure of merit; I, intensity; r.m.s., root mean square.
Data were processed with MOSFLM (Leslie, 1991) and SCALA (Collaborative Computational Project, 1994). The position of one Zn ion produced starting phases (Table 1) that were improved by density modification using a solvent content of 40% using SOLVE/RESOLVE 2.0 software (Terwilliger & Berendzen, 1999) and CNS 1.0 (Brünger et al., 1998). The resulting electron-density map was readily interpretable. Model building was performed partly manually with XtalView software (McRee, 1999) and partly automatically using the Arp/Warp package (Perrakis et al., 1999). The model was refined using CNS (Brünger et al., 1998) and Refmac (Murshudov et al., 1997) to a final crystallographic R factor of 0.227 and an Rfree of 0.268. We were not able to locate residues 1–37 on the electron-density map. In some areas of the cell, density blobs were still visible and have not been interpreted.
The quality of the final model was checked with the PROCHECK program (Laskowski et al., 1993). It has good stereochemistry, with all the conformational torsion angles in the most favoured or additionally allowed regions, and an overall G factor of 0.2.
Acknowledgments
This paper is dedicated to the memory of Eraldo Antonini. We thank the staff of beamlines ID29 and ID14-4 of ESRF and of XRD1 (Elettra) for technical assistance during data collection. This work was supported by the Ministero dell'Istruzione, dell'Universita e della Ricerca under the Programmi Ricerca Interesse Nazionale and the Fondi Investimenti Ricerca di Base and by the Italian National Research Council (CNR). Coordinates have been deposited at the Protein Data Bank (http://www.rcsb.org) for immediate release on publication (ID code 1NZE).
References
- Ananyev G.M., Zaltsman L., Vasko C. & Dismukes G.C. ( 2001) The inorganic biochemistry of photosynthetic oxygen evolution/water oxidation. Biochim. Biophys. Acta, 1503, 52–68. [DOI] [PubMed] [Google Scholar]
- Berman H.M. et al. ( 2002) The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr., 58, 899–907. [DOI] [PubMed] [Google Scholar]
- Berthold D.A., Babcock G.T. & Yocum C.F. ( 1981) A highly resolved oxygen-evolving photosystem II preparation from spinach thylakoid membranes: EPR and electron transport properties. FEBS Lett., 134, 231–234. [Google Scholar]
- Brünger A. et al. ( 1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr., 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, number 4 ( 1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., 50, 760–763. [DOI] [PubMed] [Google Scholar]
- De Vitry C., Olive J., Drapier D., Recouvreur M. & Wollman F.A. ( 1989) Posttranslational events leading to the assembly of photosystem II protein complex: a study using photosynthesis mutants from Chlamydomonas reinhardtii. J. Cell Biol., 109, 991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanotakis D.F., Topper J.N., Babcock G. & Yocum C.F. ( 1984) Water-soluble 17 and 23 kDa polypeptides restore oxygen evolution activity by creating a high-affinity binding site for Ca2+ on the oxidizing side of photosystem II. FEBS Lett., 170, 169–173. [Google Scholar]
- Hankamer B., Barber J. & Boekema E.J. ( 1997) Structure of the oxygen evolving complex of photosystem II by electron microscopy. Annu. Rev. Plant Physiol. Plant Mol. Biol., 48, 641–671. [DOI] [PubMed] [Google Scholar]
- Hankamer B., Morris E., Nield J., Gerle C. & Barber J. ( 2001) Three-dimensional structure of the photosystem II core dimer of higher plants determined by electron microscopy. J. Struct. Biol., 135, 262–269. [DOI] [PubMed] [Google Scholar]
- Holm L. & Sander C. ( 1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Kamiya N. & Shen J.-R. ( 2003) Crystal structure of oxygen-evolving photosystem II from Thermosynechoccus vulcanus at 3.7 Å resolution. Proc. Natl Acad. Sci. USA, 100, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. ( 1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S. & Thornton J.M. ( 1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 25, 283–291. [Google Scholar]
- Leslie A.G.W. ( 1991) In Crystallographic Computing V (eds Moras, D., Podjiarni, A.D. & Thierry, J.P.), 50–61. Oxford Univ. Press, Oxford, UK. [Google Scholar]
- Matthews B. ( 1968) Solvent contents of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- McRee D.E. ( 1999) XtalView/Xfit. A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol., 125, 156–165. [DOI] [PubMed] [Google Scholar]
- Merrit E.A. & Bacon D.J. ( 1997) Raster3D—photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A. & Dodson E.J. ( 1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr., 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Nanba O. & Satoh K. ( 1987) Isolation of a photosystem II reaction center consisting of D-1 and D-2 polypeptides and cytochrome b-559. Proc. Natl Acad. Sci. USA, 84, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp K.A. & Honig B. ( 1991) Protein folding and associations: insight from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet., 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Nield J., Orlova E.V., Morris E.P., Gowen B., van Heel M. & Barber J. ( 2000a) 3D map of the plant photosystem II supercomplex obtained by cryoelectron microscopy and single particle analysis. Nature Struct. Biol., 7, 44–47. [DOI] [PubMed] [Google Scholar]
- Nield J., Kruse O., Ruprecht J., da Fonseca P., Buchel P. & Barber J. ( 2000b) Three-dimensional structure of Chlamydomonas reinhardtii and Synechococcus elongatus photosystem II complexes allows for comparison of their oxygen-evolving complex organization. J. Biol. Chem., 275, 27940–27946. [DOI] [PubMed] [Google Scholar]
- Nield J., Balsera M., De Las Rivas J. & Barber J. ( 2002) Three-dimensional electron cryo-microscopy study of the extrinsic domains of the oxygen-evolving complex of spinach. J. Biol. Chem., 277, 15006–15012. [DOI] [PubMed] [Google Scholar]
- Ono T.-A. ( 2001) Metallo-radical hypothesis for photoassembly of (Mn)4-cluster of photosynthetic oxygen evolving complex. Biochim. Biophys. Acta, 1503, 40–51. [DOI] [PubMed] [Google Scholar]
- Ono T.-A. & Inoue Y. ( 1983) Mn-preserving extraction of 33, 24 and 16 kDa proteins from O2 evolving PSII particles by divalent salt washing. FEBS Lett., 164, 255–260. [Google Scholar]
- Perrakis A., Morris R.J.H. & Lamzin V.S. ( 1999) Automated protein model building combined with iterative structure refinement. Nature Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Rhee K.-H., Morris E.P., Barber J. & Külbrandt W. ( 1998) Three-dimensional structure of the plant photosystem II reaction centre at 8 Å resolution. Nature, 396, 283–286. [DOI] [PubMed] [Google Scholar]
- Seidler A. ( 1996) The extrinsic polypeptides of photosystem II. Biochim. Biophys. Acta, 1277, 35–60. [DOI] [PubMed] [Google Scholar]
- Shen J.R., Qian M., Inoue Y. & Burnap R.L. ( 1988) Functional characterization of Synechocystis sp. PCC 6803 ΔpsbU and ΔpsbV mutants reveals important roles of cytochrome c-550 in cyanobacterial oxygen evolution. Biochemistry, 37, 1551–1558. [DOI] [PubMed] [Google Scholar]
- Shen J.R., Burnap R.L. & Inoue Y. ( 1995) An independent role of cytochrome c-550 in cyanobacterial photosystem II as revealed by double-deletion mutagenesis of the psbO and psbV genes in Synechocystis sp. PCC 6803. Biochemistry, 34, 12661–12668. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. & Berendzen J. ( 1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr., 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouni A., Witt H.-T., Kern J., Fromme P., Krauss N., Saenger W. & Orth P. ( 2001) Crystal structure of photosystem II from Synechoccus elongatus at 3.8 Å resolution. Nature, 409, 739–743. [DOI] [PubMed] [Google Scholar]