

Abstract
Current models envision replicative senescence to be under dual control by the p53 and retinoblastoma (RB) tumour-suppressor pathways. The role of the p16INK4a–RB pathway is controversial, and the function of RB in human cells has not been tested directly. We used targeted homologous recombination to knock out one copy of RB in presenescent human fibroblasts. During entry into senescence, RB+/− cells underwent spontaneous loss of heterozygosity and the resultant RB−/− clones bypassed senescence. The extended lifespan phase was eventually terminated by a crisis-like state. The same phenotype was documented for p21 CIP1/WAF1 and p53 heterozygous cells, indicating that loss of function of all three genes results in failure to establish senescence. By contrast, the abolition of p16 function by the expression of a p16-insensitive cyclin-dependent kinase 4 protein or siRNA-mediated knockdown provided only minimal lifespan extension that was terminated by senescence. We propose that p53, p21 and RB act in a linear genetic pathway to regulate cell entry into replicative senescence.
Introduction
Telomere shortening is the molecular mechanism that counts elapsed divisions on an individual cell basis. In addition to replicative exhaustion, growth arrest states resembling senescence can be elicited by a variety of stresses and signalling imbalances (Drayton & Peters, 2002). With few exceptions (von Zglinicki, 2002), these premature senescence states are independent of telomere attrition, and in some cases they might assume a predominant role (Sherr & DePinho, 2000). In culture conditions that minimize extrinsic stresses, telomere attrition appears to be the main driving force behind replicative senescence of human cells (Wright & Shay, 2002). Normal human diploid fibroblasts (HDFs) senesce predominantly in response to telomere attrition even under standard culture conditions (Bodnar et al., 1998; Itahana et al., 2003).
Cell entry into senescence is correlated with the upregulation of the cyclin-dependent kinase (CDK) inhibitors p21CIP1/WAF1 and p16INK4a (hereafter p21 and p16, respectively). Early studies implicated RB, the retinoblastoma tumour-suppressor protein, and p53 in regulating this response (Shay et al., 1991) by showing that expression of viral oncogenes (such as simian virus 40 large T antigen or human papilloma virus E6 and E7 proteins) resulted in an extended lifespan that was terminated by a period of active cell death designated as crisis. The interpretation of these studies is, however, uncertain because viral oncogenes might achieve only partial ablation and are also known to have multiple targets (Funk et al., 1997). Gene targeting of p21 resulted in escape from senescence (Brown et al., 1997; Wei & Sedivy, 1999), whereas a bypass of p16 (by expression of a p16-insensitive CDK4) did not (Brookes et al., 2002; Morris et al., 2002). To analyse further the senescence regulatory circuit and to avoid the ambiguities associated with dominant-interfering proteins (viral or cellular), we chose to construct null alleles of p53 and RB by gene targeting, and to downregulate p16 by expressing short interfering RNA (siRNA).
Results and Discussion
p21+/− (heterozygous) cultures enter senescence normally, but on further incubation colonies of proliferating cells appear and grow out on top of the senescent cell monolayers (Brown et al., 1997). Examination by Southern hybridization showed loss of heterozygosity (LOH) at the p21 locus with preferential retention of the targeted (p21 null) allele, and immunoblotting confirmed loss of p21 protein. On subculture the p21−/− cells eventually underwent a proliferative decline with the characteristics of crisis, as evidenced by a high 5-bromodeoxyuridine (BrdU) labelling index (Wei & Sedivy, 1999). The p21−/− cells retain a functional RB pathway (Wei et al., 2001, 2003).
To examine the generality of the LOH phenomenon we performed an analogous experiment with p53+/− HDFs that were also generated by gene targeting (Bunz et al., 1998). A p53+/− culture was single-cell cloned after gene targeting, and individual colonies were expanded, verified for p53 status, and passaged until proliferation slowed down. In all cases proliferation resumed again, and examination revealed distinct colonies that were never observed in control dishes with p53+/+ cells (Fig. 1A). Sampling at successive passages revealed a progressive loss of the wild-type p53 allele (Fig. 1B), as well as p53 protein (Fig. 1C). Both RB and p16 proteins continued to be expressed normally (Fig. 1C), and the RB pathway appears to be functionally intact (Wei et al., 2001, 2003). Cultures were propagated until no net increase in cell number occurred for a minimum of three weeks, and BrdU labelling was performed. The high labelling index (30–35%) indicated that the cultures were in a crisis-like state. HDFs from patients with Li–Fraumeni syndrome (p53+/−) have also been documented to spontaneously give rise to clones endowed with extended lifespan (Rogan et al., 1995). These results indicate that entry into an extended lifespan is under strong selection in senescent cultures, and that LOH occurs at a high enough frequency in HDFs to make such events readily observable during routine cell culture.
Figure 1.
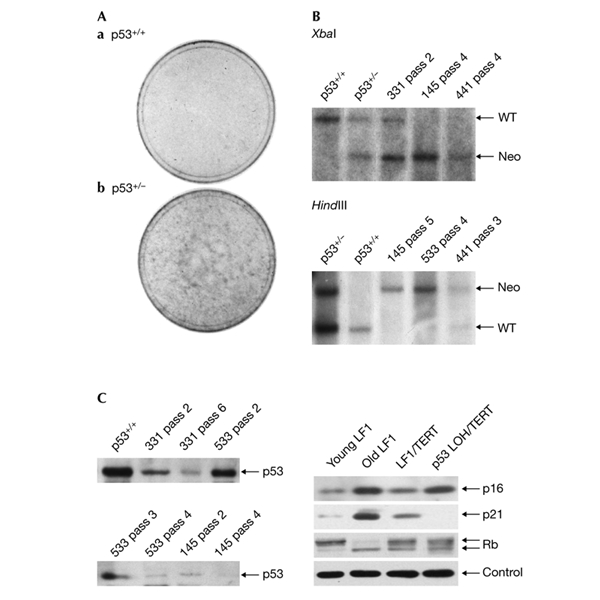
Loss of heterozygosity in p53+/− human fibroblasts. (A) Stained 10-cm culture dishes. Cultures were propagated until cell numbers ceased to increase. At this time most of the cells had a flattened morphology and stained positive for senescence-associated β-galactosidase. Dishes were then further incubated with regular media changes for 3–5 weeks. (B) Southern hybridization analysis. Dishes with colonies (Ab) were trypsinized and subjected to a regular 1:4 passaging regimen (the first trypsinization was designated as passage 1). Cell-line designations and passage numbers are indicated above the lanes. Presenescent p53+/+ and p53+/− cells were used as controls. Southern hybridization was performed with a unique chromosomal probe to the right (3′) of the targeting vector (Fig. 2B). Untargeted wild-type (WT) and Neo vector-targeted (Neo) bands are indicated by arrows. (C) Immunoblot analysis. Left panel, extracts from cells treated as in (B) were harvested at successive passages and immunoblotted with an antibody to p53 protein. Right panel, expression of p16, p21, and RB before and after p53 loss of heterozygosity. Control, actin.
To address directly the role of RB in the regulation of senescence we knocked out one gene copy (Fig. 2A,B) in the embryo lung HDF strain LF1 that was used for the knockouts of p21 and p53. As an additional control we used an independently derived embryo skin HDF strain designated SF1. In both cases we obtained targeted clones with good efficiency and verified them using PCR and Southern hybridization (Fig. 2C). Cells heterozygous for all three genes (p21, p53 and RB) grow indistinguishably from parental cells of roughly comparable age. So, at least by this criterion, all three loss-of-function alleles appear to be fully recessive.
Figure 2.
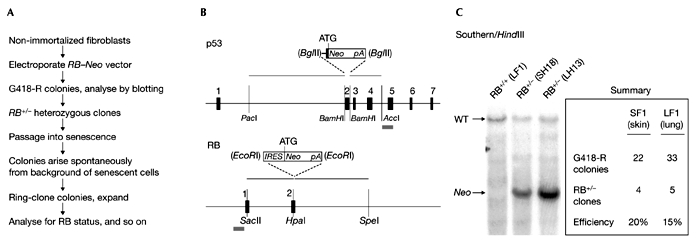
Gene targeting of the human retinoblastoma gene. (A) Overall experimental outline. Steps subsequent to gene targeting that resulted in the selection of clones that bypassed senescence are also indicated. (B) Design of targeting vectors. The genomic loci are shown underneath the vectors and are drawn approximately to scale; black boxes (numbered) represent exons. The grey bars underneath the genomic loci depict the location of Southern hybridization probes. (C) Knockout of one copy of the human RB gene. Southern hybridization was performed with a unique chromosomal probe to the left (5′) of the targeting vector. Further verification of correct chromosomal configuration was performed by PCR of genomic DNA. The summary presents the number of colonies screened and the number of hits recovered. Note that two fibroblast cell strains (SF1 and LF1) were targeted in two independent experiments. SH18 is a derivative of SF1, and LH13 is a derivative of LF1. RB, retinoblastoma.
Both strains of RB+/− cells were passaged into senescence, as evidenced by a downturn in proliferation, positive staining for senescence-associated β-galactosidase (SA-β-gal; Fig. 3A) and a low BrdU labelling index (Fig. 4A). After continued incubation, however, colonies of apparently healthy and proliferating cells were observed, very similar to what was previously seen with p53+/− and p21+/− cultures (Fig. 3A). In situ immunocytochemical detection of RB protein was negative in several independent clones that displayed lifespan extension (Fig. 3B), and loss of RB expression was confirmed by immunoblotting (Fig. 3C). Notably, LOH at the RB locus is frequently observed in vivo during tumour development (Cairns et al., 1991). The frequency of LOH, as estimated by the fraction of cultures that show LOH, and the frequency (number) of LOH colonies was in the order of p53 > p21 > RB (Brown et al., 1997; Bunz et al., 1998). Two out of three RB cultures examined underwent LOH, with colonies in the range of 5–20 per 106 cells.
Figure 3.
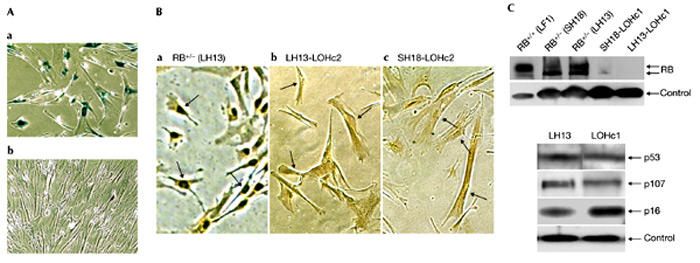
Loss of heterozygosity in RB+/− human fibroblasts. (A) Photomicrographs taken at the time when colonies began to appear. (a) Senescent monolayer of RB+/− cells; (b) a colony with proliferating cells. Both images were taken from the same plate that had been stained for senescence-associated β-galactosidase activity. (B) In situ immunohistochemical detection of RB protein. Colonies of proliferating cells were removed from the primary plates with cloning rings, placed in 24 microtitre wells and after several days of culture expanded into 6-cm dishes. Cells were processed for in situ immunodetection of RB protein after the cultures became established. (a) Presenescent, proliferating LH13 (RB+/−) cells are shown as a positive control. (b) LH13-LOHc2 is a derivative of LH13, and (c) SH18-LOHc2 is a derivative of SH18. Arrowheads point to representative nuclei. (C) Immunoblot analysis. Upper panel, colonies of proliferative cells were picked and expanded as in (B) and immunoblotted with an antibody to RB protein. Cell-line designations are indicated above the lanes. LH13-LOHc1 is a derivative of LH13, and SH18-LOHc1 is a derivative of SH18. Lower panel, expression of p53, p107 and p16 before and after RB LOH. Control, actin; LOH, loss of heterozygosity; RB, retinoblastoma.
Figure 4.
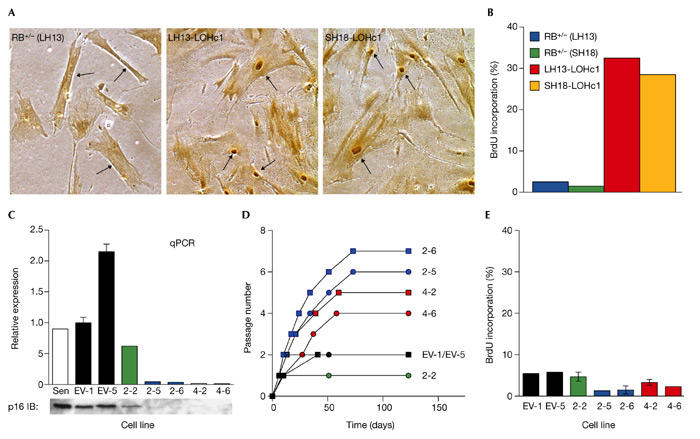
Effect of retinoblastoma loss of heterozygosity and p16 siRNA on establishment of senescence. (A) 5-Bromodeoxyuridine (BrdU) labelling of replicatively exhausted retinoblastoma loss of heterozygosity clones. Colonies were picked and expanded as indicated in Fig. 3. Cultures were incubated for 3–5 weeks after cell numbers ceased to increase before labelling. Labelling with BrdU was for 48 h. BrdU incorporation was detected immunohistochemically and scored by microscopic observation. Representative photomicrographs of stained plates are shown, with arrows indicating nuclei. (B) Quantification of BrdU incorporation presented in graphical format. (C) Quantification of p16 mRNA and protein in clones expressing p16 short-interfering (si)RNA. Bar graph, quantitative real-time reverse transcription–PCR (qPCR); lower panel, immunoblotting (IB). Clonal cell lines infected with target 2 (2-2, 2-5, 2-6) and target 4 (4-2, 4-6) siRNA are shown. The same cell lines are analysed in panels (C–E). Sen, noninfected senescent LF1 cells; EV-1, EV-5, cell lines infected with empty vector. (D) Lifespan extension of clones expressing p16 siRNA. Passage numbers were set to zero at the point when each clone was first expanded into a 10-cm dish. Each passage corresponds to approximately two population doublings. Four additional clones expressing empty vector senesced before the 10-cm dish stage and are not shown. (E) BrdU labelling of replicatively exhausted p16 siRNA-expressing clones. Experiment was performed as indicated in (A).
Clones that had entered into an extended lifespan phase eventually ceased proliferating. To address whether this downturn was caused by entry into senescence or into a crisis-like state we performed BrdU labelling. In all cases RB−/− clones displayed high BrdU labelling indexes (approximately 30%, Fig. 4A,B), which are in the same range as observed in analogous clones arising from p21+/− and p53+/− cultures. Terminally non-proliferative cultures contained cells with condensed and/or blebbing nuclei as evidenced by Hoechst dye staining, but we had insufficient material to perform more rigorous analyses of apoptosis. Cultures could not be kept for extended periods of time, and deteriorated slowly over a period of 4–6 weeks. Although these phenotypes are consistent with a crisis-like state, it is also possible that the cultures were arrested in one of the 'intermediate M' (Mint) states (Bond et al., 1999; Morris et al., 2002). The high BrdU labelling index is, however, clearly indicative of an escape from typical replicative senescence and entry into an extended lifespan phase.
We followed numerous clones (five each from LH13 and SH18 cultures) until apparent growth arrest and determined the final number of cells. Assuming that each clone was founded by a single cell that converted to RB−/− status, the population doublings (PD) required to reach the observed terminal cell number would be approximately 16 PD for eight clones, 19 PD for one clone and 23 PD for one clone. These values are minimum estimates, as it is unlikely that we recovered all the cells during ring cloning or that subsequent passaging was 100% efficient. The values are in the same range as the 20–30 PD lifespan extension previously documented for p21−/− cells.
To investigate other factors that may have contributed to escape from replicative senescence, we determined the expression of p53, p21, p16, and the RB family members p107 and p130 in the two clones (LH13-LOH-c1 and SH18-LOH-c2) that were previously analysed for RB expression. p53, p21 and p107 were expressed normally (Fig. 3C), and p130 expression was below the level of detection in all cells (including parental RB+/−) cells. As expected, p16 expression was increased in the absence of RB. Finally, we examined the expression of telomerase activity using the telomeric repeat amplification protocol (TRAP) and found no evidence for its upregulation following loss of RB expression.
We next investigated whether eliminating p16 function had the same effect as loss of RB. In spite of repeated attempts we failed to knock out p16 by gene targeting. This is because the promoter-less method of gene targeting used in somatic cells requires that the drug-selectable marker (for example, Neo) in the vector be expressed from the promoter of the chromosomal target locus (Sedivy & Dutriaux, 1999). In cases where the target gene is expressed at very low concentrations (as is p16) the homologously integrated vector does not express sufficient Neo protein. To phenocopy knockout of p16 we first expressed a fusion protein between the p16-insensitive R24C mutant of CDK4 and cyclin D1 (DK; Wei et al., 2003). Expression of DK in LF1 cells elicited a limited lifespan extension of 5–10 PD, at the end of which the cultures entered into a typical senescence-like state. LF1/DK cells were immune to the inhibitory effects of ectopic p16 expression, indicating that the expressed DK protein had biological activity (Wei et al., 2003), and the DK protein continued to be expressed in senescent cells. While this work was in progress, similar studies using R24C and wild-type CDK4, or naturally occurring point mutations in the INK4a gene, reached the same conclusions (Brookes et al., 2002; Morris et al., 2002).
Because the DK gain-of-function intervention could have unsuspected effects we sought to ablate p16 mRNA expression using siRNA. To avoid affecting the expression of the p14ARF protein also encoded by the CDKN2A locus, siRNA targets in the p16INK4a -specific exon 1α were chosen. The short hairpin RNA (shRNA)-encoding DNA templates were cloned in the retrovirus vector pRetroSuper (Brummelkamp et al., 2002) and expressed in presenescent LF1 cells. All four siRNAs elicited p16 mRNA knockdown in pools of drug-resistant cells, but to variable extents. To achieve higher and more consistent knockdown, the best two pools were single-cell cloned, and multiple clones of each were expanded into cell lines. Cells infected with empty vector and subjected to the same manipulations were used as controls. Expression of p16 siRNA elicited >95% reduction of p16 mRNA and protein (Fig. 4C) in individual clones. Reduction of p16 expression was highly correlated with an extended lifespan (Fig. 4D). Lifespan extension was modest (5–10 PD) and was terminated by typical senescence, as evidenced by low BrdU incorporation (Fig. 4E). We therefore conclude that loss of p16 expression is insufficient to allow escape from replicative senescence.
Importantly, different expression levels of p21 and p16 may elicit different cell-cycle responses, especially when combined with other proliferative or anti-proliferative signals. The ectopic expression of either p16 or p21 alone is sufficient, if the protein is present at high enough concentrations, to establish cell-cycle arrest (McConnell et al., 1998). The concentrations of p16 continued to increase in p21−/− cells during the extended lifespan of the cells to levels beyond those seen in normal senescent cells, but the cultures continued to proliferate (albeit at reduced rates) (Brown et al., 1997). However, a similar concentration of p16 was strongly inhibitory in presenescent p21+/+ cells, as were further increases in p16 concentration in p21−/− cells (Wei et al., 2001). Likewise, p21 expression increased during the short lifespan extension of LF1/DK cells, and accumulated to higher levels than those seen in normal senescent cells. So, the failure of the loss of p16 function to bypass senescence can be explained by the accumulation of p21 to levels that are sufficiently high to inhibit all G1 CDKs, whereas the failure of endogenous p16 to arrest p21−/− cells can be attributed to insufficient accumulation in the window of opportunity preceding the onset of crisis. Because in both situations the outcome is dictated by the expression levels of p16 or p21, it is easy to imagine that the situation could change in different cell types or under different culture conditions. Nevertheless, in all situations the genetic hierarchy that best accommodates existing data is to place RB downstream of both p16 and p21.
Replicative senescence, defined as the loss of proliferative capacity elicited by continuous subculture, is triggered largely by telomere shortening in HDFs, but by stress and/or signalling imbalances in mouse embryo fibroblasts (MEFs; Sherr & DePinho, 2000). In MEFs, the senescence response is regulated primarily by the Arf–p53 pathway, and neither Arf nor p53 knockout MEFs undergo senescence. Although p16 is induced in senescent MEFs, it apparently has a minor role, and p16 knockout MEFs do not immortalize with increased frequency (Sharpless et al., 2001). Initial studies indicated that disruption of the RB gene did not appreciably affect the senescence response in MEFs, with only a concurrent inactivation of all three RB family members (pRB, p107, p130) eliciting spontaneous immortalization (Sage et al., 2000). This requirement for concurrent activation is apparently caused by adaptation during embryonic development, given that an acute conditional deletion of RB in MEFs can overcome the senescence programme (Sage et al., 2003). So, in the light of recent evidence, it appears that p16 and RB influence the senescence of HDFs and MEFs in similar ways, in spite of the fact that the responses may be triggered by different stimuli.
In summary, replicative senescence has been typically described as being regulated by two pathways, the p53–p21 pathway and the p16–RB pathway, that act in concert to achieve growth arrest. On the basis of the genetic evidence presented in this communication we propose that the signalling cascade is better described by a linear relationship, with p53 at the top and RB at the bottom, and p16 forming a branch that enters at the level of RB.
Methods
LF1 and SF1 cells were cultured as previously described (Brown et al., 1997). Gene targeting and retroviral transduction of human fibroblasts have been described in detail (Hemmer et al., 2003; Wei et al., 2003). The sources of antibodies were: anti-CDK4 (sc-260), anti-p21 (sc-397), anti-p53 (sc-6243), anti-p16 (sc-759), Santa Cruz; anti-RB (G3-245), PharMingen; anti-actin (N-350), Amersham; anti-BrdU (555 627), Becton Dickinson. Immunoblotting was performed as previously described (Wei et al., 2001, 2003). Equivalent loading of lanes was determined by Coomassie or SYPRO staining of duplicate gels, and the efficiency of transfer was verified by direct staining of the membranes with Ponceau red. Where indicated, loading was further verified by immunoblotting for actin. BrdU labelling and SA-β-gal staining were performed as previously described (Wei & Sedivy, 1999). Quantitative real-time reverse transcription–PCR (qPCR) was performed using the SYBR green system in an Applied Biosystems 7700 Sequence Detector as previously described (O'Connell et al., 2003). The p16 target sequences were: target 1, CGCACCGAATAGTTACGGT; target 2, TAGTTACGGTCGGAGGCCG; target 3, GGAGCAGCATGGAGCCTTC; target 4, GTGCTCGGAGTTAATAGCA.
Acknowledgments
This work was supported by grant AG16694 from the National Institutes of Health to J.M.S.
References
- Bodnar A.G. et al. ( 1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- Bond J.A. et al. ( 1999) Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol., 19, 3103–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes S. et al. ( 2002) INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J., 21, 2936–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.P., Wei W. & Sedivy J.M. ( 1997) Bypass of senescence after disruption of p21 CIP1/WAF1 gene in normal diploid human fibroblasts. Science, 277, 831–834. [DOI] [PubMed] [Google Scholar]
- Brummelkamp T.R., Bernards R. & Agami R. ( 2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell, 2, 243–247. [DOI] [PubMed] [Google Scholar]
- Bunz F. et al. ( 1998) The induction of p21 by p53 is required for sustained G2 arrest following DNA damage. Science, 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Cairns P., Proctor A.J. & Knowles M.A. ( 1991) Loss of heterozygosity at the RB locus is frequent and correlates with muscle invasion in bladder carcinoma. Oncogene, 6, 2305–2309. [PubMed] [Google Scholar]
- Drayton S. & Peters G. ( 2002) Immortalisation and transformation revisited. Curr. Opin. Genet. Dev., 12, 98–104. [DOI] [PubMed] [Google Scholar]
- Funk J.O. et al. ( 1997) Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev., 11, 2090–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer R.M., Wei W., Dutriaux A. & Sedivy J.M. ( 2003) in Methods in Molecular Medicine, Vol. 223 (ed. El-Deiry, W.S.) 187–206. Humana, Towata, New Jersey, USA. [Google Scholar]
- Itahana K. et al. ( 2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell. Biol., 23, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell B.B., Starborg M., Brookes S. & Peters G. ( 1998) Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr. Biol., 8, 351–356. [DOI] [PubMed] [Google Scholar]
- Morris M., Hepburn P. & Wynford-Thomas D. ( 2002) Sequential extension of proliferative lifespan in human fibroblasts induced by over-expression of CDK4 or 6 and loss of p53 function. Oncogene, 21, 4277–4288. [DOI] [PubMed] [Google Scholar]
- O'Connell B.C. et al. ( 2003) A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem., 278, 12563–12573. [DOI] [PubMed] [Google Scholar]
- Rogan E.M. et al. ( 1995) Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell. Biol., 15, 4745–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J. et al. ( 2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev., 14, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J., Miller A.L., Perez-Macera P.A., Wysocki J.M. & Jacks T. ( 2003) Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature, 424, 223–227. [DOI] [PubMed] [Google Scholar]
- Sedivy J.M. & Dutriaux A. ( 1999) Gene targeting and somatic cell genetics: a rebirth or a coming of age? Trends Genet., 14, 88–90. [DOI] [PubMed] [Google Scholar]
- Sharpless N.E. et al. ( 2001) Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature, 413, 86–91. [DOI] [PubMed] [Google Scholar]
- Shay J.W., Pereira-Smith O.M. & Wright W.E. ( 1991) A role for both RB and p53 in the regulation of human senescence. Exp. Cell Res., 196, 33–39. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. & DePinho R.A. ( 2000) Cellular senescence: mitotic clock or culture shock? Cell, 102, 407–410. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T. ( 2002) Oxidative stress shortens telomeres. Trends Biochem. Sci., 27, 339–344. [DOI] [PubMed] [Google Scholar]
- Wei W. & Sedivy J.M. ( 1999) Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp. Cell Res., 253, 519–522. [DOI] [PubMed] [Google Scholar]
- Wei W., Hemmer R.M. & Sedivy J.M. ( 2001) Role of p14ARF in replicative and induced senescence of human fibroblasts. Mol. Cell. Biol., 21, 6748–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W., Jobling W.A., Chen W., Hahn W.C. & Sedivy J.M. ( 2003) Abolition of cyclin–dependent kinase inhibitors p16 Ink4a and p21 Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol. Cell. Biol., 23, 2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright W.E. & Shay J.W. ( 2002) Historical claims and current interpretations of replicative aging. Nature Biotechnol., 20, 682–688. [DOI] [PubMed] [Google Scholar]