

Abstract
The MRE11–RAD50–NBS1 (MRN) protein complex has been linked to many DNA metabolic events that involve DNA double-stranded breaks (DSBs). In vertebrate cells, all three components are encoded by essential genes, and hypomorphic mutations in any of the human genes can result in genome-instability syndromes. MRN is one of the first factors to be localized to the DNA lesion, where it might initially have a structural role by tethering together, and therefore stabilizing, broken chromosomes. This suggests that MRN could function as a lesion-specific sensor. As well as binding to DNA, MRN has other roles in both the processing and assembly of large macromolecular complexes (known as foci) that facilitate efficient DSB responses. Recently, a novel mediator protein, mediator of DNA damage checkpoint protein 1 (MDC1), was shown to co-immunoprecipitate with the MRN complex and regulate MRE11 foci formation. However, whether the initial recruitment of MRN to DSBs requires MDC1 is unclear. Here, we focus on recent developments in MRN research and propose a model for how DSBs are sensed and the cellular responses to them are mediated.
Introduction
DNA damage, such as double-stranded breaks (DSBs), poses a considerable threat to genomic integrity and cell survival. DSBs arise spontaneously during normal DNA processing (for example, during DNA replication and meiosis) and can be induced by a variety of DNA-damaging agents (for example, cancer therapeutic agents such as ionizing radiation (IR) and bleomycin). Checkpoint signalling pathways are required to initially sense the DSB, to amplify this signal and to transduce it to produce the appropriate biological responses. These responses include the induction of a transcriptional programme, the prevention of entry into S phase (the G1/S checkpoint), the slowing of ongoing DNA synthesis (the intra-S checkpoint), the triggering of checkpoints in the G2 and M phases and, in the presence of low to moderate amounts of DNA damage, the enhancement of repair mechanisms by modification and activation of specific repair factors. Alternatively, if the damage is irreparable or an excessive number of lesions is present, checkpoint signalling is also required to induce apoptotic cell death. The two main mechanisms of DSB repair are non-homologous end joining (NHEJ) and homologous recombination (HR; Barnes, 2001; van den Bosch et al., 2002). The malfunction of these mechanisms can result in the fusion of DNA ends that were originally distant from one another in the genome, which generates chromosomal rearrangements such as inversions, translocations and deletions. The resulting disruption of gene expression can perturb normal cell proliferation or result in cell death.
The MRN complex and the repair of DNA damage
The highly conserved MRE11–RAD50–NBS1 (MRN) complex is thought to have a key role in the sensing, processing and repair of DSBs, and orthologues of MRE11 and RAD50 are found in all taxonomic kingdoms. The MRE11 protein has amino-terminal phosphoesterase motifs and two DNA-binding motifs (Fig. 1A). RAD50 contains Walker A and B motifs that are required for nucleotide binding and are separated by two coiled-coil regions that are required for intramolecular interactions (de Jager et al., 2001; Hopfner et al., 2001; Figs 1A,B). Recent studies of the architecture of the human and Pyrococcus furiosis MRE11–RAD50 (MR) complexes revealed that they have a structural role in bridging DNA ends, and possibly sister chromatids, through the coiled-coil regions of RAD50. The Cys-X-X-Cys motif located in the middle of the intramolecular coiled-coil domain of RAD50 functions as a dimerization domain between two RAD50 arms (Fig. 1B; Hopfner et al., 2002). In eukaryotic cells, the MR complex interacts with a third polypeptide, which is known as Xrs2 in yeast and NBS1 in vertebrate cells. NBS1 interacts with MRE11 but its exact position within the MRN complex remains to be determined. The N-terminal region of this 95-kDa protein contains two distinct domains that are often found in cell-cycle checkpoint and DNA-damage response proteins: an FHA (fork-head associated) domain and a BRCT (BRCA1 carboxy-terminal) domain (Fig. 1A). FHA domains in other proteins have been shown to function as specific phospho-residue-binding domains and BRCT domains are also known to mediate protein–protein interactions, for example, the oligomerization of the yeast Rad9 checkpoint protein (Soulier & Lowndes, 1999).
Figure 1.
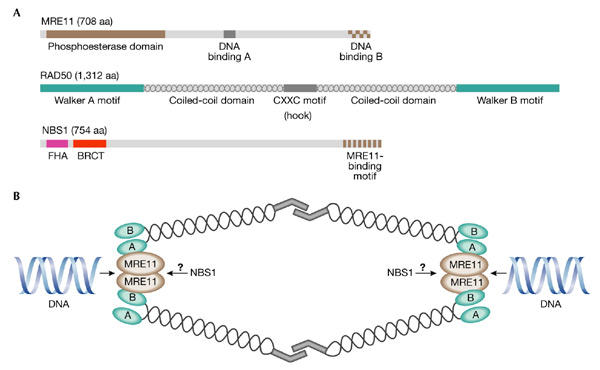
The human MRE11–RAD50–NBS1 complex. (A) The MRE11, RAD50 and NBS1 proteins. Known structural domains for each protein are indicated. Amino termini are to the left. (B) The structure of the MRE11–RAD50–NBS1 (MRN) complex. The MRN complex binds to DNA ends through the MRE11 protein, with the coiled-coil arms of RAD50 extending outwards and interacting with an MRN complex on the other side of the break through their Cys-X-X-Cys (CXXC) motifs. Two MRN complexes are shown for clarity, but many MRN molecules are thought to cluster around the DSB. The many intermolecular interactions between the RAD50 arms are collectively referred to as 'molecular Velcro' (de Jager et al., 2001). A and B, Walker A and B motifs, respectively; aa, amino acids; BRCT, BRCA1 carboxy-terminal domain; FHA, fork-head-associated domain.
In addition to the DNA binding and bridging functions of the MRN complex, in vitro studies have shown enzymatic roles for this complex. Biochemical experiments have shown that the phosphoesterase domain (Fig. 1A) of MRE11 functions as both a single- and doublestranded (ds) DNA endonuclease, as well as a 3′–5′ dsDNA exonuclease (for a review, see D'Amours & Jackson, 2002). RAD50 and NBS1/Xrs2 stimulate the enzymatic activities of MRE11, which are also regulated by ATP (Paull & Gellert, 1999). ATP binding by RAD50 promotes the binding of the MR complex to 3′ overhangs and, in addition, ATP hydrolysis is required to stimulate the cleavage of DNA hairpins, to induce DNA unwinding activity and to alter endonuclease specificity (Paull & Gellert, 1999; de Jager et al., 2002). One or more of these in vitro nuclease activities may well be relevant to the degradation of hairpin structures formed during DNA replication (Lobachev et al., 2002; Mirzoeva & Petrini, 2003).
The MRN complex and disease
The inactivation of components of the yeast equivalent of the MRN complex, known as MRX, affects a remarkable number of DNA metabolic processes, such as HR, NHEJ, telomere maintenance, the formation of meiotic DSBs, the removal of Spo11 from meiotic DSBs, the processing of DSBs and checkpoint regulation (van den Bosch et al., 2002). This suggests that this complex is involved in many, perhaps all, DNA metabolic processes involving DSBs. Therefore, it is not surprising that the disruption of Mre11, Rad50 and Nbs1 in mice results in embryonic lethality (Xiao & Weaver, 1997; Luo et al., 1999; Zhu et al., 2001). However, viable hypomorphic Rad50 and Nbs1 alleles have recently been described (Bender et al., 2002; Williams et al., 2002; Kang et al., 2002). In human cells, the idea that the MRN complex is central to the response to DSBs has been reinforced by the observations that hypomorphic mutations of MRE11 cause ataxia-telangiectasia-like disease (ATLD) and hypomorphic mutations of NBS1 cause Nijmegen breakage syndrome (NBS). Both disorders are phenotypically related to ataxia-telangiectasia (AT), which is caused by mutation of the ATM gene. ATM encodes a protein kinase, that is activated immediately after the induction of DSBs and is a central regulator of all known cellular responses to this damage (Shiloh, 2003). AT patients suffer from cerebellar degeneration—which leads to severe neuromotor dysfunction—immunodeficiency and thymic and gonadal atrophy, and they have a predisposition to cancer (particularly lymphomas). Cells derived from AT patients are extremely hypersensitive to DSB-inducing agents, and are characterized by chromosomal instability and radioresistant DNA synthesis (RDS), which is indicative of the inability to fully induce an intras-phase checkpoint. ATLD patients develop most of the hallmarks of AT, albeit at a later stage and with slower progression. NBS is characterized by immunodeficiency, small head size, mental deficiency, genomic instability, radiation sensitivity and acute predisposition to lymphoid malignancies. The NBS phenotype shows significant overlap with those of AT and ATLD, except that cerebellar degeneration does not occur. Recently, a mutation in RAD50 has been described in a patient with NBS. Thus, all three components of the MRN complex have been implicated in related cancer-prone diseases, reinforcing the central role of this complex in the maintenance of genome stability.
A functional link between the MRN complex and ATM
The similarity of the symptoms seen in ATLD, NBS and AT patients suggests a functional link between the MRN complex and ATM. However, at the cellular level, the extent of RDS is greater in AT cells than in cells from NBS and ATLD patients. This indicates either that the ATM mutations in AT are more penetrant or that ATM activates parallel pathways that are required for an efficient DSB response, one or more of which are independent of MRN. Recent evidence supporting the latter hypothesis is that ATM-dependent phosphorylation of the MRN complex is required to activate the intra-S checkpoint, whereas ATM-dependent, MRN-independent phosphorylation of checkpoint kinase 2 (CHK2) activates a parallel pathway that inhibits DNA synthesis on treatment with DSB-inducing agents (Falck et al., 2002). However, other studies have shown that under some circumstances NBS1 is required for CHK2 activation (Buscemi et al., 2001). The reason for this discrepancy is not clear, but might be related to the radiation dose used and/or the origin of the cell line. Another intriguing link between ATM and MRN is SMC1, a component of the cohesin complex that is required for sister-chromatid cohesion during S phase (Hirano, 2002). IR-induced phosphorylation of SMC1 by ATM is dependent on NBS1 and is required for inhibition of DNA synthesis (Kim et al., 2002; Yazdi et al., 2002), which suggests that modulation of sister-chromatid cohesion may be important for checkpoint regulation after the occurrence of DSBs.
The similar cellular and whole-animal phenotypes that result when either ATM or genes encoding the components of the MRN complex are mutated indicate that the MRN complex is likely to be of central importance to the DSB-specific component of ATM activation. Indeed, interactions between ATM and NBS1 have been shown (Zhao et al., 2000; Gatei et al., 2000). Moreover, ATM phosphorylates NBS1 (primarily at Ser 278 and Ser 343) in response to DNA damage, an event that is required for activation of the S-phase checkpoint (Lim et al., 2000; Zhao et al., 2000). After ATM is recruited to a DSB, it is thought to orchestrate the DSB response by phosphorylating substrates required for the G1/S, intras and G2/M checkpoints, the DSB transcriptional response, the induction of DSB repair, or, in the event of incomplete repair, apoptosis.
Foci and the DSB response
The cellular response to DSBs includes the localization of a complex array of proteins to the region surrounding the lesion in a dynamically organized and timely manner. These proteins form a very large, overlapping, multi-component assembly, known as a focus (Nelms et al., 1998). Two main classes of DSB-dependent foci have been described: the small, early foci that are generally observed in cells from 10 min to 8 h after exposure to a DNA-damaging agent; and the larger, late foci, which start to appear after 4 h, and persist until at least 24 h after treatment (Maser et al., 1997). At present, the physiological role of damage-induced foci is not clear, but the sequestration of proteins in the vicinity of the DSB might be important for amplifying the damage-induced checkpoint signal and for facilitating the repair of persistent damage. Late foci in particular might mark the location of lesions that are particularly long-lasting and difficult to repair.
Members of the recently defined class of mediator proteins, which includes the prototypical yeast checkpoint protein, Rad9, as well as breast cancer associated protein 1 (BRCA1), p53-binding protein 1(53BP1) and the newly identified mediator of DNA damage checkpoint protein 1 (MDC1; Fig. 2), are among the first proteins to be recruited to foci at sites of DSBs. They might function initially as scaffolds for the recruitment of other proteins (for example, factors required for repair and/or for the amplification of the DNA-damage signal), as well as providing a structural link between the DNA ends. It is now clear that as well as being phosphorylated by ATM, each of these mediators colocalizes with phosphorylated H2AX (γ-H2AX), a histone variant that is distributed throughout most of the genome. H2AX contains a highly conserved serine residue in its C terminus (Fig. 2) that is rapidly phosphorylated (Rogakou et al., 1998) after the occurrence of DSBs in an ATM-dependent fashion (Burma et al., 2001; Fernandez-Capetillo et al., 2002). Note, however, that phosphorylation of H2AX is seen in ATM−/− cells at high doses of IR, which suggests the existence of redundant pathways that activate H2AX in the presence of numerous DSBs (Fernandez-Capetillo et al., 2002). After it is modified, this variant histone is crucial for facilitating the focal recruitment of many different proteins to the region of the DNA lesion (Paull et al., 2000).
Figure 2.
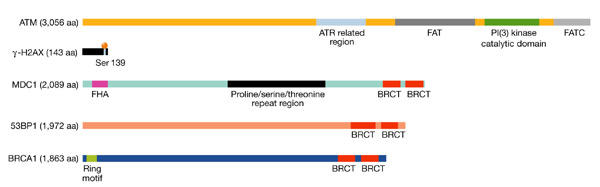
The human ATM, γ-H2AX and mediator proteins. Key molecules involved in the double-stranded-break (DSB) response. Known structural domains for each protein are indicated. The ATM and RAD3-related (ATR)-related region in ATM is of unknown function. 53BP1, p53-binding protein 1; ATM, ataxia telangiectasia mutated; BRCA1, breast-cancer-associated protein 1; BRCT, BRCA1 carboxy terminal; FAT, FRAP/ATM/TRRAP; FATC, FAT carboxy terminal; FHA, fork-head associated; MDC1, mediator of DNA damage checkpoint protein 1; PI(3) kinase, phosphatidylinositol-3-OH kinase.
The race to the break
Most microscopic techniques that are used to examine foci are unable to detect single or small numbers of molecules. Therefore, the initial recruitment of proteins to a DSB might often be missed and the appearance of microscopically visible foci could, in fact, reflect later events in the DSB response. For this reason, it is difficult to determine which of the components are the first to function at the lesion. In undamaged cells, ATM molecules are held together as dimers (or perhaps higher-order multimers) in the nucleoplasm, with the kinase domain of each molecule bound and inactivated by the FRAP/ATM/TRRAP (FAT) domain of another ATM molecule (Bakkenist & Kastan, 2003; see Figs 2 and 3A). DSBspecific alterations to chromatin structure (perhaps due to the unravelling of the torsionally restrained chromosome loop that contains the DSB) have been proposed to activate the soluble, oligomeric ATM kinase (Fig. 3B). This activation can also be achieved by exposing cells to chromatin-modifying drugs or hypotonic stress, treatments that are not believed to result in DSBs. Importantly, the mech-anism by which perturbations in chromatin structure lead to the activation of soluble ATM has yet to be fully described, although it is known to involve intermolecular autophosphorylation of ATM at Ser 1,981 and the consequent disruption of the inactive ATM multimers (Bakkenist & Kastan, 2003). Active, soluble ATM monomers are then free to migrate within the nucleoplasm, phosphorylate any soluble substrates (such as p53) and bind to the ends of broken chromosomes. The then insoluble, chromatin-bound ATM can interact with substrates already at the DSB (such as H2AX, and also NBS1 if the initial localization of the MRN complex to the break is extremely rapid), as well as with other substrates that are subsequently recruited to the DSB (for example, MDC1, 53BP1 and BRCA1; Fig. 3C–E). An important observation is that NBS1 phosphorylation by ATM in response to DNA damage is required for activation of the intras checkpoint, but ATM does not influence the recruitment of MRN to sites of damage in response to DSB induction (Mirzoeva & Petrini, 2001). This indicates an ATM-independent ability of the MRN complex to bind to DSBs (Fig. 3C). Another possibility is that the MR complex is recruited early at the sites of damage and that NBS1 is phosphorylated and subsequently brought to the complex, where it regulates its activity. Thus, at least the initial binding of MR or MRN to the lesion may coincide with, or even precede, that of ATM. The DNA-binding abilities of MRE11 and the structural design of the MR complex are ideally suited to an important early role in sensing and tethering broken chromosomes (Fig. 1B). As suggested by studies in Saccharomyces cerevisiae, the nuclease activities of Mre11 might be important for the initial processing of the break and therefore facilitate its repair (Sugawara & Haber, 1992), although they are not required for the recombinational repair of DSBs (Bressan et al., 1999; Moreau et al., 1999). The subsequent recruitment of MRN to the vicinity of the lesion might be associated with the growing focus rather than the initial sensing of the DSB.
Figure 3.
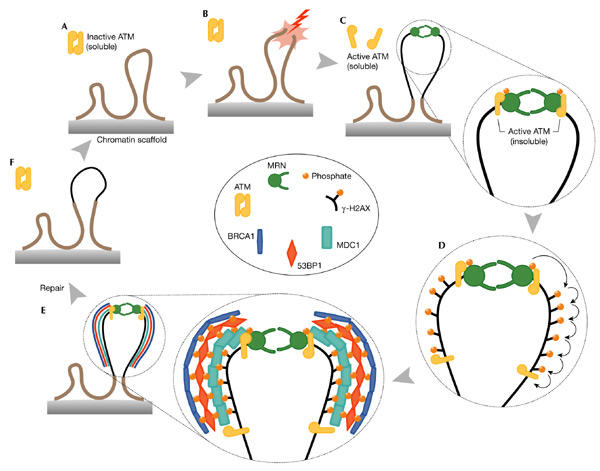
Model of the double-stranded-break response cycle. (A) Undamaged section of a chromosome, showing two chromatin loops and an inactive ATM dimer. (B,C) Induction of a DNA double-stranded break (DSB), modification of chromatin, activation of ATM and recruitment of both ATM and MRE11/RAD50/NBS1 (MRN). The possibility that MRN binds before ATM is shown, but the exact order of events is unknown. The thin black line indicates modified chromatin. (D,E) A wave of H2AX phosphorylation is followed by recruitment of mediators (mediator of DNA damage checkpoint protein 1 (MDC1), p53-binding protein 1 (53BP1) and breast-cancer-associated protein 1 (BRCA1)) to the growing focus, and their ATM-dependent phosphorylation. The molecular architecture of the focus is unknown. (F) Disassembly of the focus, ATM inactivation and chromatin remodelling. The model suggests at least two distinct forms of soluble ATM: an inactive oligomer and an active monomer, and at least two distinct active, insoluble forms: one directly at the lesion and another integral to the growing focus. Note that the MRN complex is also a component of the growing focus but, for clarity, has been omitted here. Complex, persistent lesions are thought to be more difficult to repair, and this is reflected in the size attained by the growing focus. See text for further details. The inset contains a colour-coded key to the molecules shown.
ATM that has been recruited to a broken chromosome can phosphorylate histone H2AX over a distance of several megabases on both sides of the break (Rogakou et al., 1999; Fig. 3D). Recently, it has been shown that loss of H2ax in mice results in genomic instability and in defective recruitment and concentration of Nbs1 and mediator proteins such as 53bp1 and Brca1 to IR-induced nuclear foci (Celeste et al., 2002). However, the observation that cell-cycle checkpoints are largely intact in H2AX−/− cells indicates that the MRN complex and ATM can still detect lesions and signal to downstream effectors in the absence of H2AX phosphorylation. Similarly, phosphorylation of the conserved serine residue in yeast H2a is not required for checkpoint activation (Downs et al., 2000). Nevertheless, the fact that H2AX leads to genome instability and radiation sensitivity (Celeste et al., 2002) suggests that this histone variant is important for the efficient repair of, if not signalling from, DSBs. Thus, the dynamic assembly of large and ever-growing foci may be required for the repair of some lesions, particularly those that are complex and difficult to repair, rather than for the initial DSB signalling. The inability of H2AX−/− cells to concentrate repair proteins in the vicinity of the DSB, which would facilitate efficient repair, might be the critical defect of these cells with respect to genome instability. Alternatively, recent studies in budding yeast suggest that although this histone modification facilitates the efficient repair of DSBs, it is particularly important for the repair of topoisomerase-I-mediated DNA damage (Redon et al., 2003).
The recently identified MRN-interacting protein MDC1 is likely to be involved in the recruitment of other proteins to DSBs, as it is phosphorylated in an ATM-dependent manner and forms foci that colocalize extensively with γ-H2AX within minutes of exposure to IR (Goldberg et al., 2003; Lou et al., 2003; Stewart et al., 2003). Importantly, the downregulation of MDC1 inhibited the formation of foci containing NBS1, 53BP1 and BRCA1, but not the phosphorylation of NBS1. With respect to focus formation, these observations suggest that MDC1 functions upstream of the above-mentioned proteins and is required to regulate their focal accumulation. Therefore, MDC1 facilitates the loading of NBS1, 53BP1, BRCA1, and presumably other proteins, in the immediate vicinity of the DSB. It therefore seems to mediate the assembly of an expanding structure of damage-response proteins that are nucleated laterally on either side of the lesion after a wave of ATM-dependent H2AX phosphorylation (Fig. 3E). At present, it is unclear which proteins are subsequently recruited to MDC1 in the growing nucleoprotein structure around the DSB. However, Wang and colleagues (2002) have provided evidence that BRCA1 does not localize to foci in 53BP1 mutant cells, which suggests a hierarchical order in the recruitment of proteins to the lesion site (Fig. 3E). Similarly, BRCA1 is required for ATM- dependent phosphorylation of NBS1 and CHK2 after exposure to IR, but dispensable for H2AX phoshorylation (Foray et al., 2003). In the enlarging focus, ATM phosphorylates the mediator proteins (for example, MDC1, 53BP1 and BRCA1), which can in turn recruit further damage-response proteins involved in repair, signal transduction or both (for example, RAD51, CHK1, CHK2, and so on).
When repair is complete, disassembly of the focus, presumably involving the removal of some modifications (such as certain phosphorylations), the addition of new modifications (for example, ubiquitination), proteolysis, chromatin remodelling, and so on, is likely to be as tightly regulated as the initial formation of the focus (Fig. 3F).
Conclusions
Chromosome breaks can result from both endogenous and exogenous sources and are a particularly dangerous type of genetic lesion. Emerging data strongly support crucial roles for the MRN complex in preventing the genotoxic consequences of broken chromosomes. As a DSB-specific 'guardian of the genome', the MRN complex is thought to function in the suppression of transformation and malignancy. Perhaps the best indicator of the central importance of the MRN complex in maintaining genome stability is the observation that variants of this complex are found in all taxonomic kingdoms and that, in higher eukaryotes, cell viability is MRN-dependent. Moreover, a pivotal role for the MRN complex in the DSB response is suggested by the observation that adenovirus targets this particular host complex for degradation to facilitate its integration into the host genome (Stracker et al., 2002). By inactivating the MRN complex of the host cell, the virus suppresses the DSB response of the host, thereby allowing time for its integration.
Despite the wealth of recent data on the MRN complex, its precise molecular mechanisms of action remain unclear. For example, is the intact MRN complex always recruited to the DSB or is there a role for MR subcomplexes? Perhaps an MR subcomplex binds to the DNA break and NBS1 is subsequently recruited. Does MDC1 have a role in the initial recruitment of the MR or MRN complexes to the break or is it more important for the subsequent loading of these complexes into the growing focus? Are the enzymatic activities that have been ascribed to MRN components from in vitro studies important for the DSB response? Perhaps the enzymatic activities are more relevant to the suggested role of MRN in DNA replication. Clearly, much remains to be explained.

Acknowledgments
We apologize to our colleagues whose work could not be cited, or was cited indirectly, due to space restrictions. We thank the members of the Genome Stability Laboratory and C. Morrison and A. Pastink for their comments. Research in the Lowndes laboratory is supported by grants from the Health Research Board and the Higher Education Authority.
References
- Bakkenist C.J. & Kastan M.B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature, 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Barnes D.E. (2001) Non-homologous end joining as a mechanism of DNA repair. Curr. Biol., 11, R455–R457. [DOI] [PubMed] [Google Scholar]
- Bender C.F. et al. (2002) Cancer predisposition and hematopoietic failure in Rad50S/S mice. Genes Dev., 16, 2237–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan D.A., Baxter B.K. & Petrini J.H. (1999) The Mre11–Rad50–Xrs2 protein complex facilitates homologous recombination-based doublestrand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 7681–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Murphy M., Kurimasa A. & Chen D.J. (2001) ATM phosphorylates histone H2AX in response to DNA doublestrand breaks. J. Biol. Chem., 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- Buscemi G. et al. (2001) Chk2 activation dependence on Nbs1 after DNA damage. Mol. Cell. Biol., 21, 5214–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A. et al. (2002) Genomic instability in mice lacking histone H2AX. Science, 296, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amours D. & Jackson S.P. (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nature Rev. Mol. Cell Biol., 3, 317–327. [DOI] [PubMed] [Google Scholar]
- de Jager M., van Noort J., van Gent D.C., Dekker C., Kanaar R. & Wyman C. (2001) Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell, 8, 1129–1135. [DOI] [PubMed] [Google Scholar]
- de Jager M., Wyman C., van Gent D.C. & Kanaar R. (2002) DNA end-binding specificity of human Rad50/Mre11 is influenced by ATP. Nucleic Acids Res., 30, 4425–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J.A., Lowndes N.F. & Jackson S.P. (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature, 408, 1001–1004. [DOI] [PubMed] [Google Scholar]
- Falck J., Petrini J.H., Williams B.R., Lukas J. & Bartek J. (2002) The DNA damage-dependent intras phase checkpoint is regulated by parallel pathways. Nature Genet., 30, 290–294. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O. et al. (2002) DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nature Cell Biol., 4, 993–997. [DOI] [PubMed] [Google Scholar]
- Foray N. et al. (2003) A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J., 22, 2860–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatei M. et al. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nature Genet., 25, 115–119. [DOI] [PubMed] [Google Scholar]
- Goldberg M. et al. (2003) MDC1 is required for the intras-phase DNA damage checkpoint. Nature, 421, 952–956. [DOI] [PubMed] [Google Scholar]
- Hirano T. (2002) The ABCs of SMC proteins: two-armed ATPases for chromosome condensation, cohesion, and repair. Genes Dev., 16, 399–414. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Karcher A., Craig L., Woo T.T., Carney J.P. & Tainer J.A. (2001) Structural biochemistry and interaction architecture of the DNA doublestrand break repair Mre11 nuclease and Rad50-ATPase. Cell, 105, 473–485. [DOI] [PubMed] [Google Scholar]
- Hopfner K.P. et al. (2002) The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature, 418, 562–566. [DOI] [PubMed] [Google Scholar]
- Kang J., Bronson R.T. & Xu Y. (2002) Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J., 21, 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.T., Xu B. & Kastan M.B. (2002) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev., 16, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.S. et al. (2000) ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature, 404, 613–617. [DOI] [PubMed] [Google Scholar]
- Lobachev K.S., Gordenin D.A. & Resnick M.A. (2002) The Mre11 complex is required for repair of hairpin-capped doublestrand breaks and prevention of chromosome rearrangements. Cell, 108, 183–193. [DOI] [PubMed] [Google Scholar]
- Lou Z., Chini C.C., Minter-Dykhouse K. & Chen J. (2003) Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem., 278, 13599–13602. [DOI] [PubMed] [Google Scholar]
- Luo G. et al. (1999) Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl Acad. Sci. USA, 96, 7376–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser R.S., Monsen K.J., Nelms B.E. & Petrini J.H. (1997) hMRE11 and hRad50 nuclear foci are induced during the normal cellular response to DNA doublestrand breaks. Mol. Cell. Biol., 17, 6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O.K. & Petrini J.H. (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol., 21, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva O.K. & Petrini J.H. (2003) DNA replication-dependent nuclear dynamics of the mre11 complex. Mol. Cancer Res., 1, 207–218. [PubMed] [Google Scholar]
- Moreau S., Ferguson J.R. & Symington L.S. (1999) The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol., 19, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms B.E., Maser R.S., MacKay J.F., Lagally M.G. & Petrini J.H. (1998) In situ visualization of DNA doublestrand break repair in human fibroblasts. Science, 280, 590–592. [DOI] [PubMed] [Google Scholar]
- Paull T.T. & Gellert M. (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev., 13, 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M. & Bonner W.M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol., 10, 886–895. [DOI] [PubMed] [Google Scholar]
- Redon C., Pilch D.R., Rogakou E.P., Orr A.H., Lowndes N.F. & Bonner W.M. (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep., 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S. & Bonner W.M. (1998) DNA doublestranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem., 273, 5858–68. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Boon C., Redon C. & Bonner W.M. (1999) Megabase chromatin domains involved in DNA doublestrand breaks in vivo. J. Cell Biol., 146, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. (2003) ATM and related protein kinases: safeguarding genome integrity. Nature Rev. Cancer, 3, 155–168. [DOI] [PubMed] [Google Scholar]
- Soulier J. & Lowndes N.F. (1999) The BRCT domain of the S. cerevisiae checkpoint protein Rad9 mediates a Rad9–Rad9 interaction after DNA damage. Curr. Biol., 9, 551–554. [DOI] [PubMed] [Google Scholar]
- Stewart G.S., Wang B., Bignell C.R., Taylor A.M. & Elledge S.J. (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature, 421, 961–966. [DOI] [PubMed] [Google Scholar]
- Stracker T.H., Carson C.T. & Weitzman M.D. (2002) Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature, 418, 348–352. [DOI] [PubMed] [Google Scholar]
- Sugawara N. & Haber J.E. (1992) Characterization of doublestrand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol., 12, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch M., Lohman P.H. & Pastink A. (2002) DNA doublestrand break repair by homologous recombination. Biol. Chem., 383, 873–892. [DOI] [PubMed] [Google Scholar]
- Wang B., Matsuoka S., Carpenter P.B. & Elledge S.J. (2002) 53BP1, a mediator of the DNA damage checkpoint. Science, 298, 1435–1438. [DOI] [PubMed] [Google Scholar]
- Williams B.R., Mirzoeva O.K., Morgan W.F., Lin J., Dunnick W. & Petrini J.H. (2002) A murine model of Nijmegen breakage syndrome. Curr. Biol., 12, 648–653. [DOI] [PubMed] [Google Scholar]
- Xiao Y. & Weaver D.T. (1997) Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the doublestrand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res., 25, 2985–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi P.T., Wang Y., Zhao S., Patel N., Lee E.Y. & Qin J. (2002) SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev., 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. et al. (2000) Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature, 405, 473–477. [DOI] [PubMed] [Google Scholar]
- Zhu J., Petersen S., Tessarollo L. & Nussenzweig A. (2001) Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol., 11, 105–109. [DOI] [PubMed] [Google Scholar]