

Abstract
In eukaryotes, a family of six homologous minichromosome maintenance (MCM) proteins has a key function in ensuring that DNA replication occurs only once before cell division. Whereas all eukaryotes have six paralogues, in some Archaea a single protein forms a homomeric assembly. The complex is likely to function as a helicase during DNA replication. We have used electron microscopy to obtain a three-dimensional reconstruction of the full-length MCM from Methanobacterium thermoautotrophicum. Six monomers are arranged around a sixfold axis, generating a ring-shaped molecule with a large central cavity and lateral holes. The channel running through the molecule can easily accommodate double-stranded DNA. The crystal structure of the amino-terminal fragment of MCM and a model for the AAA+ hexamer have been docked into the map, whereas additional electron density suggests that the carboxy-terminal domain is located at the interface between the two domains. The structure suggests that the MCM complex is likely to act in a different manner to traditional hexameric helicases and is likely to bear more similarity to the SV40 large T antigen or to double-stranded DNA translocases.
Introduction
Eukaryotic DNA replication is a highly coordinated and tightly regulated process. A complex network of proteins, under strict control of the cell cycle, is required to ensure that each origin is used only once and that no segment of DNA is left unreplicated or undergoes multiple rounds of replication. A key function is performed by the minichromosome maintenance (MCM) proteins, a family of six homologous polypeptides (MCM2–7) conserved in all eukaryotic genomes (Kearsey & Labib, 1998; Tye, 1999). The regeneration of replication competence is associated with the rebinding of MCMs to chromatin at the end of mitosis. Not only do MCM proteins have a function during the initiation of DNA replication, but also they are essential in the elongation step (Labib et al., 2000).
Within the MCM homologues, a central region of about 230 amino-acid residues (Fig. 1) shows the highest degree of conservation and belongs to the AAA+ superfamily of ATPases (Neuwald et al., 1999). AAA+ proteins share a common structure, consisting of an amino-terminal mononucleotide-binding fold and a carboxy-terminal helical domain. In several cases, they have been demonstrated to form oligomeric assemblies, showing a strong preference for hexameric structures with the nucleotide-binding pocket at the interface between subunits.
Figure 1.
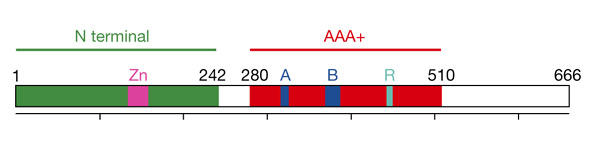
A schematic representation of the Methanobacterium thermoauto-trophicum MCM sequence. The sequence of the MtMCM consists of 666 amino-acid residues and can be divided into an amino-terminal domain (in green, amino acids 1–242), including a Zn motif (in magenta), and an AAA+ ATPases domain (in red, amino acids 280–510), which includes the Walker A and B motifs (A and B, in blue) and the arginine finger (R, in cyan). The ticks on the scale bar correspond to 100 amino-acid residues.
A growing body of evidence suggests that the principal enzymatic activity of the MCM complex is unwinding DNA, making it the primary candidate for the elusive replicative helicase (Labib & Diffley, 2001). The most direct piece of evidence that MCM proteins have helicase activity comes from studies of the homologous proteins in Archaea. Whereas eukaryotes all have six different, albeit homologous, MCM proteins, their number in Archaea is variable, with several species having only a single paralogue. Three independent studies have shown that it is possible to overexpress in a bacterial system the MCM protein from Methanobacterium thermoautotrophicum (MtMCM) and that the purified recombinant protein has a processive helicase activity (Kelman et al., 1999; Chong et al., 2000; Shechter et al., 2000). The sequences of the archaeal proteins are closest to those of the eukaryotic MCM4 paralogues, with 35% sequence identity between MtMCM and the human and yeast MCM4.
The crystal structure of the N-terminal domain of MtMCM has been determined to 3.0-Å resolution (Fletcher et al., 2003) and shows a dodecameric architecture, with two hexameric rings juxtaposed in a head-to-head configuration. A three-dimensional (3D) reconstruction obtained from negatively stained particles revealed unexpected heptameric rings (Yu et al., 2002).
Here, we present a 23-Å 3D reconstruction of the full-length MtMCM from negatively stained particles. The electron density map shows a hexameric architecture, consistent with the crystallographic and biochemical results, with the MtMCM monomers arranged symmetrically around a sixfold axis. The crystal structure of one hexamer of the N-terminal domain could be clearly fitted into the electron density. A model of the hexameric AAA+ ATPase ring has also been built, and additional density can be assigned to the C-terminal region, which is sandwiched between the N-terminal and AAA+ domains. The large channel that is present in the N-terminal domain continues throughout the entire structure and is wide enough to accommodate double-stranded DNA (dsDNA). A large central cavity in the middle of the complex and the presence of lateral holes on the side bear a striking resemblance to the recently determined structure of the large T antigen from the SV40 simian virus (Li et al., 2003), indicating that these two complexes might act in a similar way despite the lack of sequence homology.
Results
Overall structure
Negatively stained samples of MtMCM were examined by electron microscopy (EM). An initial reference-free alignment and multivariate statistical analysis of images identified the presence of clear sixfold (C6) symmetry (Fig. 2A). An initial recontruction performed in the absence of any symmetry constraints confirmed the presence of a sixfold axis (Fig. 2B), which was then applied throughout the refinement procedure (Fig. 2C–F). The final reconstruction includes 2,400 particles and has a nominal resolution of 23 Å.
Figure 2.

Overview of the three-dimensional reconstruction procedure. (A) The three predominant eigenimages. The first closely resembles the total average of the data set used as a first reference for the translational alignment; the two subsequent eigenimages illustrate the most important differences existing within the data set, providing clear evidence of sixfold symmetry. (B) Average of 100 top-view images rotationally aligned to the first class averages at the beginning of the image processing analysis, without (left) and with (right) sixfold symmetry imposed. (C) Examples of original images of MtMCM stained with uranyl acetate. These images are members of the class averages shown in the row below (protein is white). (D) Class averages (characteristic views) obtained by multi-reference alignment and classification. (E) Reprojections of the 3D structure in the Euler-angle directions found for the class averages in (C). (F) Surface representations of the 3D reconstruction viewed from directions identical to the Euler directions assigned to the corresponding class averages in (C). The far-left images correspond to the top view; the far-right images to the bottom view. Scale bar, 100 Å.
MtMCM forms hexameric rings having a roughly cylindrical shape, with a maximum diameter of 130 Å and a height of 100 Å (Fig. 3). The overall architecture is consistent with six MtMCM monomers arranged around a sixfold axis with a wide central channel running throughout the entire molecule. The channel has a diameter of 45–60 Å in the centre of the molecule and is constricted at both ends to about 25 Å. Despite the overall cylindrical shape, there is a clear asymmetry between the top and bottom halves of the molecule (Fig. 3). At the bottom, the central channel is encircled by a rim, surrounded by a groove, whereas the top half of the molecule is dome-shaped.
Figure 3.
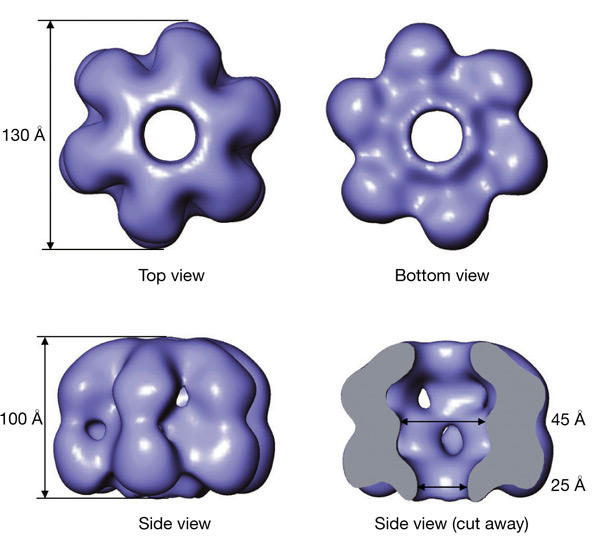
Three-dimensional reconstruction of MtMCM at 23-Å resolution. A surface representation including 100% of the expected volume of the electron density obtained from the three-dimensional reconstruction of the MtMCM complex is shown in different orientations. The overall dimensions for the complex are given. The protein monomers assemble into a hexameric ring around a wide central channel, with a clear asymmetry between the top and bottom views. A side view has been cut open to reveal the large central chamber and the channel spanning the entire length of the molecule.
Model fitting
The asymmetry of the molecule permits the unambiguous identification of the locations of the N-terminal and C-terminal domains of the protein. The crystal structure of one hexamer of the N-terminal domain (Fletcher et al., 2003) fits well into the bottom half of the density, with the ridge corresponding to the Zn motifs (Fig. 4). The large central channel, which in the crystal structure is highly positively charged, extends throughout the entire molecule, with both ends wide enough to accommodate dsDNA.
Figure 4.
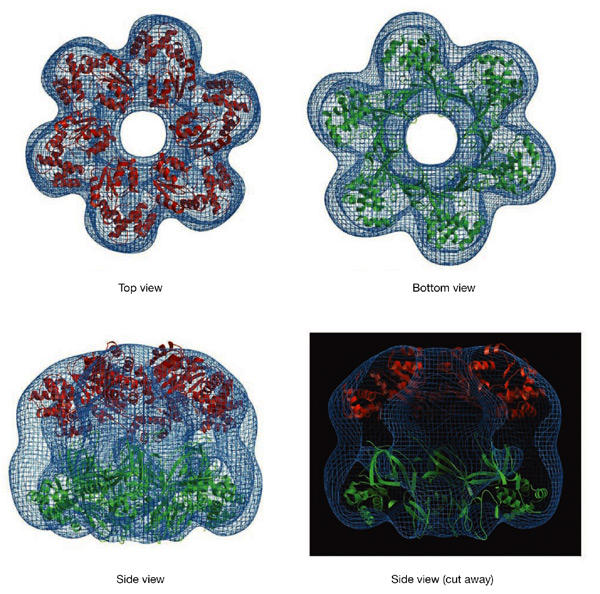
Model fitting. The electron density obtained for the MtMCM reconstruction is shown in blue. The crystal structure of the amino-terminal domain of MtMCM, shown in green, has been placed in the bottom half of the reconstruction, with the Zn motifs fitting into the ridge surrounding the bottom aperture of the central channel. A hexameric model of the core AAA+ domain of RuvB has been placed in the top half of the density (shown in red).
Because no crystal structure is available for the ATPase domain of an MCM protein, the atomic structure of another AAA+ domain was used as an approximate model for the top half of the map. Only a handful of AAA+ proteins have been crystallized in their hexameric forms, and in each case the size of the central channel is smaller than that observed in our 3D reconstruction. We obtained a good fit to our electron density map by using the monomeric structure of RuvB (Putnam et al., 2001) arranged in a hexamer, based on the oligomeric structure of p97 (Zhang et al., 2000). Only the AAA+ structural core (domains I and II, residues 30–256) was used. This generates a hexamer that mimics the dome-like shape of the top half of the MtMCM reconstruction and with a similar-sized central hole (Fig. 4). In this orientation the N terminus of the AAA+ domain is facing towards the C terminus of the MtMCM N-terminal domain.
The atomic coordinates of the N-terminal domain of MtMCM and the modelled RuvB hexamer were used to generate calculated electron density maps to 20-Å resolution. A comparison between the EM 3D reconstruction and these maps reveals additional density forming a belt around the interface between the two domains (Fig. 5A). This residual density probably accommodates the connection between the two domains and the C-terminal domain consisting of about 150 amino-acid residues (Fig. 1). In our model the C terminus of the modelled AAA+ domain points towards the residual density, which is consistent with this assignment. We therefore propose that the C-terminal domain of MtMCM is located at the interface between the two domains (Fig. 5B).
Figure 5.
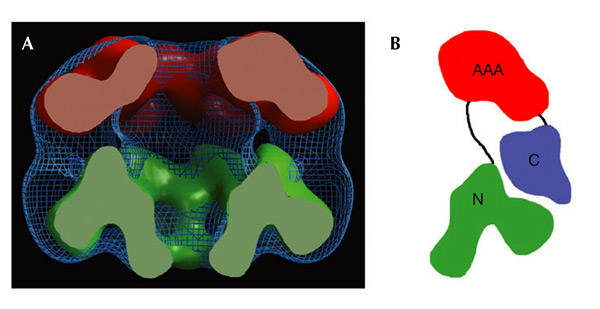
Location of the MtMCM carboxy-terminal domain. (A) The three-dimensional reconstruction of the full-length MtMCM (in blue) is compared with electron density maps calculated to 20 Å from the atomic coordinates of the amino-terminal domain (in green) and the AAA+ domain (in red). A 'belt' of unassigned electron density is located at the interface between the two domains and could accommodate the C-terminal 150 amino-acid residues (Fig. 1). (B) Schematic diagram showing the proposed arrangement of the C-terminal domain (in blue) in a MtMCM monomer.
Discussion
Symmetry and stoichiometry of the MCM complex
Despite the wealth of information on the MCM complex, there is still considerable uncertainty about the composition and stoichiometry of the active species and the relative roles of the six paralogues. Most of the results on the eukaryotic MCM complex are consistent with the presence in vivo of an active complex containing one copy of each of the six subunits. However, the failure to detect helicase activity in vitro in the presence of the hexameric complex and the weak non-processive activity associated with the MCM4/6/7 subcomplex led to the proposal of a structural model within which the MCM2/3/5 and MCM4/6/7 subunits are arranged as two staggered trimers (Schwacha & Bell, 2001). In these, three subunits in a trimer are responsible for the activity and the other three, arranged in a second trimer, have a regulatory function.
Although the situation is simpler in archaeal cells containing a single MCM protein, the MtMCM has been shown by size-exclusion chromatography to have a size corresponding to a dodecamer (Kelman et al., 1999; Chong et al., 2000; Shechter et al., 2000), whereas the protein from Sulfolobus solfataricus forms single hexamers in solution (Carpentieri et al., 2002). The crystal structure of the N-terminal domain of MtMCM revealed a dodecameric architecture (Fletcher et al., 2003), whereas a 3D reconstruction showed an unexpected sevenfold arrangement around a central axis (Yu et al., 2002), which is not consistent with biochemical studies.
Our EM reconstruction unambiguously shows six monomers arranged in a hexameric ring around a large central channel (Figs 3,4). The construct used to express the MtMCM was provided by T. Gautier and the purification protocol was similar to that previously reported (Shechter et al., 2000). Size-exclusion chromatography consistently shows the presence of a major peak corresponding to a dodecamer and a minor peak corresponding to a hexamer (supplementary information online figure 1). However, under the conditions used for the EM imaging, the sample seemed to be largely hexameric. A possible explanation might be the different concentration of the sample, affecting the hexamer–dodecamer equilibrium, because the EM studies were performed on a sample that was far more dilute. A dodecameric model of the full-length MtMCM can easily be extrapolated from the dodecameric crystal structure of the N-terminal domain (supplementary information online figure 2).
The discrepancy between the hexameric and dodecameric architectures is therefore likely to be the result of a dynamic equilibrium. It is more difficult to reconcile the heptameric structure obtained in the 3D reconstruction of MtMCM (Yu et al., 2002). In contrast, proteins making oligomeric rings with six or more subunits have occasionally been observed to give rise to unusual stoichiometries, which can easily be achieved with minor adjustments of the subunit–subunit interface. Small differences in the expression or purification protocols might well shift the equilibrium towards one of these forms. The fact that only 20% of the assemblies could be used for the reconstruction in Yu et al. (2002) is another indication that the sample used was probably not homogeneous and was subject to a high degree of polymorphism. In addition, the approach by Penczek et al. (1992) used by Yu et al. (2002) to determine the symmetry, instead of the technically more sound unbiased global symmetry analysis approach used here (Dube et al., 1993; van Heel et al., 2000), might have exacerbated the problem and biased the results towards the heptameric subpopulation in the sample.
The structure presented here displays an exact sixfold symmetry as we exploited the presence of the rotational symmetry from an early stage in the refinement procedure by imposing C6 symmetry. This might have masked any subtle departure from sixfold symmetry in the hexamer. Such deviations from exact sixfold symmetry have indeed been observed in the crystal structure of the bacteriophage T7 gene 4 helicase and have been proposed to have functional implications (Singleton et al., 2000). A biochemical analysis of ATP binding and hydrolysis suggests that the eukaryotic MCM2–7 hexamer has ATP-binding modes with high, medium and low affinities (Schwacha & Bell, 2001). Although in the eukaryotic complex there is an intrinsic asymmetry due to the presence of six different, albeit homologous, subunits, it is unlikely that the kinetic behaviour of the archaeal hexamer is markedly different.
Mechanism of helicase activity
The exact details of the mechanisms of action of hexameric helicases are not completely understood, but one proposal is that the hexameric ring encircles single-stranded DNA (ssDNA), and DNA unwinding is caused by the ATP-driven unidirectional translocation of the ring along one of the DNA strands, with the consequent displacement of the other strand (Patel & Picha, 2000). In MtMCM, a significant ATPase activity is detected only in the presence of DNA and is stimulated by either ssDNA or dsDNA (Kelman et al., 1999; Chong et al., 2000; Shechter et al., 2000). Furthermore, the N-terminal domain of MtMCM was shown to bind dsDNA in a gel-shift assay, and this binding was abolished by mutation of residues facing the central channel (Fletcher et al., 2003).
Despite the evidence suggesting that the MCM complex is responsible for DNA unwinding during replication, a few findings are puzzling, giving rise to the 'MCM paradox' (Hyrien et al., 2003). MCM proteins seem not to move with the replication fork but are instead located on unreplicated DNA. Moreover, each origin recruits 10–40 MCM complexes that become distributed over a large region of DNA, so the number of chromatin-bound MCM complexes far exceeds the number of replication origins. This led to the proposal that the helicase activity might be achieved through a novel mechanism of action, in which many MCM hexameric rings translocate along dsDNA, are then anchored to a fixed nuclear structure, and act as a rotary pump twisting and unwinding the spooled dsDNA (Laskey & Madine, 2003).
A major difference between the classical hexameric helicase mechanism of action and the rotary-pump model is that the latter requires MCM to be loaded and translocated along dsDNA. Our 3D reconstruction shows the presence of a very large central channel (Fig. 3), far wider than the channel previously seen in hexameric ring helicases such as T7 gene 4 protein, RepA and Rho helicases (Patel & Picha, 2000). The size of the channel varies between 25 and 60 Å and can easily accommodate duplex DNA. We have found a good fit to the EM map for a hexameric model of the AAA+ domain of RuvB, a motor protein that helps in the remodelling of Holliday junctions with a mechanism reminiscent of the proposed MCM rotary-pump model.
A similar but distinct mode of action has been proposed for the large T antigen helicase in the simian virus SV40. The crystal structure of a fragment of the SV40 helicase (Li et al., 2003) reveals a hexameric architecture with a large, internal positively charged 'chamber' in the centre of the molecule and 'holes' between monomers on the side walls of the central chamber. The mechanism of action proposed involves two head-to-head hexamers pumping dsDNA into the central chamber, whereas ssDNA is extruded through the lateral holes. Both the central chamber and the lateral holes are present in MtMCM (Fig. 3; supplementary information online figure 3), suggesting that the two helicases might function in a similar manner.
Methods
Protein expression and purification.
In the expression construct used for producing the recombinant protein, the M. thermoautotrophicum gene encoding MCM was subcloned into a pET21b vector (Novagen) to produce the full-length polypeptide fused to a non-cleavable C-terminal histidine tag. Protein expression and purification were performed as described by Shechter et al. (2000).
Electron microscopy data acquisition and image processing.
Samples of MtMCM to a concentration of 0.1–0.2 μM were applied to carbon-coated grids and negatively stained with 3% uranyl acetate. Images were recorded under a low dose condition on a Philips CM100 electron microscope at an accelerating voltage of 100 kV and a magnification of ×51,000. Good micrographs were digitized on a patchwork densitometer (Image Science Software, GmbH, Berlin, Germany) leading to a pixel size of 2.35 Å on the specimen scale. All image analysis was performed with the IMAGIC-5 package (van Heel et al., 1996). Eigenimages of the multivariate statistical analysis of translationally centred images reveal a clear sixfold symmetry (Fig. 2A; Dube et al., 1993), which was applied throughout the subsequent analysis. Further details are given in supplementary information online.
Fitting of X-ray structures.
Fitting of one hexamer of the N-terminal MtMCM (Protein Database (PDB) accession code ) into the map was performed automatically by the real-space correlation algorithm ESSENS (Kleywegt & Jones, 1997), which led to a unique position. An arbitrary choice was made during image processing about the handedness of the structure; although the final reconstruction does not show a strong asymmetry at this resolution, the fit of the crystal structure of the MtMCM N-terminal domain agrees slightly better for the map obtained than for the corresponding mirror image.
The model for the hexameric AAA+ domain was built by placing residues 30–256 of a monomer of RuvB (PDB, ; Putnam et al., 2001) on a hexagonal framework based on the crystal structure of p97 (PDB, ; Zhang et al., 2000). This model was fitted into the EM electron density map as described for the N-terminal domain.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor7400010-s1.pdf).
Supplementary Material
Supplementary information online figures 1, 2, 3 and 4
Acknowledgments
We thank J. Gautier (Columbia University, New York) for providing the MtMCM expression construct; X. Chen for making available to us the coordinates of the N-terminal domain of MtMCM before release by PDB; and P. Brick for critical reading of the manuscript. This work was supported by a Wellcome Trust Grant to S.O. and by BBSRC grants to M.v.H.
References
- Carpentieri F. De Felice M. De Falco M. Rossi M. & Pisani F.M. ( 2002) Physical and functional interaction between the mini-chromosome maintenance-like DNA helicase and the single-stranded DNA binding protein from the crenarchaeon Sulfolobus solfataricus. J. Biol. Chem. 277, 12118–12127. [DOI] [PubMed] [Google Scholar]
- Chong J.P. Hayashi M.K. Simon M.N. Xu R.M. & Stillman B. ( 2000) A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc. Natl Acad. Sci. USA 97, 1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube P. Tavares P. Lurz R. & van Heel M. ( 1993) The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J. 12, 1303–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher R.J. Bishop B.E. Leon R.P. Sclafani R.A. Ogata C.M. & Chen X.S. ( 2003) The structure and function of MCM from archaeal M. thermoautotrophicum. Nature Struct. Biol. 10, 160–167. [DOI] [PubMed] [Google Scholar]
- Hyrien O. Marheineke K. & Goldar A. ( 2003) Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. BioEssays, 25, 116–125. [DOI] [PubMed] [Google Scholar]
- Kearsey S.E. & Labib K. ( 1998) MCM proteins: evolution, properties, and role in DNA replication. Biochim. Biophys. Acta, 1398, 113–136. [DOI] [PubMed] [Google Scholar]
- Kelman Z. Lee J.K. & Hurwitz J. ( 1999) The single minichromosome maintenance protein of Methanobacterium thermoautotrophicum DeltaH contains DNA helicase activity. Proc. Natl Acad. Sci. USA 96, 14783–14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt G.J. & Jones T.A. ( 1997) Template convolution to enhance or detect structural features in macromolecular electron-density maps. Acta Crystallogr. D 53, 179–185. [DOI] [PubMed] [Google Scholar]
- Labib K. & Diffley J.F. ( 2001) Is the MCM2–7 complex the eukaryotic DNA replication fork helicase? Curr. Opin. Genet. Dev 11, 64–70. [DOI] [PubMed] [Google Scholar]
- Labib K. Tercero J.A. & Diffley J.F. ( 2000) Uninterrupted MCM2–7 function required for DNA replication fork progression. Science, 288, 1643–1647. [DOI] [PubMed] [Google Scholar]
- Laskey R.A. & Madine M.A. ( 2003) A rotary pumping model for helicase function of MCM proteins at a distance from replication forks. EMBO Rep., 4, 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. Zhao R. Lilyestrom W. Gai D., Zhang R DeCaprio J.A. Fanning E. Jochimiak A. Szakonyl G. & Chen X.S. ( 2003) Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature, 423, 512–518. [DOI] [PubMed] [Google Scholar]
- Neuwald A.F. Aravind L. Spouge J.L. & Koonin E.V. ( 1999) AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res., 9, 27–43. [PubMed] [Google Scholar]
- Patel S.S. & Picha K.M. ( 2000) Structure and function of hexameric helicases. Annu. Rev. Biochem., 69, 651–697. [DOI] [PubMed] [Google Scholar]
- Penczek P. Radermacher M. & Frank J. ( 1992) Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy, 40, 33–53. [PubMed] [Google Scholar]
- Putnam C.D. Clancy S.B. Tsuruta H. Gonzalez S. Wetmur J.G. & Tainer J.A. ( 2001) Structure and mechanism of the RuvB Holliday junction branch migration motor. J. Mol. Biol., 311, 297–310. [DOI] [PubMed] [Google Scholar]
- Schwacha A. & Bell S.P. ( 2001) Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol. Cell, 8, 1093–1104. [DOI] [PubMed] [Google Scholar]
- Shechter D.F. Ying C.Y. & Gautier J. ( 2000) The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum delta H minichromosome maintenance protein. J. Biol. Chem., 275, 15049–15059. [DOI] [PubMed] [Google Scholar]
- Singleton M.R. Sawaya M.R. Ellenberger T. & Wigley D.B. ( 2000) Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell, 101, 589–600. [DOI] [PubMed] [Google Scholar]
- Tye B.K. ( 1999) MCM proteins in DNA replication. Annu.Rev. Biochem., 68, 649–686. [DOI] [PubMed] [Google Scholar]
- van Heel M. Harauz G. Orlova E.V. Schmidt R. & Schatz M. ( 1996) A new generation of the IMAGIC image processing system. J. Struct. Biol., 116, 17–24. [DOI] [PubMed] [Google Scholar]
- van Heel M. et al. ( 2000) Single-particle electron cryo-microscopy: towards atomic resolution. Q. Rev. Biophys., 33, 307–369. [DOI] [PubMed] [Google Scholar]
- Yu X. VanLoock M.S. Poplawski A. Kelman Z. Xiang T. Tye B.K. & Egelman E.H. ( 2002) The Methanobacterium thermoautotrophicum MCM protein can form heptameric rings. EMBO Rep., 3, 792–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. ( 2000) Structure of the AAA ATPase p97. Mol.Cell 6, 1473–1484. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information online figures 1, 2, 3 and 4